GWAS Trait Categorisation
Isobel Beasley
2025-08-24
Last updated: 2026-03-25
Checks: 7 0
Knit directory:
genomics_ancest_disease_dispar/
This reproducible R Markdown analysis was created with workflowr (version 1.7.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220216) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 2dc80c9. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rproj.user/
Ignored: .venv/
Ignored: Aus_School_Profile.xlsx
Ignored: BC2GM/
Ignored: BioC.dtd
Ignored: FormatConverter.jar
Ignored: FormatConverter.zip
Ignored: SeniorSecondaryCompletionandAchievementInformation_2025.xlsx
Ignored: analysis/.DS_Store
Ignored: ancestry_dispar_env/
Ignored: code/.DS_Store
Ignored: code/full_text_conversion/.DS_Store
Ignored: data/.DS_Store
Ignored: data/RCDCFundingSummary_01042026.xlsx
Ignored: data/cdc/
Ignored: data/cohort/
Ignored: data/epmc/
Ignored: data/europe_pmc/
Ignored: data/gbd/.DS_Store
Ignored: data/gbd/IHME-GBD_2021_DATA-d8cf695e-1.csv
Ignored: data/gbd/IHME-GBD_2023_DATA-73cc01fd-1.csv
Ignored: data/gbd/gbd_2019_california_percent_deaths.csv
Ignored: data/gbd/ihme_gbd_2019_global_disease_burden_rate_all_ages.csv
Ignored: data/gbd/ihme_gbd_2019_global_paf_rate_percent_all_ages.csv
Ignored: data/gbd/ihme_gbd_2021_global_disease_burden_rate_all_ages.csv
Ignored: data/gbd/ihme_gbd_2021_global_paf_rate_percent_all_ages.csv
Ignored: data/gwas_catalog/
Ignored: data/icd/.DS_Store
Ignored: data/icd/2025AA/
Ignored: data/icd/IHME_GBD_2019_COD_CAUSE_ICD_CODE_MAP_Y2020M10D15.XLSX
Ignored: data/icd/IHME_GBD_2019_NONFATAL_CAUSE_ICD_CODE_MAP_Y2020M10D15.XLSX
Ignored: data/icd/IHME_GBD_2021_COD_CAUSE_ICD_CODE_MAP_Y2024M05D16.XLSX
Ignored: data/icd/IHME_GBD_2021_NONFATAL_CAUSE_ICD_CODE_MAP_Y2024M05D16.XLSX
Ignored: data/icd/UK_Biobank_master_file.tsv
Ignored: data/icd/cdc_valid_icd10_Sep_23_2025.xlsx
Ignored: data/icd/cdc_valid_icd9_Sep_23_2025.xlsx
Ignored: data/icd/hp_umls_mapping.csv
Ignored: data/icd/lancet_conditions_icd10.xlsx
Ignored: data/icd/manual_disease_icd10_mappings.xlsx
Ignored: data/icd/mondo_umls_mapping.csv
Ignored: data/icd/phecode_international_version_unrolled.csv
Ignored: data/icd/phecode_to_icd10_manual_mapping.xlsx
Ignored: data/icd/semiautomatic_ICD-pheno.txt
Ignored: data/icd/semiautomatic_ICD-pheno_UKB_subset.txt
Ignored: data/icd/umls-2025AA-mrconso.zip
Ignored: doccano_venv/
Ignored: figures/
Ignored: output/.DS_Store
Ignored: output/abstracts/
Ignored: output/doccano/
Ignored: output/fulltexts/
Ignored: output/gwas_cat/
Ignored: output/gwas_cohorts/
Ignored: output/icd_map/
Ignored: output/pubmedbert_entity_predictions.csv
Ignored: output/pubmedbert_entity_predictions.jsonl
Ignored: output/pubmedbert_predictions.csv
Ignored: output/pubmedbert_predictions.jsonl
Ignored: output/supplement/
Ignored: output/text_mining_predictions/
Ignored: output/trait_ontology/
Ignored: population_description_terms.txt
Ignored: pubmedbert-cohort-ner-model/
Ignored: pubmedbert-cohort-ner/
Ignored: renv/
