Sample IDs and Relative Abundance Misc.
Last updated: 2020-12-17
Checks: 6 1
Knit directory: esoph-micro-cancer-workflow/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200916) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version cf91029. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/
Untracked files:
Untracked: analysis/waterfall-plot.Rmd
Untracked: waterfall-plots.Rmd
Unstaged changes:
Modified: analysis/data_processing_tcga.Rmd
Modified: analysis/test-of-replication.Rmd
Modified: code/get_cleaned_data.R
Modified: code/get_data.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/test-of-replication.Rmd) and HTML (docs/test-of-replication.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | cf91029 | noah-padgett | 2020-12-02 | updated analyses |
| html | cf91029 | noah-padgett | 2020-12-02 | updated analyses |
- Goal is to replicate Fisher’s exact test.
Data mung
First, we need to format the data for the analyses.
# transform to relative abundances
phylo.data.nci.umd <- transform_sample_counts(phylo.data.nci.umd, function(x){x / sum(x)})
phylo.data.tcga.RNAseq <- transform_sample_counts(phylo.data.tcga.RNAseq, function(x){x / sum(x)})
phylo.data.tcga.WGS <- transform_sample_counts(phylo.data.tcga.WGS, function(x){x / sum(x)})
# melt data down for use
dat.16s <- psmelt(phylo.data.nci.umd)
dat.rna <- psmelt(phylo.data.tcga.RNAseq)
dat.wgs <- psmelt(phylo.data.tcga.WGS)
# fix otu formatting
dat.rna$otu2 <- "a"
dat.wgs$otu2 <- "a"
i <- 1
for(i in 1:nrow(dat.rna)){
dat.rna$otu2[i] <- str_split(dat.rna$OTU[i], ";")[[1]][7]
}
for(i in 1:nrow(dat.wgs)){
dat.wgs$otu2[i] <- str_split(dat.wgs$OTU[i], ";")[[1]][7]
}
# subset to fuso. nuc. only
# Streptococcus sanguinis
# Campylobacter concisus
# Prevotella spp.
dat.16s <- filter(
dat.16s,
OTU %in% c(
"Fusobacterium_nucleatum",
unique(dat.16s$OTU[dat.16s$OTU %like% "Streptococcus_"]),
unique(dat.16s$OTU[dat.16s$OTU %like% "Campylobacter_"]),
"Prevotella_melaninogenica")
)
dat.rna <- filter(
dat.rna,
otu2 %in% c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Streptococcus oralis",
"Streptococcus mitis",
"Streptococcus pneumoniae",
"Streptococcus parasanguinis",
"Streptococcus salivarius",
"Campylobacter concisus",
"Prevotella melaninogenica")
)
dat.wgs <- filter(
dat.wgs,
otu2 %in% c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Streptococcus oralis",
"Streptococcus mitis",
"Streptococcus pneumoniae",
"Streptococcus parasanguinis",
"Streptococcus salivarius",
"Campylobacter concisus",
"Prevotella melaninogenica")
)
# new names
dat.16s$OTU1 <- factor(
dat.16s$OTU,
levels = c(
"Fusobacterium_nucleatum",
"Streptococcus_dentisani:Streptococcus_infantis:Streptococcus_mitis:Streptococcus_oligofermentans:Streptococcus_oralis:Streptococcus_pneumoniae:Streptococcus_pseudopneumoniae:Streptococcus_sanguinis",
"Campylobacter_rectus:Campylobacter_showae",
"Prevotella_melaninogenica"
),
labels = c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Campylobacter concisus",
"Prevotella melaninogenica"
)
)
dat.rna$OTU1 <- factor(
