Figure 5
Florian Wuennemann
Last updated: 2024-03-21
Checks: 7 0
Knit directory: mi_spatialomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230612) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4c4da66. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/deprecated/.DS_Store
Ignored: analysis/molecular_cartography_python/.DS_Store
Ignored: analysis/seqIF_python/.DS_Store
Ignored: analysis/seqIF_python/pixie/.DS_Store
Ignored: analysis/seqIF_python/pixie/cell_clustering/
Ignored: annotations/.DS_Store
Ignored: annotations/SeqIF/.DS_Store
Ignored: annotations/molkart/.DS_Store
Ignored: annotations/molkart/Figure1_regions/.DS_Store
Ignored: annotations/molkart/Supplementary_Figure4_regions/.DS_Store
Ignored: data/.DS_Store
Ignored: data/140623.calcagno_et_al.seurat_object.rds
Ignored: data/Calcagno2022_int_logNorm_annot.h5Seurat
Ignored: data/IC_03_IF_CCR2_CD68 cell numbers.xlsx
Ignored: data/Traditional_IF_absolute_cell_counts.csv
Ignored: data/Traditional_IF_relative_cell_counts.csv
Ignored: data/pixie.cell_table_size_normalized_cell_labels.csv
Ignored: data/results_cts_100.sqm
Ignored: data/seqIF_regions_annotations/
Ignored: data/seurat/
Ignored: omnipathr-log/
Ignored: output/.DS_Store
Ignored: output/mol_cart.harmony_object.h5Seurat
Ignored: output/molkart/
Ignored: output/proteomics/
Ignored: output/results_cts.lowres.125.sqm
Ignored: output/seqIF/
Ignored: pipeline_configs/.DS_Store
Ignored: plots/
Ignored: references/.DS_Store
Ignored: renv/.DS_Store
Ignored: renv/library/
Ignored: renv/staging/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/figures.Figure5.Rmd) and
HTML (docs/figures.Figure5.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 008f74a | FloWuenne | 2024-03-21 | Fixed links in data analysis and figures pages. |
Load data
pca_res <- readRDS("./output/proteomics/proteomics.pca_res.rds")
vsn_mat <- fread("./output/proteomics/proteomics.vsn_norm_proteins.tsv")
limma_res <- fread("./output/proteomics/proteomics.limma.full_statistics.tsv")
mi_pathways <- fread("./output/proteomics/proteomics.pathway_results.MIiz_MIremote.tsv")Subfigure A
Subfigure B - Principal component analysis
pcs <- as.data.frame(pca_res$x)
pcs$sample <- colnames(vsn_mat[,1:11])
pcs <- pcs %>%
mutate("group" = if_else(grepl("control",sample),"control",
if_else(grepl("MI_IZ",sample),"MI_IZ","MI_remote"))
)
## Set order of groups
pcs$group <- factor(pcs$group,
levels = c("control","MI_remote","MI_IZ"))
## Plot PCs
pca_plot <- ggplot(pcs,aes(PC1,PC2)) +
geom_point(size = 5,pch = 21,color = "black", aes(fill = group)) +
ggforce::geom_mark_ellipse(color = "white",aes(fill = group)) +
expand_limits(y = c(-50, 40),
x = c(-40,80)) +
scale_fill_manual(values = proteome_palette,
labels = c("Control","MI_remote","MI_IZ")) +
labs(color = "Group") +
guides(fill=guide_legend(title="Group")) +
theme(legend.position = "none")