Ignored: spacy_venv_requirements.txt
Ignored: spacyr_venv/
Untracked files:
Untracked: code/full_text_conversion/html_to_xml.R
Untracked: code/text_mining_models/tokenise_data.py
Untracked: schools.R
Unstaged changes:
Modified: analysis/disease_inves_by_ancest.Rmd
Modified: analysis/get_dbgap_ids.Rmd
Modified: analysis/group_cancer_diseases.Rmd
Modified: analysis/gwas_to_gbd.Rmd
Modified: analysis/map_trait_to_icd10.Rmd
Modified: analysis/replication_ancestry_bias.Rmd
Modified: analysis/text_for_cohort_labels.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/trait_ontology_categorization.Rmd) and HTML
(docs/trait_ontology_categorization.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2dc80c9 | IJbeasley | 2026-03-25 | Fixing disease paper selection mistakes |
| html | 1d36e53 | IJbeasley | 2026-03-20 | Build site. |
| Rmd | 86b7c26 | IJbeasley | 2026-03-20 | Update trait ontology categorization |
| html | 31383df | IJbeasley | 2026-01-12 | Build site. |
| Rmd | 23ef969 | IJbeasley | 2026-01-12 | Update initial trait categorisation |
| html | 2ddf5ba | IJbeasley | 2026-01-05 | Build site. |
| Rmd | 21f31d9 | IJbeasley | 2026-01-05 | Update filtering of GWAS traits |
| html | 522a96e | IJbeasley | 2026-01-03 | Build site. |
| Rmd | 0a0162d | IJbeasley | 2026-01-03 | Removing non-specific disease terms |
| html | 19fb675 | IJbeasley | 2026-01-03 | Build site. |
| Rmd | 767dda0 | IJbeasley | 2026-01-03 | Update fixing of trait mapping |
| html | 34fc448 | IJbeasley | 2025-12-29 | Build site. |
| Rmd | e56780f | IJbeasley | 2025-12-29 | Fixing commas |
| html | 1f666c4 | IJbeasley | 2025-12-29 | Build site. |
| Rmd | 6cbdd3c | IJbeasley | 2025-12-29 | Fixing some comma-induced errors in trait mapping |
| html | 99a061d | IJbeasley | 2025-12-29 | Build site. |
| Rmd | 206b33c | IJbeasley | 2025-12-29 | Updating identifying disease studies step to keep STUDY column |
| html | 8212b8b | IJbeasley | 2025-12-29 | Build site. |
| Rmd | f1b9ff4 | IJbeasley | 2025-12-29 | Updating identifying disease studies step |
| html | 5e4dc04 | IJbeasley | 2025-09-14 | Build site. |
| Rmd | 6846cca | IJbeasley | 2025-09-14 | Fixing typos on intital trait categorization .. again |
| html | cba6936 | IJbeasley | 2025-09-14 | Build site. |
| Rmd | 14b700b | IJbeasley | 2025-09-14 | Fixing typos on intital trait categorization |
| html | 6a8f9cc | IJbeasley | 2025-09-10 | Build site. |
| Rmd | 63bd79a | IJbeasley | 2025-09-10 | Update cancer grouping |
| html | 2853e61 | IJbeasley | 2025-09-10 | Build site. |
| Rmd | a7e2f7c | IJbeasley | 2025-09-10 | Fixing / re-formatting of initial trait categorization |
1 Set up
knitr::opts_chunk$set(echo = TRUE,
message = FALSE,
warning = FALSE
)
library(data.table)
library(dplyr)
library(ggplot2)
library(stringr)1.1 Get data.frame of GWAS traits
gwas_study_info <- fread(here::here("data/gwas_catalog/gwas-catalog-v1.0.3.1-studies-r2025-07-21.tsv"))
gwas_study_info =
gwas_study_info |>
rename_all(~gsub(" ", "_", .x))
gwas_study_info <-