dat.rna$otu2,
levels = c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Campylobacter concisus",
"Prevotella melaninogenica"),
labels = c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Campylobacter concisus",
"Prevotella melaninogenica")
)
dat.wgs$OTU1 <- factor(
dat.wgs$otu2,
levels = c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Campylobacter concisus",
"Prevotella melaninogenica"),
labels = c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Campylobacter concisus",
"Prevotella melaninogenica")
)
# rename bacteria
dat.16s$OTU <- factor(
dat.16s$OTU,
levels = c(
"Fusobacterium_nucleatum",
"Streptococcus_dentisani:Streptococcus_infantis:Streptococcus_mitis:Streptococcus_oligofermentans:Streptococcus_oralis:Streptococcus_pneumoniae:Streptococcus_pseudopneumoniae:Streptococcus_sanguinis",
"Campylobacter_rectus:Campylobacter_showae",
"Prevotella_melaninogenica"
),
labels = c(
"Fusobacterium nucleatum",
"Streptococcus spp.",
"Campylobacter concisus",
"Prevotella melaninogenica"
)
)
dat.rna$OTU <- factor(
dat.rna$otu2,
levels = c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Streptococcus oralis",
"Streptococcus mitis",
"Streptococcus pneumoniae",
"Streptococcus parasanguinis",
"Streptococcus salivarius",
"Campylobacter concisus",
"Prevotella melaninogenica"),
labels = c(
"Fusobacterium nucleatum",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Campylobacter concisus",
"Prevotella melaninogenica")
)
dat.wgs$OTU <- factor(
dat.wgs$otu2,
levels = c(
"Fusobacterium nucleatum",
"Streptococcus sanguinis",
"Streptococcus oralis",
"Streptococcus mitis",
"Streptococcus pneumoniae",
"Streptococcus parasanguinis",
"Streptococcus salivarius",
"Campylobacter concisus",
"Prevotella melaninogenica"),
labels = c(
"Fusobacterium nucleatum",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Streptococcus spp.",
"Campylobacter concisus",
"Prevotella melaninogenica")
)
# make tumor vs normal variable
dat.16s$tumor.cat <- factor(dat.16s$tissue, levels=c("BO", "N", "T"), labels = c("Non-Tumor", "Non-Tumor", "Tumor"))
dat.rna$tumor.cat <- factor(dat.rna$SampleType_Level2, levels=c("Normal", "Tumor"), labels = c("Non-Tumor", "Tumor"))
dat.wgs$tumor.cat <- factor(dat.wgs$SampleType_Level2, levels=c("Normal", "Tumor"), labels = c("Non-Tumor", "Tumor"))
# dataset id
dat.16s$source <- "16s"
dat.rna$source <- "rna"
dat.wgs$source <- "wgs"
# plotting ids
dat.16s$X <- paste0(dat.16s$source, "-", dat.16s$tumor.cat)
dat.rna$X <- paste0(dat.rna$source, "-", dat.rna$tumor.cat)
dat.wgs$X <- paste0(dat.wgs$source, "-", dat.wgs$tumor.cat)
# relabel as (0/1) for analysis
dat.16s$tumor <- as.numeric(factor(dat.16s$tissue, levels=c("BO", "N", "T"), labels = c("Non-Tumor", "Non-Tumor", "Tumor"))) - 1
dat.rna$tumor <- as.numeric(factor(dat.rna$SampleType_Level2, levels=c("Normal", "Tumor"), labels = c("Non-Tumor", "Tumor"))) - 1
dat.wgs$tumor <- as.numeric(factor(dat.wgs$SampleType_Level2, levels=c("Normal", "Tumor"), labels = c("Non-Tumor", "Tumor"))) - 1