pca_plot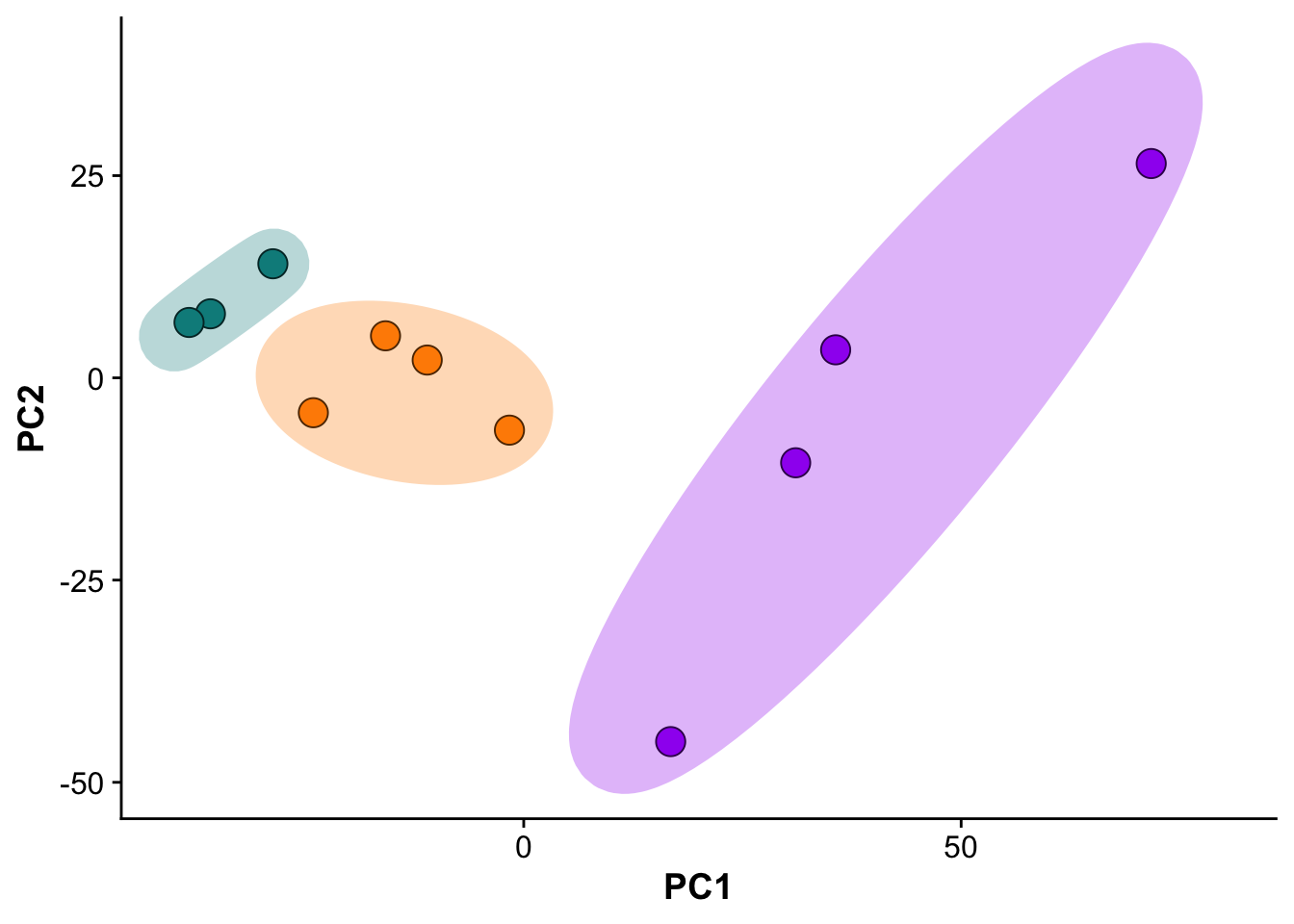
save_plot(filename = "./plots/Figure_5.pca_plot.pdf",
plot = pca_plot,
base_asp = 1,
base_height = 4)Subfigure C - Volcano plot: Remote vs control
limma_mi_remote <- subset(limma_res,analysis == "MI_IZ_vs_MI_remote")
limma_remote_control <- subset(limma_res,analysis == "MI_remote_vs_control")
limma_mi_control <- subset(limma_res,analysis == "MI_IZ_vs_control")
## Which proteins are differentially expressed in MI vs remote but not in MI vs remote but not in remote vs control?
iz_uniq <- setdiff(subset(limma_mi_remote,adj.P.Val < 0.05)$gene,subset(limma_remote_control,adj.P.Val < 0.05)$gene)
limma_mi_remote_uniq <- subset(limma_mi_remote, gene %in% iz_uniq) %>%
subset(adj.P.Val < 0.05) %>%
arrange(desc(logFC))
## Get proteins from Coagulation pathway from pathway analysis results to highlight on volcano plot
mh_gsea_net <- readRDS("./references/mh.all.v2023.1.Mm.symbols.sets.rds")
pathway <- 'HALLMARK_COAGULATION'
df <- mh_gsea_net %>%
filter(source == pathway) %>%
arrange(target)
path_de_inter <- sort(intersect(limma_mi_remote$gene,df$target))# top_10_proteins <- limma_mi_remote %>%
# arrange(desc(logFC)) %>%
# top_n(wt = logFC, 10)
# top_10_proteins <- top_10_proteins$gene
# bottom_10_proteins <- limma_mi_remote %>% arrange(desc(logFC))
# bottom_10_proteins <- tail(bottom_10_proteins,n=10)
manual_labeled_proteins <- c("Thbd","Vwf","Coro1a","Thbs1")
limma_mi_remote <- limma_mi_remote %>%
# mutate("label_protein" = if_else(gene %in% path_de_inter & adj.P.Val < 0.05 & (logFC > 1.25 | logFC < 0), gene, ""))
mutate("label_protein" = if_else(gene %in% manual_labeled_proteins,gene,""))
limma_mi_remote$label_protein <- gsub("Vwf","vWF",limma_mi_remote$label_protein)
volc_limma_IZ_remote <- plot_pretty_volcano(limma_mi_remote,
pt_size = 2,
plot_title = "",
sig_thresh = 0.05,
col_pos_logFC = proteome_palette[['MI_IZ']],
col_neg_logFC = proteome_palette[['MI_remote']]) +
# geom_point(data = subset(limma_mi_remote, gene %in% path_de_inter),pch = 21, color = "black", size = 4) +
geom_label_repel(box.padding = 0.5, max.overlaps = Inf) +
geom_vline(xintercept= 0 , linetype = 2)
## Interactive plotly plot to view genes on points
# plot_ly(data = limma_mi_remote, x = ~logFC, y = ~-log10(adj.P.Val),
# text = ~paste("Gene: ", gene))
save_plot(filename = "./plots/Figure_5.volcano_plot.pdf",
plot = volc_limma_IZ_remote,
base_asp = 1.3,
base_height = 3.25)Warning: Removed 58 rows containing missing values (`geom_label_repel()`).## Volcano plot for schema
volc_limma_remote_control <- ggplot(data=limma_remote_control,
aes(x= logFC, y= -log10(pval))) +
geom_point(size = 3, color = "black")+
theme(axis.title = element_text(size =20),
axis.text = element_blank(),
axis.ticks = element_blank()) +
geom_vline(xintercept = 0, linetype = 2)
volc_limma_remote_controlWarning: Removed 114 rows containing missing values (`geom_point()`).
