Filter proteomics data
FloWuenne
2023-06-12
Last updated: 2023-06-13
Checks: 6 1
Knit directory: mi_spatialomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230612) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 59f6eae. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: references/
Ignored: renv/library/
Ignored: renv/staging/
Untracked files:
Untracked: analysis/proteomics.filter_proteomic_data.Rmd
Unstaged changes:
Modified: analysis/data_analysis.Rmd
Deleted: analysis/filter_proteomic_data.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
Introduction
Here we will process and analyze the data generated via laser-microdissection coupled with high-sensitiviy proteomics of the endocardial layer from control hearts and hearts one day after myocardial infarction. We have a total of 3 groups in this dataset, all representing laser microdissected regions of endocardium from mouse hearts: 1) control = Mouse hearts without infarct 2) MI_IZ = Mouse hearts 1 day post MI. Endocardial region adjacent to the infarct. 3) MI_remote = Mouse hearts 1 day post MI. Endocardial region remote to the infarct.
Load data
First, let’s load the raw data and the associated metadata for all samples.
prot_res <- fread("./data/proteomics_endocardial_layer.tsv")
metadata <- fread("./data/metadata.proteomics_endocardial_layer.txt")Analysis
Calculate missingness per sample (# NA values)
As a first QC, we will calculate the amount of missing data (NA values) per sample in the proteomics data.
missing <- colSums(is.na(prot_res))[6:ncol(prot_res)]
non_missing <-colSums(!is.na(prot_res))[6:ncol(prot_res)]
missingness <- missing / (non_missing + missing)
missingness <- as.data.frame(missingness)
missingness$sample <- rownames(missingness)
## Add group to counts
missingness <- missingness %>%
mutate("group" = if_else(grepl("control",sample),"control",
if_else(grepl("MI_IZ",sample),"MI_IZ","MI_remote")))
## Set order of groups
missingness$group <- factor(missingness$group,
levels = c("control","MI_remote","MI_IZ"))
avg_missingness <- mean(missingness$missingness)
ggplot(missingness,aes(sample,missingness,fill = group)) +
geom_bar(stat = "identity") +
coord_flip() +
scale_fill_npg() +
labs(x = "Samples",
y = "% missing values") +
geom_hline(yintercept = avg_missingness, linetype = 2)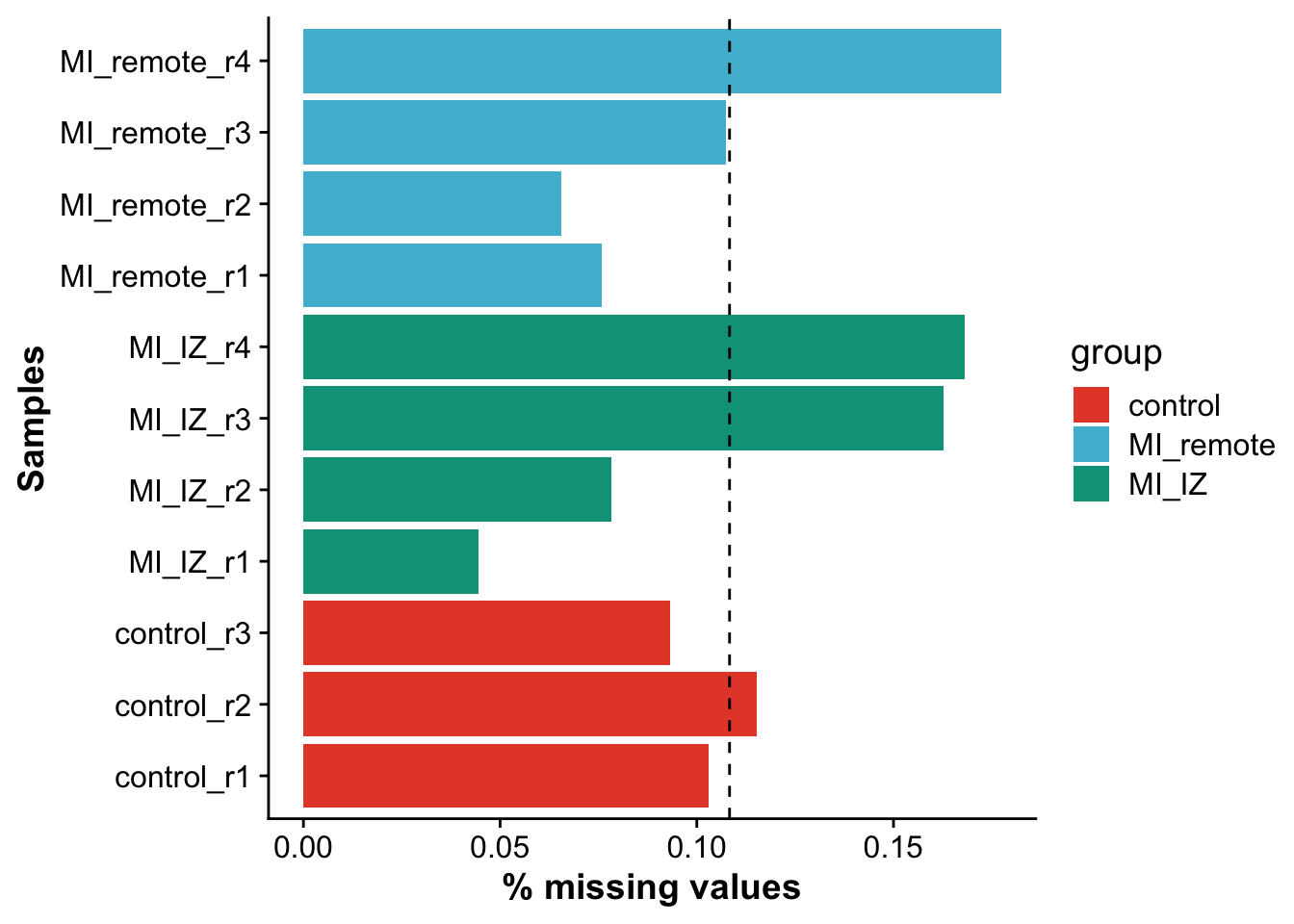
