Preliminary Enrichment Fine-mapping Results
Nicholas Knoblauch
2019-07-02
Last updated: 2019-07-10
Checks: 7 0
Knit directory: ptb_workflowr/
This reproducible R Markdown analysis was created with workflowr (version 1.4.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190313) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: (dellxps's conflicted copy 2019-06-19).Rhistory
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .drake/
Ignored: analysis/PTB_features (dellxps's conflicted copy 2019-06-19).Rmd
Ignored: cache/
Ignored: data/NA_character_.RDS
Ignored: data/ag_df.RDS
Ignored: data/annotations.h5
Ignored: data/big_hic_t.tsv.gz
Ignored: data/eqtl_gwas_file.tsv.gz
Ignored: data/exons.bed
Ignored: data/fat_eqtl.tsv.gz
Ignored: data/gencode.v28.annotation.gff3.gz
Ignored: data/genes.bed
Ignored: data/gwas_file.tsv.gz
Ignored: data/gwas_ptb_file.tsv.gz
Ignored: data/gwas_ptb_file_i.tsv.zstd
Ignored: data/gwas_scz_file.tsv.gz
Ignored: data/hic_t.tsv.gz
Ignored: data/little_gwas_i.tsv.zstd
Ignored: data/new_cache_log.RDS
Ignored: data/ngwas_df.h5
Ignored: data/old_cache_log.RDS
Ignored: data/retd.RDS
Ignored: data/shuffeqtl_gwas_file.tsv.gz
Ignored: data/split_d.h5
Ignored: data/sub_gwas_ptb.tsv.gz
Ignored: data/susie_r.RDS
Ignored: data/ut_eqtl.tsv.gz
Ignored: old_index/
Untracked files:
Untracked: #.Rprofile#
Untracked: #make.R#
Untracked: HDF5.ttl
Untracked: R/#handlers.R#
Untracked: analysis/23_and_me.Rmd
Untracked: analysis/awd.RData
Untracked: analysis/dataset_SNPs_mismatches-reference.txt
Untracked: analysis/enrichment_analysis.Rmd
Untracked: analysis/ldshrink_drake.Rmd
Untracked: analysis/mws.RData
Untracked: analysis/test_knit_code.Rmd
Untracked: analysis/test_knit_code2.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/.snakemake/
Untracked: code/Snakefile
Untracked: code/Untitled.ipynb
Untracked: code/bed_monet.R
Untracked: config/#packages.yaml#
Untracked: config/#workflow_params_gardner.yaml#
Untracked: config/workflow_params_desktop.yaml
Untracked: ptb_cache/
Untracked: sub_drake.sh
Unstaged changes:
Modified: R/config.R
Modified: R/functions.R
Modified: R/plan.R
Modified: analysis/PTB_features.Rmd
Modified: analysis/multifeature.Rmd
Deleted: code/README.md
Modified: slurm_clustermq.tmpl
Modified: torque_clustermq.tmpl
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 48698b5 | CreRecombinase | 2019-07-10 | wflow_publish(“analysis/multifeature_update.Rmd”) |
| html | 791ea4a | Nicholas Knoblauch | 2019-07-01 | Build site. |
| Rmd | d61a9ee | Nicholas Knoblauch | 2019-07-01 | wflow_publish(“analysis/multifeature_update.Rmd”) |
The loci