# presence- absence
dat.16s$pres <- ifelse(dat.16s$Abundance > 0, 1, 0)
dat.16s$pres[is.na(dat.16s$pres)] <- 0
dat.rna$pres <- ifelse(dat.rna$Abundance > 0, 1, 0)
dat.rna$pres[is.na(dat.rna$pres)] <- 0
dat.wgs$pres <- ifelse(dat.wgs$Abundance > 0, 1, 0)
dat.wgs$pres[is.na(dat.wgs$pres)] <- 0
# blood exclusions
dat.16s$bloodWGS <- 0
dat.rna$bloodWGS <- dat.rna$WGS_Blood_Normal
dat.wgs$bloodWGS <- dat.wgs$WGS_Blood_Normal
# merge data
cls <- c("OTU", "OTU1", "Sample", "Abundance", "tumor.cat", "tumor", "source", "X", "pres", "bloodWGS")
mydata <- full_join(dat.16s[,cls], dat.rna[,cls])Joining, by = c("OTU", "OTU1", "Sample", "Abundance", "tumor.cat", "tumor", "source", "X", "pres", "bloodWGS")mydata <- full_join(mydata, dat.wgs[,cls])Joining, by = c("OTU", "OTU1", "Sample", "Abundance", "tumor.cat", "tumor", "source", "X", "pres", "bloodWGS")Replicating the Analysis
Plot
p <- ggplot(mydata, aes(x=X, y=Abundance)) +
geom_violin() +
geom_jitter(alpha=0.5) +
facet_wrap(.~OTU)+
scale_y_continuous(
trans = "sqrt",
breaks=c(0.002, 0.01, 0.05, 0.1, 0.25, 0.5, 0.8)) +
labs(x=NULL, y="Relative Abundance")+
theme_classic() +
theme(
axis.text.x = element_text(angle = 30,vjust=0.95, hjust=0.95)
)
pWarning: Removed 1107 rows containing non-finite values (stat_ydensity).Warning: Removed 1107 rows containing missing values (geom_point).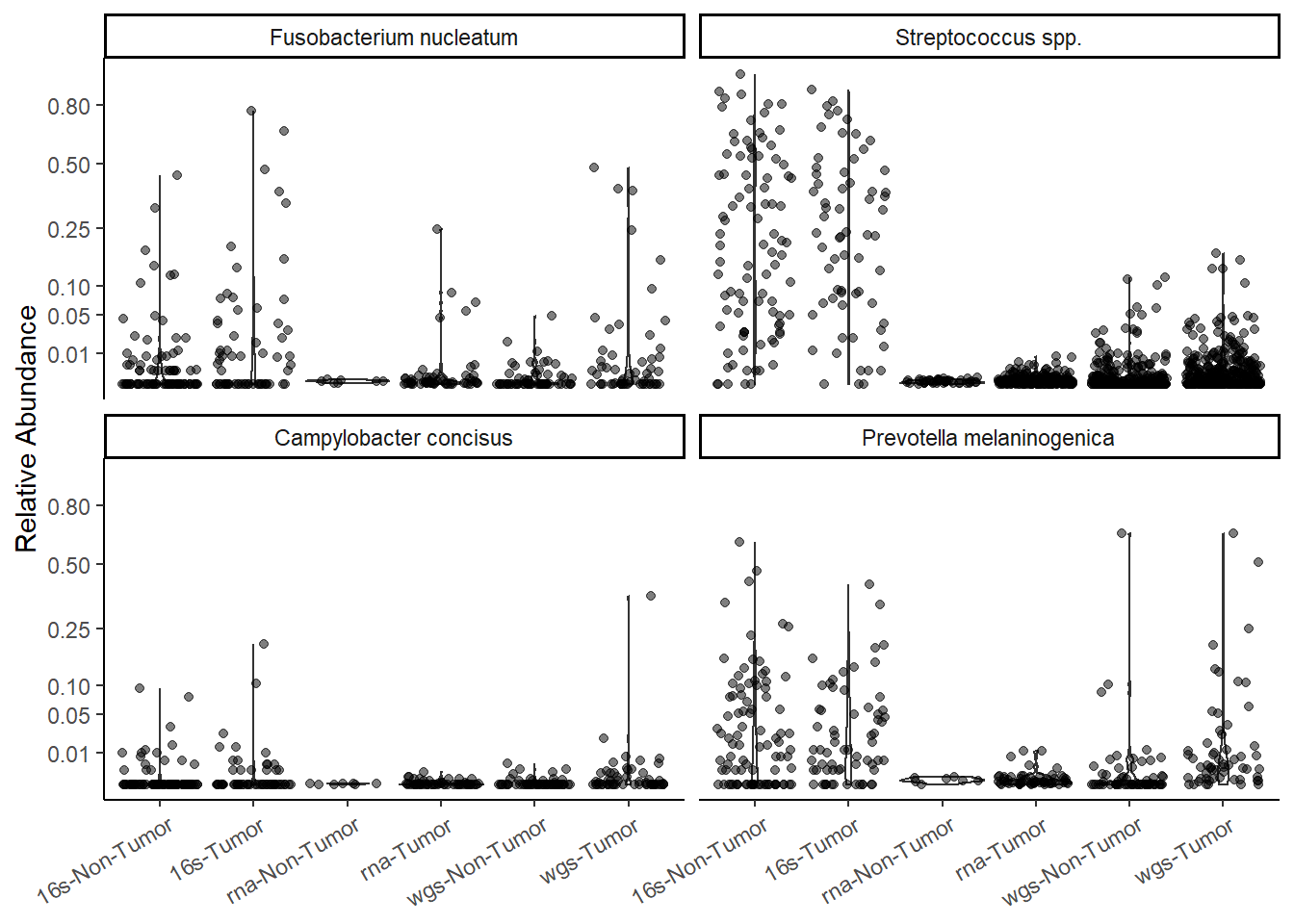
Statistical Tests
Fusobacterium nucleatum
# No additional subsetting/exclusions
d <- mydata %>%
filter(OTU == "Fusobacterium nucleatum",
source == "16s")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.1487
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.8126368 3.2144776
sample estimates:
odds ratio
1.610386 d <- mydata %>%
filter(OTU == "Fusobacterium nucleatum",
source == "wgs")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.02533
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
1.065529 4.759083
sample estimates:
odds ratio
2.232631 d <- mydata %>%
filter(OTU == "Fusobacterium nucleatum",
source == "rna")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.04707
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.05593801 1.12690374
sample estimates:
odds ratio
0.2723903 # Excluding blood != 0
d <- mydata %>%
filter(OTU == "Fusobacterium nucleatum",
source == "wgs",
bloodWGS==0)
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.7626
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.3753539 5.6058176
sample estimates:
odds ratio
1.432327 Streptococcus spp.
difficult to determine which species were used
# No additional subsetting/exclusions
d <- mydata %>%
filter(OTU == "Streptococcus spp.",
source == "16s")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 1
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.2189448 7.8326544
sample estimates:
odds ratio
1.173049 d <- mydata %>%
filter(OTU == "Streptococcus spp.",
source == "wgs")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 1.413e-06
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
1.487867 2.643073
sample estimates:
odds ratio
1.981098 d <- mydata %>%