save_plot(filename = "./plots/Figure_4.volcano_schema.pdf",
plot = volc_limma_remote_control,
base_asp = 1.4,
base_height = 3)Warning: Removed 114 rows containing missing values (`geom_point()`).Subfigure D - Pathway enrichment for MI_IZ vs MI_remote
sig_pathways_mi <- subset(mi_pathways,p_value <= 0.05) %>%
arrange(desc(score)) %>%
dplyr::select(-statistic,-condition) %>%
subset(score > 3 | score < -3)
sig_pathways_mi$source <- gsub("HALLMARK_","",sig_pathways_mi$source)
sig_pathways_mi$source <- gsub("_"," ",sig_pathways_mi$source)
path_plot <- ggplot(sig_pathways_mi, aes(x = reorder(source, score), y = score)) +
geom_bar(aes(fill = score),color = "black", stat = "identity") +
scale_fill_gradient2(low = "darkorange", high = "purple",
mid = "white", midpoint = 0) +
# scale_fill_viridis(option = "F", direction = 1) +
theme(axis.title = element_text(face = "bold", size = 20),
axis.text.x = element_text(hjust = 1, size =20, face= "bold"),
axis.text.y = element_text(size =16),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
legend.text = element_text(size =20),
legend.title = element_text(size =20)) +
xlab("Pathways") +
coord_flip()
path_plot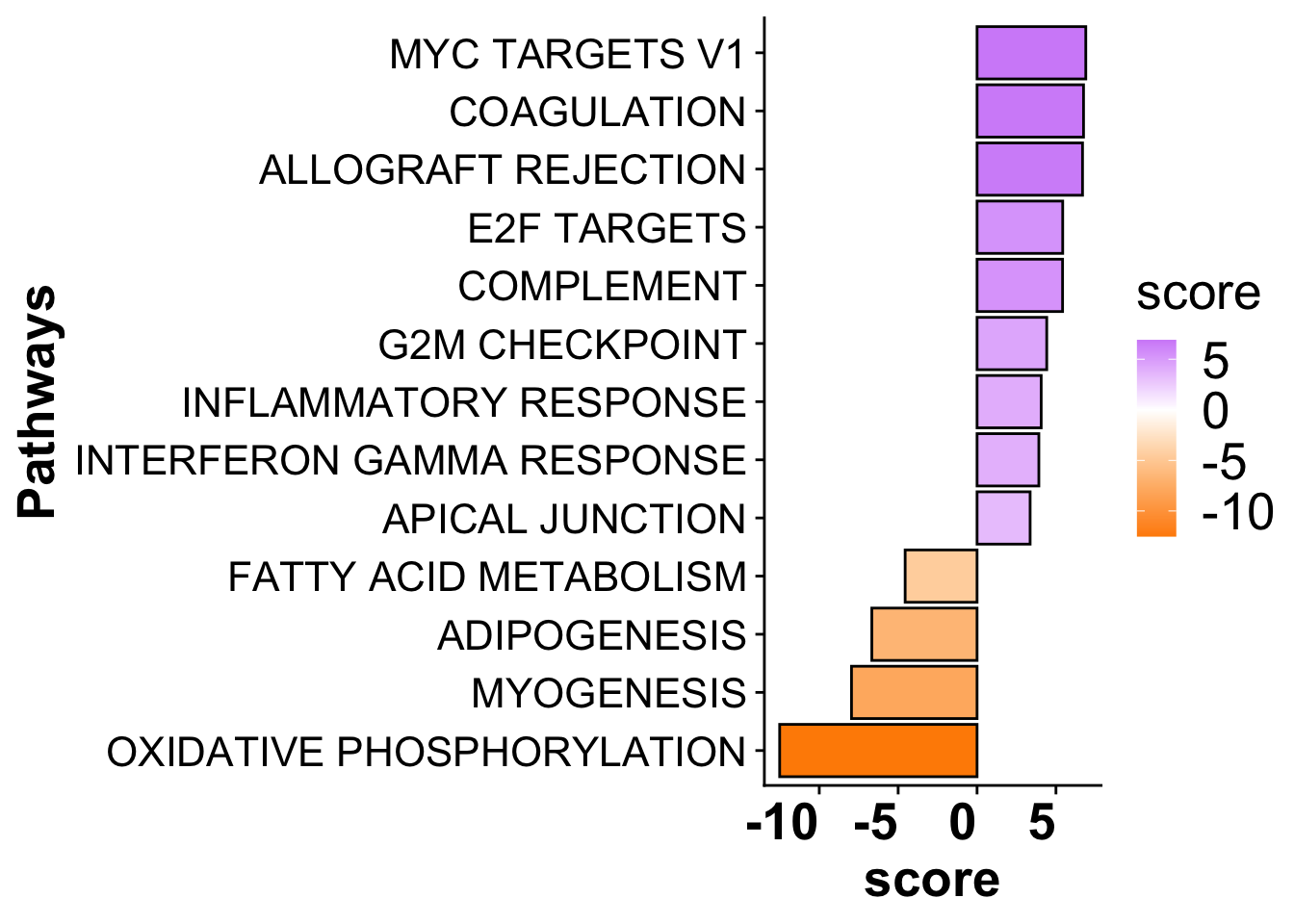
save_plot(filename = "./plots/Figure_5.pathway_plot.pdf",
plot = path_plot,
base_asp = 2,
base_height = 4)Subfigure E - Vwf specificity for Endocardial cells