Filter out proteins that are identified in < 3 MI samples
Then we are going to calculate the missingness per sample group (the treatment group and area that was excised by laser microdissection) and exclude proteins that are only identified in a subset of samples per group. For controls, we expect a protein to be measured in at least 2/3 samples (33% missingness allowed). For MI samples, we expect a protein to be measured in at least 2/4 samples (50% missingness allowed)
na_mi <- prot_res %>%
pivot_longer(control_r1:MI_remote_r4,
names_to = "sample",
values_to = "protein_exp") %>%
mutate("group" = if_else(grepl("control",sample),"control",
if_else(grepl("MI_IZ",sample),"MI_IZ","MI_remote"))) %>%
group_by(Protein_Ids,Genes,group) %>%
summarise(na_count = sum(is.na(protein_exp))) %>%
ungroup() %>%
mutate("percent_na" = if_else(group == "control",na_count / 3, na_count / 4)) %>%
select(-na_count) %>% ## control: n = 3 and MI: n = 4
pivot_wider(names_from = "group",values_from = percent_na) %>%
mutate("retain" = if_else(control <= 0.3 | MI_remote <= 0.25 | MI_IZ <= 0.25, "yes","no"))`summarise()` has grouped output by 'Protein_Ids', 'Genes'. You can override
using the `.groups` argument.prot_res_filt <- subset(prot_res,Protein_Ids %in% subset(na_mi,retain == "yes")$Protein_Ids)Exclude contaminants (keratins etc.)
After filtering based on missingness, we will filter out any proteions that are considered contaminants based on know contaminations based on Frankenfeld 2022
## Count number of detected proteins with contaminants
prot_res_quant_cont <- prot_res_filt %>%
select(control_r1:MI_remote_r4) %>%
summarise_all(funs(sum(!is.na(.))))Warning: `funs()` was deprecated in dplyr 0.8.0.
ℹ Please use a list of either functions or lambdas:
# Simple named list: list(mean = mean, median = median)
# Auto named with `tibble::lst()`: tibble::lst(mean, median)
# Using lambdas list(~ mean(., trim = .2), ~ median(., na.rm = TRUE))
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.## Count number of detected proteins without contaminants
prot_res_filt <- prot_res_filt %>%
mutate("contaminant" = if_else(grepl("Cont",Protein_Ids),"yes","no"))
prot_res_quant <- prot_res_filt %>%
subset(contaminant == "no") %>%
select(control_r1:MI_remote_r4) %>%
summarise_all(funs(sum(!is.na(.))))Warning: `funs()` was deprecated in dplyr 0.8.0.