filter(OTU == "Streptococcus spp.",
source == "rna")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 8.651e-07
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.1589282 0.4818080
sample estimates:
odds ratio
0.2795264 # Excluding blood != 0
d <- mydata %>%
filter(OTU == "Streptococcus spp.",
source == "wgs",
bloodWGS==0)
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 1
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.5998557 1.6670677
sample estimates:
odds ratio
1 Streptococcus sanguinis
difficult to determine which species were used
# No additional subsetting/exclusions
d <- mydata %>%
filter(OTU1 == "Streptococcus sanguinis",
source == "16s")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 1
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.2189448 7.8326544
sample estimates:
odds ratio
1.173049 d <- mydata %>%
filter(OTU1 == "Streptococcus sanguinis",
source == "wgs")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.4639
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.6098689 2.9683346
sample estimates:
odds ratio
1.339246 d <- mydata %>%
filter(OTU1 == "Streptococcus sanguinis",
source == "rna")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.04205
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.05280152 1.06581611
sample estimates:
odds ratio
0.2573433 # Excluding blood != 0
d <- mydata %>%
filter(OTU1 == "Streptococcus sanguinis",
source == "wgs",
bloodWGS==0)
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 1
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.2622982 6.4033751
sample estimates:
odds ratio
1.267969 Campylobacter concisus
# No additional subsetting/exclusions
d <- mydata %>%
filter(OTU == "Campylobacter concisus",
source == "16s")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.2325
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.7306314 3.9724225
sample estimates:
odds ratio
1.698461 d <- mydata %>%
filter(OTU == "Campylobacter concisus",
source == "wgs")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.008873
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
1.225362 6.656564
sample estimates:
odds ratio
2.797326 d <- mydata %>%
filter(OTU == "Campylobacter concisus",
source == "rna")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.04188
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.05364598 1.20067358
sample estimates:
odds ratio
0.235494 # Excluding blood != 0
d <- mydata %>%
filter(OTU == "Campylobacter concisus",
source == "wgs",
bloodWGS==0)
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 1
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.2461498 4.0625664
sample estimates:
odds ratio
1 Prevotella melaninogenica
# No additional subsetting/exclusions
d <- mydata %>%
filter(OTU == "Prevotella melaninogenica",
source == "16s")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.1531
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.790056 3.924226
sample estimates:
odds ratio
1.728747 d <- mydata %>%
filter(OTU == "Prevotella melaninogenica",