The genome was broken in to 1703 regions (using Pickrell’s ldetect). Regions were broken into subregions if they contained greater than 5000 SNPs, unless this would result in a subregion with fewer than 50 SNPs. This means that region size varied from 50 SNPs to 5049 SNPs. The functional enrichment program torus was run individually on each of the following features:
Warning in bind_rows_(x, .id): Unequal factor levels: coercing to characterWarning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vectorFine mapping with susie was then run on the top 5 loci, after generating priors using torus. susie was run assuming 1 causal variant per locus.
The genomic annotations
A variable selection procedure (forward selection) was used to come up with a set of four features to use to generate a per-variant prior for susie.
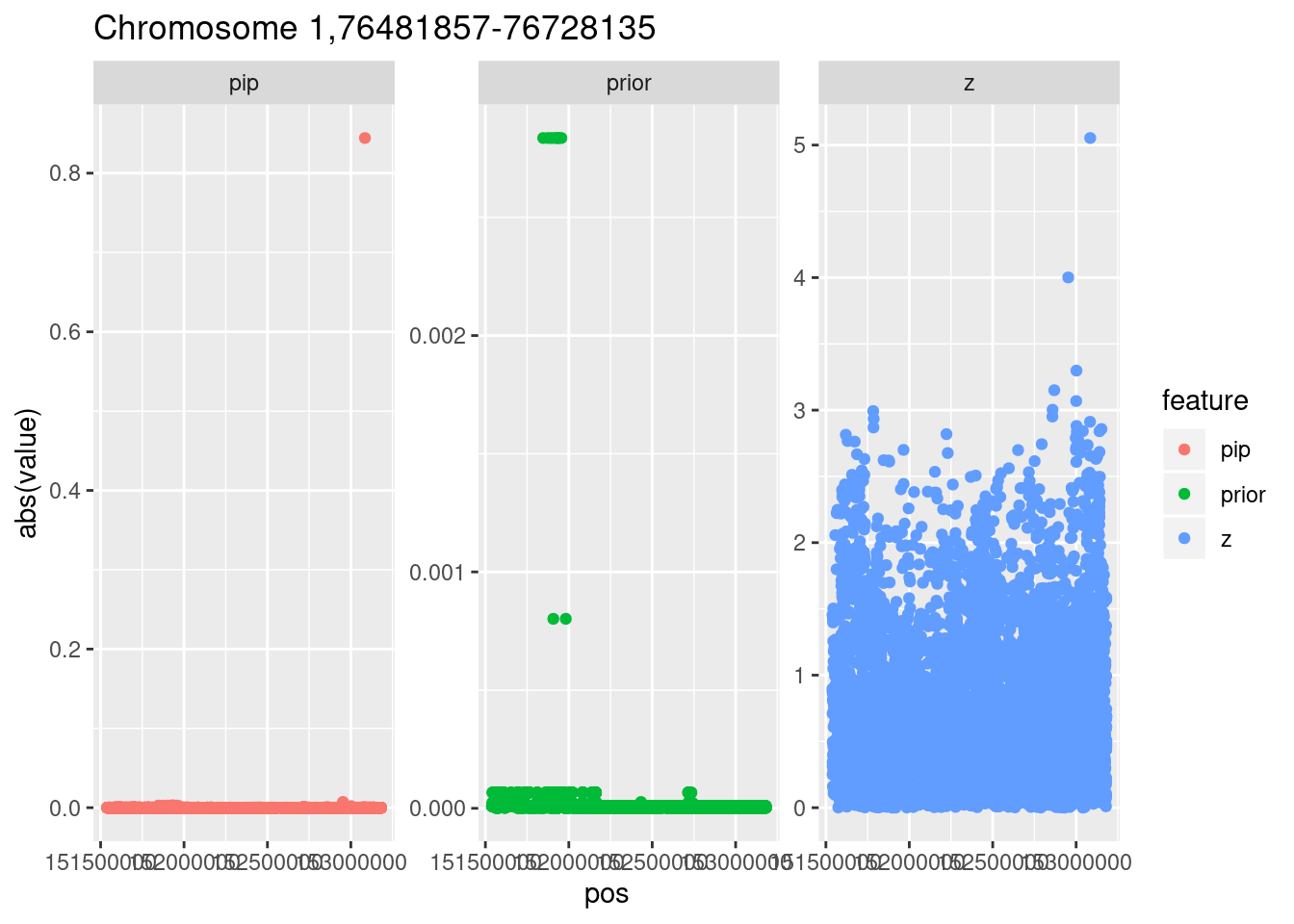
Locus 1
The top hit is on chromosome 1.
| Version | Author | Date |
|---|---|---|
| 791ea4a | Nicholas Knoblauch | 2019-07-01 |
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Manjaro Linux
Matrix products: default
BLAS/LAPACK: /usr/lib/libopenblas_haswellp-r0.3.6.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] tidyselect_0.2.5 RSSp_0.9.0.9000 ldmap_0.0.0.9000
[4] daprcpp_1.0 ldshrink_1.0-1 archive_1.0.0
[7] vroom_1.0.2.9000 RSQLite_2.1.1 glue_1.3.1
[10] drake_7.4.0.9000 fs_1.3.1 susieR_0.8.1.0525
[13] here_0.1 forcats_0.4.0 stringr_1.4.0
[16] dplyr_0.8.3 purrr_0.3.2 readr_1.3.1
[19] tidyr_0.8.3 tibble_2.1.3 ggplot2_3.2.0
[22] tidyverse_1.2.1 dbplyr_1.4.2 MonetDBLite_0.6.1
[25] DT_0.7
loaded via a namespace (and not attached):
[1] nlme_3.1-140 bitops_1.0-6 lubridate_1.7.4
[4] bit64_0.9-7 filelock_1.0.2 httr_1.4.0
[7] rprojroot_1.3-2 GenomeInfoDb_1.20.0 tools_3.6.1
[10] backports_1.1.4 R6_2.4.0 DBI_1.0.0
[13] lazyeval_0.2.2 BiocGenerics_0.30.0 colorspace_1.4-1
[16] wavethresh_4.6.8 withr_2.1.2 bit_1.1-14
[19] compiler_3.6.1 git2r_0.26.1 cli_1.1.0
[22] rvest_0.3.4 xml2_1.2.0 labeling_0.3
[25] scales_1.0.0 digest_0.6.20 txtq_0.1.3
[28] rmarkdown_1.13 XVector_0.24.0 pkgconfig_2.0.2
[31] htmltools_0.3.6 htmlwidgets_1.3 rlang_0.4.0.9000
[34] readxl_1.3.1 rstudioapi_0.10 shiny_1.3.2
[37] generics_0.0.2 jsonlite_1.6 crosstalk_1.0.0
[40] RCurl_1.95-4.12 magrittr_1.5 GenomeInfoDbData_1.2.1
[43] Matrix_1.2-17 Rcpp_1.0.1 munsell_0.5.0
[46] S4Vectors_0.22.0 stringi_1.4.3 whisker_0.3-2
[49] yaml_2.2.0 MASS_7.3-51.4 storr_1.2.2
[52] zlibbioc_1.30.0 grid_3.6.1 blob_1.1.1
[55] promises_1.0.1 parallel_3.6.1 crayon_1.3.4
[58] lattice_0.20-38 haven_2.1.0 hms_0.4.2
[61] knitr_1.23 pillar_1.4.2 igraph_1.2.4.1
[64] GenomicRanges_1.36.0 base64url_1.4 codetools_0.2-16
[67] stats4_3.6.1 evaluate_0.14 RcppParallel_4.4.3
[70] modelr_0.1.4 httpuv_1.5.1 cellranger_1.1.0
[73] gtable_0.3.0 assertthat_0.2.1 xfun_0.7
[76] mime_0.7 xtable_1.8-4 RcppEigen_0.3.3.5.0
[79] broom_0.5.2 later_0.8.0 memoise_1.1.0
[82] IRanges_2.18.1 workflowr_1.4.0