ℹ Please use a list of either functions or lambdas:
# Simple named list: list(mean = mean, median = median)
# Auto named with `tibble::lst()`: tibble::lst(mean, median)
# Using lambdas list(~ mean(., trim = .2), ~ median(., na.rm = TRUE))
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.final_counts <- t(rbind(prot_res_quant_cont,
prot_res_quant))Merge metadata with protein counts per sample
Here, we will plot the number of proteins identified per group.
colnames(final_counts) <- c("w_cont","wo_cont")
final_counts <- as.data.frame(final_counts) %>%
mutate("sample" = rownames(final_counts))
## Add protein counts to metadata
metadata_counts <- left_join(metadata,final_counts, by = "sample")
## Add group to counts
metadata_counts <- metadata_counts %>%
mutate("group" = if_else(grepl("control",sample),"control",
if_else(grepl("MI_IZ",sample),"MI_IZ","MI_remote")))
## Set order of groups
metadata_counts$group <- factor(metadata_counts$group,
levels = c("control","MI_remote","MI_IZ"))
## Plot number of proteins without contaminants per sample
ggbarplot(metadata_counts,
x = "group",
y = "wo_cont",
fill = "group",
add = c("mean_se"),
error.plot = "upper_errorbar",
color = "black",
palette = "npg") +
theme_minimal_hgrid() +
labs (x = "Treatment group",
y = "# Proteins identified") +
font("xlab", size = 16, color = "black", face = "bold") +
font("ylab", size = 16, color = "black", face = "bold") +
scale_y_continuous(
expand = expansion(mult = c(0, 0.05))
) +
rremove("legend")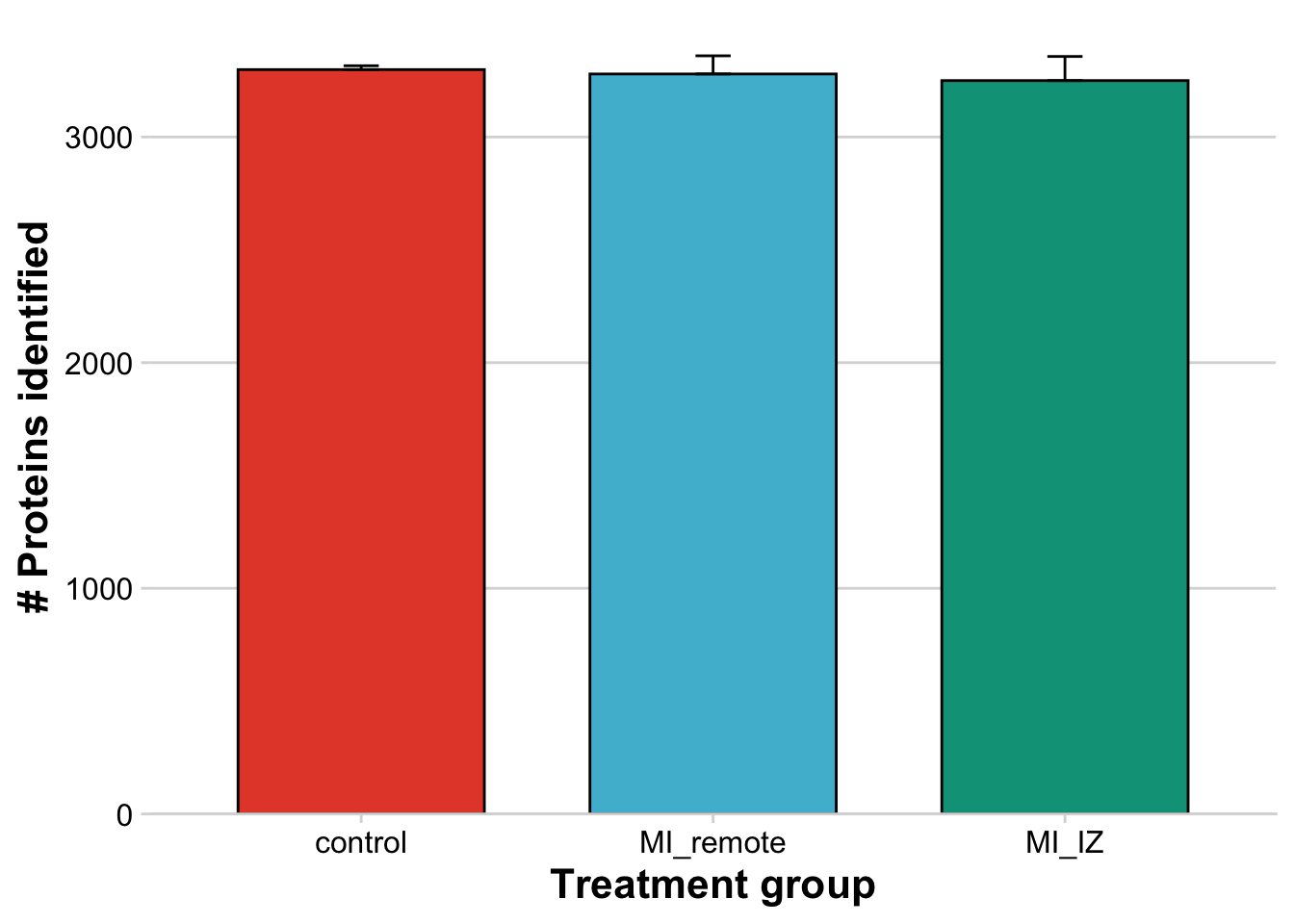
Create final filtered protein table