gwas_study_info |>
mutate(MAPPED_TRAIT = tolower(MAPPED_TRAIT),
MAPPED_BACKGROUND_TRAIT = tolower(MAPPED_BACKGROUND_TRAIT)
)
gwas_study_info <-
gwas_study_info |>
mutate(YEAR = lubridate::year(DATE))1.2 Add mapped ontology terms for studies with unmapped traits:
# some traits are not mapped:
print("Before fixing, how many unmapped traits are there?")[1] "Before fixing, how many unmapped traits are there?"gwas_study_info |>
filter(is.na(MAPPED_TRAIT) | MAPPED_TRAIT == "") |>
nrow()[1] 17# by Zoom, Anomalous atrioventricular excitation
# -> Anomalous atrioventricular excitation (disorder)
# http://snomed.info/id/17869006
unmapped_traits <-
data.frame("DISEASE/TRAIT" = "Anomalous atrioventricular excitation (PheCode 426.4)",
MAPPED_TRAIT = "anomalous atrioventricular excitation (disorder)",
MAPPED_TRAIT_URI = "http://snomed.info/id/17869006",
stringsAsFactors = FALSE
) |>
rename(`DISEASE/TRAIT` = "DISEASE.TRAIT")
# by Zooma, Pilocytic astrocytoma -> MONDO_0016691 (http://purl.obolibrary.org/obo/MONDO_0016691)
unmapped_traits <-
unmapped_traits |>
add_row(`DISEASE/TRAIT` = "Pilocytic astrocytoma",
MAPPED_TRAIT = "pilocytic astrocytoma",
MAPPED_TRAIT_URI = "http://purl.obolibrary.org/obo/MONDO_0016691"
)
# by Zooma, Pilocytic astrocytoma and optic pathway glioma
# -> http://purl.obolibrary.org/obo/MONDO_0016167, http://purl.obolibrary.org/obo/MONDO_0016691
unmapped_traits =
unmapped_traits |>
add_row(`DISEASE/TRAIT` = "Pilocytic astrocytoma and optic pathway glioma",
MAPPED_TRAIT = "optic pathway glioma, pilocytic astrocytoma",
MAPPED_TRAIT_URI = "http://purl.obolibrary.org/obo/MONDO_0016167, http://purl.obolibrary.org/obo/MONDO_0016691"
)
# by searching ontology lookup service:
# Leukotriene levels (480.2454_0.351) & Leukotriene levels (337.1632_0.339)
# -> Fatty Acid Measurement
# http://purl.obolibrary.org/obo/NCIT_C80157
unmapped_traits =
unmapped_traits |>
add_row(`DISEASE/TRAIT` = c("Leukotriene levels (480.2454_0.351)",
"Leukotriene levels (337.1632_0.339)"
),
MAPPED_TRAIT = c("fatty acid measurement",
"fatty acid measurement"
),
MAPPED_TRAIT_URI = c("http://purl.obolibrary.org/obo/NCIT_C80157",
"http://purl.obolibrary.org/obo/NCIT_C80157"
)
)
# by searching ontology lookup service:
# X-11244 levels
# X-11255 levels
# to be mapped to: http://www.ebi.ac.uk/efo/EFO_0004725
# metabolite measurement
unmapped_traits =
unmapped_traits |>
add_row(`DISEASE/TRAIT` = c("X-11244 levels",
"X-11255 levels"
),
MAPPED_TRAIT = c("metabolite measurement",
"metabolite measurement"
),
MAPPED_TRAIT_URI = c("http://www.ebi.ac.uk/efo/EFO_0004725",
"http://www.ebi.ac.uk/efo/EFO_0004725"
)
)
# by searching ontology lookup service:
# N-acetylornithine levels, & N-acetylornithine levels in chronic kidney disease
# to be mapped to http://www.ebi.ac.uk/efo/EFO_0021538
# N-acetylornithine measurement
unmapped_traits =
unmapped_traits |>
add_row(`DISEASE/TRAIT` = c("N-acetylornithine levels",
"N-acetylornithine levels in chronic kidney disease"
),