snrna_prot <- fread("./output/proteomics/proteomics.snRNAseq_comp.tsv")
snrna_prot <- snrna_prot %>%
mutate("label_gene" = if_else(gene %in% c("Cdh11","Thbd","Vcam1"),gene,
if_else(gene == "Vwf","vWF",""))) %>%
subset(pct.1 > 0.05)
endo_proteomic_corr <- ggplot(snrna_prot,aes(avg_log2FC,logFC,
label = label_gene)) +
geom_hline(yintercept = 0, linetype = 2) +
geom_point(data =subset(snrna_prot,gene != "Vwf"), size =3, fill = "darkgrey", pch = 21) +
geom_point(data = subset(snrna_prot,gene %in% c("Vwf","Vcam1")),size = 4, fill = "purple", pch = 21) +
geom_point(data = subset(snrna_prot,gene %in% c("Thbd")),size = 4, fill = "darkorange", pch = 21) +
geom_point(data = subset(snrna_prot,gene %in% c("Cdh11")),size = 4, fill = "grey20", pch = 21) +
geom_label_repel(size = 5.5, max.overlaps = 20,force = 3) +
labs(x = "Specificity endocard. cells (snRNA-seq)",
y = "Log2 fold-change (Proteomics)")
endo_proteomic_corrWarning: Removed 1598 rows containing missing values (`geom_point()`).Warning: Removed 1598 rows containing missing values (`geom_label_repel()`).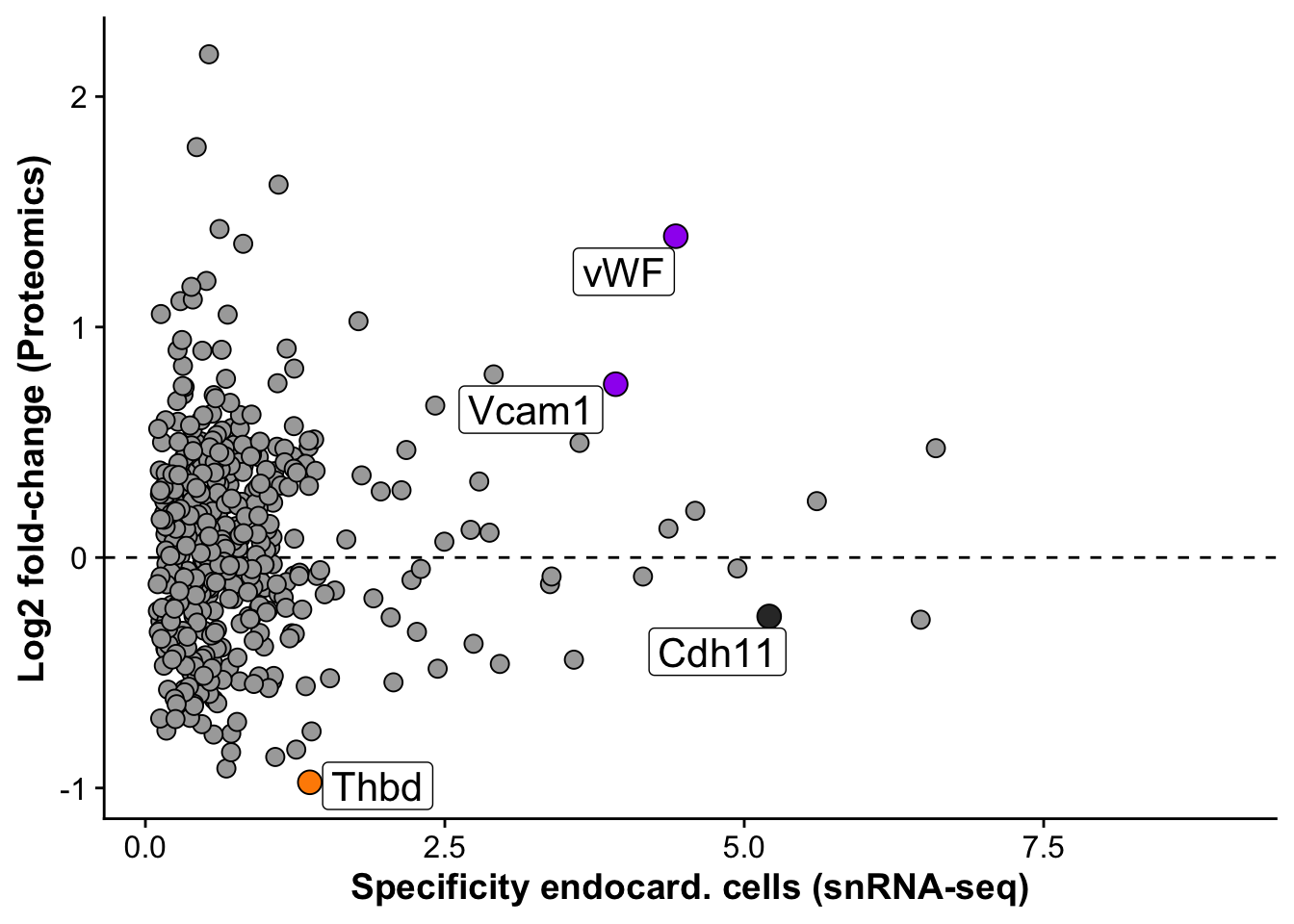
save_plot(filename = "./plots/Figure_5.vwf_specificity_plot.pdf",
plot = endo_proteomic_corr,
base_asp = 1.75,
base_height = 3.5)Warning: Removed 1598 rows containing missing values (`geom_point()`).