Finally, we will create a final filtered protein table, that we will use for imputation.
prot_res_final <- subset(prot_res_filt,contaminant == "no") %>%
select(-c(contaminant,First_Protein_Description))
write.table(prot_res_final,
file = "./output/proteomics.filtered_proteins.tsv",
sep = "\t",
col.names = TRUE,
row.names = FALSE,
quote = FALSE)
sessionInfo()R version 4.2.3 (2023-03-15)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] ggsci_3.0.0 here_1.0.1 cowplot_1.1.1 ggpubr_0.6.0
[5] data.table_1.14.8 lubridate_1.9.2 forcats_1.0.0 stringr_1.5.0
[9] dplyr_1.1.2 purrr_1.0.1 readr_2.1.4 tidyr_1.3.0
[13] tibble_3.2.1 ggplot2_3.4.2 tidyverse_2.0.0 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.10 getPass_0.2-2 ps_1.7.4 rprojroot_2.0.3
[5] digest_0.6.31 utf8_1.2.3 R6_2.5.1 backports_1.4.1
[9] evaluate_0.21 highr_0.10 httr_1.4.6 pillar_1.9.0
[13] rlang_1.1.1 rstudioapi_0.14 whisker_0.4.1 car_3.1-2
[17] callr_3.7.3 jquerylib_0.1.4 rmarkdown_2.21 labeling_0.4.2
[21] munsell_0.5.0 broom_1.0.5 compiler_4.2.3 httpuv_1.6.11
[25] xfun_0.39 pkgconfig_2.0.3 htmltools_0.5.5 tidyselect_1.2.0
[29] fansi_1.0.4 crayon_1.5.2 tzdb_0.4.0 withr_2.5.0
[33] later_1.3.1 grid_4.2.3 jsonlite_1.8.4 gtable_0.3.3
[37] lifecycle_1.0.3 git2r_0.32.0 magrittr_2.0.3 scales_1.2.1
[41] cli_3.6.1 stringi_1.7.12 cachem_1.0.8 carData_3.0-5
[45] farver_2.1.1 renv_0.17.3 ggsignif_0.6.4 fs_1.6.2
[49] promises_1.2.0.1 bslib_0.4.2 generics_0.1.3 vctrs_0.6.2
[53] tools_4.2.3 glue_1.6.2 hms_1.1.3 processx_3.8.0
[57] abind_1.4-5 fastmap_1.1.1 yaml_2.3.7 timechange_0.2.0
[61] colorspace_2.1-0 rstatix_0.7.2 knitr_1.42 sass_0.4.6