source == "wgs")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 3.232e-06
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
2.492672 12.298866
sample estimates:
odds ratio
5.430828 d <- mydata %>%
filter(OTU == "Prevotella melaninogenica",
source == "rna")
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.1884
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.09378767 1.68318325
sample estimates:
odds ratio
0.4075071 # Excluding blood != 0
d <- mydata %>%
filter(OTU == "Prevotella melaninogenica",
source == "wgs",
bloodWGS==0)
fisher.test(d$pres, d$tumor)
Fisher's Exact Test for Count Data
data: d$pres and d$tumor
p-value = 0.747
alternative hypothesis: true odds ratio is not equal to 1
95 percent confidence interval:
0.355727 6.731657
sample estimates:
odds ratio
1.509201
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 18363)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252
[2] LC_CTYPE=English_United States.1252
[3] LC_MONETARY=English_United States.1252
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.1252
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] car_3.0-8 carData_3.0-4 gvlma_1.0.0.3 patchwork_1.0.1
[5] viridis_0.5.1 viridisLite_0.3.0 gridExtra_2.3 xtable_1.8-4
[9] kableExtra_1.1.0 plyr_1.8.6 data.table_1.13.0 readxl_1.3.1
[13] forcats_0.5.0 stringr_1.4.0 dplyr_1.0.1 purrr_0.3.4
[17] readr_1.3.1 tidyr_1.1.1 tibble_3.0.3 ggplot2_3.3.2
[21] tidyverse_1.3.0 lmerTest_3.1-2 lme4_1.1-23 Matrix_1.2-18
[25] vegan_2.5-6 lattice_0.20-41 permute_0.9-5 phyloseq_1.32.0
[29] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] minqa_1.2.4 colorspace_1.4-1 rio_0.5.16
[4] ellipsis_0.3.1 rprojroot_1.3-2 XVector_0.28.0
[7] fs_1.5.0 rstudioapi_0.11 farver_2.0.3
[10] fansi_0.4.1 lubridate_1.7.9 xml2_1.3.2
[13] codetools_0.2-16 splines_4.0.2 knitr_1.29
[16] ade4_1.7-15 jsonlite_1.7.0 nloptr_1.2.2.2
[19] broom_0.7.0 cluster_2.1.0 dbplyr_1.4.4
[22] BiocManager_1.30.10 compiler_4.0.2 httr_1.4.2
[25] backports_1.1.7 assertthat_0.2.1 cli_2.0.2
[28] later_1.1.0.1 htmltools_0.5.0 tools_4.0.2
[31] igraph_1.2.5 gtable_0.3.0 glue_1.4.1
[34] reshape2_1.4.4 Rcpp_1.0.5 Biobase_2.48.0
[37] cellranger_1.1.0 vctrs_0.3.2 Biostrings_2.56.0
[40] multtest_2.44.0 ape_5.4 nlme_3.1-148
[43] iterators_1.0.12 xfun_0.19 openxlsx_4.1.5
[46] rvest_0.3.6 lifecycle_0.2.0 statmod_1.4.34
[49] zlibbioc_1.34.0 MASS_7.3-51.6 scales_1.1.1
[52] hms_0.5.3 promises_1.1.1 parallel_4.0.2
[55] biomformat_1.16.0 rhdf5_2.32.2 curl_4.3
[58] yaml_2.2.1 stringi_1.4.6 S4Vectors_0.26.1
[61] foreach_1.5.0 BiocGenerics_0.34.0 zip_2.0.4
[64] boot_1.3-25 rlang_0.4.7 pkgconfig_2.0.3
[67] evaluate_0.14 Rhdf5lib_1.10.1 tidyselect_1.1.0
[70] magrittr_1.5 R6_2.4.1 IRanges_2.22.2
[73] generics_0.0.2 DBI_1.1.0 foreign_0.8-80
[76] pillar_1.4.6 haven_2.3.1 whisker_0.4
[79] withr_2.2.0 mgcv_1.8-31 abind_1.4-5
[82] survival_3.2-3 modelr_0.1.8 crayon_1.3.4
[85] rmarkdown_2.5 grid_4.0.2 blob_1.2.1
[88] git2r_0.27.1 reprex_0.3.0 digest_0.6.25
[91] webshot_0.5.2 httpuv_1.5.4 numDeriv_2016.8-1.1
[94] stats4_4.0.2 munsell_0.5.0