Removed 1598 rows containing missing values (`geom_label_repel()`).Subfigure F - Vwf is upregulated in MI_IZ
source("./code/functions.R")
yaxis_limits <- c(11,17)
vsn_matrix <- fread("./output/proteomics/proteomics.vsn_norm_proteins.tsv")
colnames(vsn_matrix)[1:11] <- paste("s",1:11,colnames(vsn_matrix)[1:11],sep="_")
protein_sub <- vsn_matrix %>%
dplyr::select(1:11,gene) %>%
pivot_longer(1:11,names_to = "sample", values_to = "exp") %>%
mutate("group" = if_else(grepl("control",sample),"control",
if_else(grepl("MI_IZ",sample),
"MI_IZ","MI_remote"))
)
protein_sub$group <- gsub("control","Control",protein_sub$group)
protein_sub$group <- factor(protein_sub$group,
levels = c("Control","MI_remote","MI_IZ"))
## Barplot with points as alternative.
# goi <- "Vwf"
# vwf_plot_bar <- plot_proteomics_boxplot(norm_table = protein_sub,
# protein = goi,
# style = "bar") +
# geom_signif(comparisons = list(c("MI_IZ","MI_remote")),
# tip_length = 0, annotation = "0.0057", y_position = 15.5) +
# geom_signif(comparisons = list(c("MI_IZ","Control")),
# tip_length = 0, annotation = "0.0022", y_position = 16.5) +
# expand_limits(y = c(13, 17)) +
# theme(axis.text.x = element_text(angle = 90, vjust = 1, hjust=1)) +
# labs(x = "")
## Median plot with points
goi <- "Cdh11"
npr3_plot <- plot_proteomics_boxplot(norm_table = protein_sub,
protein = goi,
style = "mean") +
theme(axis.text.x = element_text(angle = 90, vjust = 1, hjust=1)) +
labs(x = "") +
ylim(yaxis_limits) +
labs(y = "") +
scale_x_discrete(labels=c("Control" = "Control",
"MI_remote" = "MI remote",
"MI_IZ" = "MI IZ"))Warning: The `fun.y` argument of `stat_summary()` is deprecated as of ggplot2 3.3.0.
ℹ Please use the `fun` argument instead.
This warning is displayed once every 8 hours.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
ℹ Please use `linewidth` instead.
This warning is displayed once every 8 hours.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.## Mean plot with points
goi <- "Thbd"
thbd_plot <- plot_proteomics_boxplot(norm_table = protein_sub,
protein = goi,
style = "mean") +
theme(axis.text.x = element_text(angle = 90, vjust = 1, hjust=1)) +
geom_signif(comparisons = list(c("MI_IZ","MI_remote")),
tip_length = 0, annotation = "0.001", y_position = 16.5) +
geom_signif(comparisons = list(c("MI_IZ","Control")),
tip_length = 0, annotation = "0.023", y_position = 15.5) +
labs(x = "") +
ylim(yaxis_limits) +
labs(y = "") +
scale_x_discrete(labels=c("Control" = "Control",
"MI_remote" = "MI remote",
"MI_IZ" = "MI IZ"))
## Median plot with points
goi <- "Vcam1"
vcam1_plot <- plot_proteomics_boxplot(norm_table = protein_sub,
protein = goi,
style = "mean") +
theme(axis.text.x = element_text(angle = 90, vjust = 1, hjust=1)) +
geom_signif(comparisons = list(c("MI_IZ","MI_remote")),
tip_length = 0, annotation = "6.7e-4", y_position = 16.5) +
geom_signif(comparisons = list(c("MI_IZ","Control")),
tip_length = 0, annotation = "0.0078", y_position = 15.5) +
labs(x = "") +
ylim(yaxis_limits) +
labs(y = "") +
scale_x_discrete(labels=c("Control" = "Control",
"MI_remote" = "MI remote",
"MI_IZ" = "MI IZ"))
## Median plot with points
goi <- "Vwf"
vwf_plot <- plot_proteomics_boxplot(norm_table = protein_sub,
protein = goi,
style = "mean") +
geom_signif(comparisons = list(c("MI_IZ","MI_remote")),
tip_length = 0, annotation = "0.0057", y_position = 16.5) +
geom_signif(comparisons = list(c("MI_IZ","Control")),
tip_length = 0, annotation = "0.0022", y_position = 15.5) +
theme(axis.text.x = element_text(angle = 90, vjust = 1, hjust=1)) +
labs(x = "") +
ylim(yaxis_limits) +
scale_x_discrete(labels=c("Control" = "Control",
"MI_remote" = "MI remote",
"MI_IZ" = "MI IZ"))
# save_plot(filename = "./figures/Figure_5.vwf_expression_plot.pdf",
# plot = vwf_plot,
# base_asp = 0.5,
# base_height = 4)
joined_plot <- npr3_plot + thbd_plot + vcam1_plot + vwf_plot + plot_layout(nrow = 1,axis_titles = "collect") & labs(y = "Normalized protein level")
save_plot(filename = "./plots/Figure_5.expression_plot.pdf",
plot = joined_plot,
base_asp = 2.5,
base_height = 4)Warning: The dot-dot notation (`..y..`) was deprecated in ggplot2 3.4.0.