MAPPED_TRAIT = c("n-acetylornithine measurement",
"n-acetylornithine measurement"
),
MAPPED_TRAIT_URI = c("http://www.ebi.ac.uk/efo/EFO_0021538",
"http://www.ebi.ac.uk/efo/EFO_0021538"
)
)
# by searching ontology lookup service:
# Scleritis and episcleritis (PheCode 379.1)
# map to: Scleritis and episcleritis (disorder)
# http://snomed.info/id/267659002
unmapped_traits =
unmapped_traits |>
add_row(`DISEASE/TRAIT` = "Scleritis and episcleritis (PheCode 379.1)",
MAPPED_TRAIT = "scleritis and episcleritis (disorder)",
MAPPED_TRAIT_URI = "http://snomed.info/id/267659002"
)
# Add the unmapped traits to the gwas study info df, by matching on DISEASE/TRAIT
# and making unmmaped traits are NA
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(MAPPED_TRAIT == "",
NA,
MAPPED_TRAIT))
gwas_study_info =
gwas_study_info |>
rows_patch(unmapped_traits,
by = c("DISEASE/TRAIT"),
unmatched = "ignore"
)
# yay all mapped now
print("After fixing, how many unmapped traits remain?")[1] "After fixing, how many unmapped traits remain?"gwas_study_info |>
filter(is.na(MAPPED_TRAIT) | MAPPED_TRAIT == "") |>
nrow()[1] 01.3 Add DISEASE/TRAIT description for studies where this is missing
print("Before fixing, how many studies have missing DISEASE/TRAIT?")[1] "Before fixing, how many studies have missing DISEASE/TRAIT?"gwas_study_info |>
filter(`DISEASE/TRAIT` == ""| is.na(`DISEASE/TRAIT`)) |>
nrow()[1] 6# pubmed id: 35240980
gwas_study_info |>
filter(PUBMED_ID == 35240980) |>
select(`DISEASE/TRAIT`,
MAPPED_TRAIT,
MAPPED_BACKGROUND_TRAIT) DISEASE/TRAIT MAPPED_TRAIT MAPPED_BACKGROUND_TRAIT
<char> <char> <char>
1: cognitive impairment premature birth# DISEASE/TRAIT == "Cognitive impairment among children born extremely preterm"
gwas_study_info =
gwas_study_info |>
mutate(`DISEASE/TRAIT` = ifelse(PUBMED_ID == 35240980,
"Cognitive impairment among children born extremely preterm",
`DISEASE/TRAIT`)
)
# study accession: GCST90624363
gwas_study_info |>
filter(STUDY_ACCESSION == "GCST90624363") |>
select(`DISEASE/TRAIT`,
MAPPED_TRAIT,
MAPPED_BACKGROUND_TRAIT) DISEASE/TRAIT MAPPED_TRAIT MAPPED_BACKGROUND_TRAIT
<char> <char> <char>
1: lyme disease # DISEASE/TRAIT == "Lyme borreliosis"
gwas_study_info =
gwas_study_info |>
mutate(`DISEASE/TRAIT` = ifelse(STUDY_ACCESSION == "GCST90624363",
"Lyme borreliosis",
`DISEASE/TRAIT`)
)
# pubmed id: 38509478
# Nausea and vomiting during pregnancy
gwas_study_info |>
filter(PUBMED_ID == 38509478) |>
select(`DISEASE/TRAIT`,
MAPPED_TRAIT,
MAPPED_BACKGROUND_TRAIT) DISEASE/TRAIT
<char>
1:
2:
3:
4:
5: Severity of nausea and vomiting of pregnancy
MAPPED_TRAIT
<char>
1: nausea and vomiting, hyperemesis gravidarum
2: hyperemesis gravidarum
3: hyperemesis gravidarum
4: hyperemesis gravidarum
5: nausea and vomiting of pregnancy severity measurement
MAPPED_BACKGROUND_TRAIT
<char>
1:
2:
3:
4:
5: gwas_study_info =
gwas_study_info |>
mutate(`DISEASE/TRAIT` = ifelse(PUBMED_ID == 38509478,
"Nausea and vomiting during pregnancy",
`DISEASE/TRAIT`)
)