ℹ Please use `after_stat(y)` instead.
This warning is displayed once every 8 hours.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.Combine plots for figure
# # Combine plots
# comb_plot <- (pca_plot + volc_limma_IZ_remote + path_plot) / (endo_proteomic_corr + wrap_plots(npr3_plot,vwf_plot))
#
# save_plot(filename = "./figures/Figure_4.proteomics_combined.pdf",
# plot = comb_plot,
# base_asp = 2.5,
# base_height = 12)Subfigure H - human CITE-seq umap
library(Seurat)Loading required package: SeuratObjectLoading required package: sp'SeuratObject' was built with package 'Matrix' 1.6.3 but the current
version is 1.6.5; it is recomended that you reinstall 'SeuratObject' as
the ABI for 'Matrix' may have changed
Attaching package: 'SeuratObject'The following object is masked from 'package:base':
intersectlibrary(SCpubr)
── SCpubr 2.0.0.9000 ───────────────────────────────────────────────────────────
ℹ Have a look at extensive tutorials in SCpubr's book.
✔ If you use SCpubr in your research, please cite it accordingly.
★ If the package is useful to you, consider leaving a Star in the GitHub repository.
! Keep track of the package updates on Twitter (@Enblacar) or in the Official NEWS website.
♥ Happy plotting!
── Package version ──
CRAN: 2.0.2
Installed: 2.0.0.9000
⚠ There is a new version available onCRAN!
── Required packages ──
✔ AnnotationDbi 1.64.1 | 1.58.0 ✔ assertthat 0.2.1 | 0.2.1 ✖ AUCell
✔ circlize 0.4.15 | 0.4.16 ✔ cluster 2.1.6 | 2.1.6 ✖ clusterProfiler
✔ colorspace 2.1.0 | 2.1-0 ✔ decoupleR 2.8.0 | 2.2.2 ✔ dplyr 1.1.4 | 1.1.4
✖ enrichplot ✔ forcats 1.0.0 | 1.0.0 ✖ ggalluvial
✔ ggbeeswarm 0.7.2 | 0.7.2 ✖ ggdist ✖ ggExtra
✖ ggnewscale ✔ ggplot2 3.4.4 | 3.5.0 ✔ ggplotify 0.1.2 | 0.1.2
✔ ggrastr 1.0.2 | 1.0.2 ✔ ggrepel 0.9.5 | 0.9.5 ✔ ggridges 0.5.5 | 0.5.6
✔ ggsignif 0.6.4 | 0.6.4 ✔ labeling 0.4.3 | 0.4.3 ✖ liana
✔ magrittr 2.0.3 | 2.0.3 ✔ MASS 7.3.60.0.1 | 7.3-60.0.1 ✔ Matrix 1.6.5 | 1.6-5
✔ Nebulosa 1.12.0 | 1.6.0 ✔ patchwork 1.2.0 | 1.2.0 ✔ pbapply 1.7.2 | 1.7-2
✔ plyr 1.8.9 | 1.8.9 ✔ RColorBrewer 1.1.3 | 1.1-3 ✔ rlang 1.1.3 | 1.1.3
✔ scales 1.3.0 | 1.3.0 ✔ scattermore 1.2 | 1.2 ✔ Seurat 5.0.1 | 5.0.3
✔ SeuratObject 5.0.1 | 5.0.1 ✔ stringr 1.5.1 | 1.5.1 ✔ svglite 2.1.3 | 2.1.3
✔ tibble 3.2.1 | 3.2.1 ✔ tidyr 1.3.0 | 1.3.1 ✖ UCell
✔ viridis 0.6.4 | 0.6.5 ✔ withr 2.5.2 | 3.0.0
ℹ Installed packages are denoted by a tick (✔) and missing packages by a cross (✖).
ℹ Installed packages that still require an update to correctly run SCpubr have an exclamation mark (!).
ℹ Packages version are displayed as: Installed | Available.
── Available functions ──
✔ do_AffinityAnalysisPlot | DEV ✖ do_AlluvialPlot ✔ do_BarPlot
✔ do_BeeSwarmPlot ✔ do_BoxPlot ✖ do_CellularStatesPlot
✔ do_ChordDiagramPlot ✔ do_ColorPalette ✖ do_CopyNumberVariantPlot
✔ do_CorrelationPlot ✔ do_DiffusionMapPlot | DEV ✔ do_DimPlot
✔ do_DotPlot ✖ do_EnrichmentHeatmap ✔ do_ExpressionHeatmap
✔ do_FeaturePlot ✖ do_FunctionalAnnotationPlot ✖ do_GeyserPlot
✖ do_GroupedGOTermPlot ✔ do_GroupwiseDEPlot ✖ do_LigandReceptorPlot | DEV
✔ do_LoadingsPlot ✔ do_MetadataPlot | DEV ✔ do_NebulosaPlot
✔ do_PathwayActivityPlot ✔ do_RidgePlot ✖ do_SCEnrichmentHeatmap | DEV
✔ do_SCExpressionHeatmap | DEV ✔ do_TermEnrichmentPlot ✔ do_TFActivityPlot
✔ do_ViolinPlot ✔ do_VolcanoPlot ✔ save_Plot | DEV
ℹ Functions tied to development builds of SCpubr are marked by the (| DEV) tag.
ℹ You can install development builds of SCpubr by following the instructions in the Releases page.
ℹ Check the package requirements function-wise with: SCpubr::check_dependencies()
── Tips! ──
ℹ To adjust package messages to dark mode themes, use: options("SCpubr.darkmode" = TRUE)
ℹ To remove the white and black end from continuous palettes, use: options("SCpubr.ColorPaletteEnds" = FALSE)
✖ To suppress this startup message, use: suppressPackageStartupMessages(library(SCpubr))
✖ Alternatively, you can also set the following option: options("SCpubr.verbose" = FALSE)
And then load the package normally (and faster) as: library(SCpubr)
────────────────────────────────────────────────────────────────────────────────human_citeseq <- readRDS("../public_data/Amrute_et_al/final_global_annotated.rds")DefaultAssay(human_citeseq) <- "SCT"
#Choose endocardial cluster
Idents(human_citeseq) <- "annotation.0.1"
human_endocardium <- subset (human_citeseq, idents = "Endocardium")
#Choose Donor and AMI only
Idents(human_endocardium) <- "HF.etiology"
human_endocardium <- subset (human_endocardium, idents = c("Donor", "AMI"))
#plot VWF expression
plot3 <- VlnPlot (human_endocardium, feature = c("VWF"), cols = c("#008000", "#CD1076"))
#plot Umap embedding using SCpubr package
named_colors <- c("Fibroblast" = "#1f77b4",
"B Cells" = "#d62728",
"Plasma Cells" = "#ff7f0e",
"Endocardium" = "#17becf",
"Endothelium" = "#8c564b",
"Lymphatics" = "lightgrey",
"T_NK Cells" = "#bcbd22",
"Myeloid" = "#2ca02c",
"Glia" = "#9467bd",
"SMC_Pericyte" = "#e377c2",
"Mast Cells" = "darkred")
human_cite_umap <- SCpubr::do_DimPlot(sample = human_citeseq,
label = FALSE, label.box= TRUE,
group.by = "annotation.0.1",
repel = TRUE,
legend.position = "right", plot_cell_borders = TRUE,
plot_density_contour = FALSE,
plot.axes = FALSE, raster.dpi = 300,
shuffle = FALSE,
pt.size = 0.4, reduction = "rna.umap",
legend.icon.size = 5,
legend.byrow = TRUE, colors.use = named_colors) +
theme(legend.position = "none")
save_plot(human_cite_umap,
file = "./plots/Figure4.human_citeseq_umap.png",
base_height = 3,
base_asp = 1.3)Subfigure I - Violin plot for vWF
sub_human_cite <- subset(human_endocardium,HF.etiology %in% c("Donor","AMI"))
sub_human_cite$disease_group <- sub_human_cite$HF.etiology
## Quick DE analysis between Donor and AMI
sub_human_cite_pb <- AggregateExpression(sub_human_cite,
return.seurat = T,
group.by = c("sample","disease_group"))Centering and scaling data matrixIdents(sub_human_cite_pb) <- "disease_group"
sub_human_cite_pb_de <- FindMarkers(object = sub_human_cite_pb,
ident.1 = "Donor",
ident.2 = "AMI",
test.use = "DESeq2",
min.pct = 0.1)converting counts to integer modegene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatessub_human_cite_pb_de$gene <- rownames(sub_human_cite_pb_de)
sub_human_cite_pb_vwf <- subset(sub_human_cite_pb_de,gene == "VWF")
pvalue <- sub_human_cite_pb_vwf$p_val
pvalue[1] 1.405446e-05Idents(sub_human_cite) <- sub_human_cite$disease_group
vwf_vlnpot <- SCpubr::do_ViolinPlot(sample = sub_human_cite,
features = "VWF",
group.by = "disease_group",
line_width = 1,
legend.position = "none",
legend.title = "",
font.size = 25,
ylab = "Expression level",
xlab = "",
colors.use = c("Donor" = "#008000",
"AMI" = "#CD1076",
"ICM" = "white",
"NICM" = "white"))
vwf_vlnpot <- vwf_vlnpot + theme(plot.margin = margin(t=10, r=10, b=-25, l=10, unit="pt"))
save_plot(vwf_vlnpot,
file = "./plots/Figure4.human_citeseq_vlnplot.pdf",
base_height = 4)Save differential protein expression results for Table 3
table3 <- limma_res %>%
select(-c(label_protein,"P.Value"))
colnames(table3) <- gsub("\\.","_",colnames(table3))
colnames(table3) <- gsub("adj_P_Val","ajusted_pval",colnames(table3))
table3 <- table3 %>%
select(analysis,logFC,AveExpr,t,pval,ajusted_pval,B,gene,protein_ids) %>%
arrange(desc(logFC)) %>%
drop_na()
write.table(table3,
file = "./output/proteomics/Table3.tsv",
sep = "\t",
quote = F,
row.names = F,
col.names = TRUE)
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.1.2
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Europe/Berlin
tzcode source: internal
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] SCpubr_2.0.0.9000 Seurat_5.0.1 SeuratObject_5.0.1 sp_2.1-2
[5] RColorBrewer_1.1-3 ggsci_3.0.0 cowplot_1.1.2 ggforce_0.4.1
[9] patchwork_1.2.0 ggsignif_0.6.4 ggbeeswarm_0.7.2 ggrepel_0.9.5
[13] here_1.0.1 data.table_1.14.10 lubridate_1.9.3 forcats_1.0.0
[17] stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2 readr_2.1.5
[21] tidyr_1.3.0 tibble_3.2.1 ggplot2_3.4.4 tidyverse_2.0.0
[25] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] fs_1.6.3 matrixStats_1.2.0
[3] spatstat.sparse_3.0-3 bitops_1.0-7
[5] httr_1.4.7 tools_4.3.1
[7] sctransform_0.4.1 utf8_1.2.4
[9] R6_2.5.1 lazyeval_0.2.2
[11] uwot_0.1.16 withr_2.5.2
[13] gridExtra_2.3 progressr_0.14.0
[15] cli_3.6.2 Biobase_2.62.0
[17] textshaping_0.3.7 spatstat.explore_3.2-5
[19] fastDummies_1.7.3 labeling_0.4.3
[21] sass_0.4.8 mvtnorm_1.2-4
[23] spatstat.data_3.0-3 ggridges_0.5.5
[25] pbapply_1.7-2 systemfonts_1.0.5
[27] yulab.utils_0.1.3 svglite_2.1.3
[29] parallelly_1.36.0 rstudioapi_0.15.0
[31] RSQLite_2.3.4 generics_0.1.3
[33] gridGraphics_0.5-1 shape_1.4.6
[35] ica_1.0-3 spatstat.random_3.2-2
[37] Matrix_1.6-5 fansi_1.0.6
[39] S4Vectors_0.40.2 abind_1.4-5
[41] lifecycle_1.0.4 whisker_0.4.1
[43] yaml_2.3.8 SummarizedExperiment_1.32.0
[45] SparseArray_1.2.3 Rtsne_0.17
[47] grid_4.3.1 blob_1.2.4
[49] promises_1.2.1 crayon_1.5.2
[51] miniUI_0.1.1.1 lattice_0.22-5
[53] KEGGREST_1.42.0 pillar_1.9.0
[55] knitr_1.45 GenomicRanges_1.54.1
[57] future.apply_1.11.1 codetools_0.2-19
[59] leiden_0.4.3.1 glue_1.7.0
[61] getPass_0.2-4 vctrs_0.6.5
[63] png_0.1-8 spam_2.10-0
[65] gtable_0.3.4 assertthat_0.2.1
[67] ks_1.14.2 cachem_1.0.8
[69] xfun_0.41 S4Arrays_1.2.0
[71] mime_0.12 pracma_2.4.4
[73] survival_3.5-7 SingleCellExperiment_1.24.0
[75] ellipsis_0.3.2 fitdistrplus_1.1-11
[77] ROCR_1.0-11 nlme_3.1-164
[79] bit64_4.0.5 RcppAnnoy_0.0.21
[81] GenomeInfoDb_1.38.5 rprojroot_2.0.4
[83] bslib_0.6.1 irlba_2.3.5.1
[85] vipor_0.4.7 KernSmooth_2.23-22
[87] colorspace_2.1-0 BiocGenerics_0.48.1
[89] DBI_1.2.0 DESeq2_1.42.0
[91] ggrastr_1.0.2 tidyselect_1.2.0
[93] processx_3.8.3 bit_4.0.5
[95] compiler_4.3.1 git2r_0.33.0
[97] DelayedArray_0.28.0 plotly_4.10.4
[99] scales_1.3.0 lmtest_0.9-40
[101] callr_3.7.3 digest_0.6.34
[103] goftest_1.2-3 spatstat.utils_3.0-4
[105] rmarkdown_2.25 XVector_0.42.0
[107] decoupleR_2.8.0 htmltools_0.5.7
[109] pkgconfig_2.0.3 MatrixGenerics_1.14.0
[111] highr_0.10 fastmap_1.1.1
[113] rlang_1.1.3 GlobalOptions_0.1.2
[115] htmlwidgets_1.6.4 shiny_1.8.0
[117] farver_2.1.1 jquerylib_0.1.4
[119] zoo_1.8-12 jsonlite_1.8.8
[121] BiocParallel_1.36.0 mclust_6.0.1
[123] RCurl_1.98-1.14 magrittr_2.0.3
[125] GenomeInfoDbData_1.2.11 ggplotify_0.1.2
[127] dotCall64_1.1-1 munsell_0.5.0
[129] Rcpp_1.0.12 viridis_0.6.4
[131] reticulate_1.34.0 stringi_1.8.3
[133] zlibbioc_1.48.0 MASS_7.3-60.0.1
[135] plyr_1.8.9 parallel_4.3.1
[137] listenv_0.9.0 deldir_2.0-2
[139] Biostrings_2.70.1 splines_4.3.1
[141] tensor_1.5 hms_1.1.3
[143] circlize_0.4.15 locfit_1.5-9.8
[145] ps_1.7.6 igraph_1.6.0
[147] spatstat.geom_3.2-7 RcppHNSW_0.5.0
[149] reshape2_1.4.4 stats4_4.3.1
[151] evaluate_0.23 Nebulosa_1.12.0
[153] renv_1.0.3 BiocManager_1.30.22
[155] tzdb_0.4.0 tweenr_2.0.2
[157] httpuv_1.6.14 RANN_2.6.1
[159] polyclip_1.10-6 future_1.33.1
[161] scattermore_1.2 xtable_1.8-4
[163] RSpectra_0.16-1 later_1.3.2
[165] viridisLite_0.4.2 ragg_1.2.7
[167] memoise_2.0.1 beeswarm_0.4.0
[169] AnnotationDbi_1.64.1 IRanges_2.36.0
[171] cluster_2.1.6 timechange_0.2.0
[173] globals_0.16.2