print("After fixing, how many studies have missing DISEASE/TRAIT?")[1] "After fixing, how many studies have missing DISEASE/TRAIT?"gwas_study_info |>
filter(`DISEASE/TRAIT` == ""| is.na(`DISEASE/TRAIT`)) |>
nrow()[1] 01.4 Correcting some MAPPED_TRAIT (MAPPED to ontology terms by GWAS Catalog)
1.4.1 Re-map some traits based on DISEASE/TRAIT
1.4.1.1 Non-specific disease mapping
# STUDY ACCESSION: GCST90043814
# MAPPED_TRAIT == "disease"
# replace with otitis media
# EFO_0004992
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(STUDY_ACCESSION == "GCST90043814" &
MAPPED_TRAIT == "disease",
"otitis media",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(STUDY_ACCESSION == "GCST90043814" &
MAPPED_TRAIT_URI == "http://www.ebi.ac.uk/efo/EFO_0000408",
"http://www.ebi.ac.uk/efo/EFO_0004992",
MAPPED_TRAIT_URI)
)
# GCST90244761
# remove MAPPED_BACKGROUND_TRAIT and MAPPED_BACKGROUND_TRAIT_URI
# (which were just 'disease')
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(STUDY_ACCESSION == "GCST90244761",
"",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(STUDY_ACCESSION == "GCST90244761",
"",
MAPPED_BACKGROUND_TRAIT_URI)
)1.4.1.3 TB
# PUBMED_ID: 33661925
# `DISEASE/TRAIT contains resistance to mycobacterium tuberculosis infection
# replace MAPPED_TRAIT == "decreased susceptibility to bacterial infection"
# with "tuberculosis"
# MAPPED_TRAIT_URI from "http://www.ebi.ac.uk/efo/EFO_0008322"
# to "http://purl.obolibrary.org/obo/MONDO_0018076"
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(PUBMED_ID == 33661925 &
MAPPED_TRAIT == "decreased susceptibility to bacterial infection",
"tuberculosis",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(PUBMED_ID == 33661925 &
MAPPED_TRAIT_URI == "http://www.ebi.ac.uk/efo/EFO_0008322",
"http://purl.obolibrary.org/obo/MONDO_0018076",
MAPPED_TRAIT_URI)
)
# for PUBMED = 28628665
# MAPPED_TRAIT = "decreased susceptibility to bacterial infection"
# MAPPED_TRAIT_URI = "http://www.ebi.ac.uk/efo/EFO_0008322"
# replace MAPPED_TRAIT to "tuberculosis"
# MAPPED_TRAIT_URI to "http://purl.obolibrary.org/obo/MONDO_0018076"
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(PUBMED_ID == 28628665 &
MAPPED_TRAIT == "tuberculin skin test reactivity measurement, decreased susceptibility to bacterial infection",
"tuberculosis",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(PUBMED_ID == 28628665 &
MAPPED_TRAIT_URI == "http://www.ebi.ac.uk/efo/EFO_0008307, http://www.ebi.ac.uk/efo/EFO_0008322",
"http://purl.obolibrary.org/obo/MONDO_0018076",
MAPPED_TRAIT_URI)
)1.4.1.4 Other bad mappings
## pubmed id: 34737426
# mapped_trait = benign neoplasm
# but should be cervical carcinoma, EFO_0001061
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(PUBMED_ID == 34737426 &
MAPPED_TRAIT == "benign neoplasm",
"cervical carcinoma",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(PUBMED_ID == 34737426 &
MAPPED_TRAIT_URI == "http://purl.obolibrary.org/obo/MONDO_0021230",
"http://www.ebi.ac.uk/efo/EFO_0001061",
MAPPED_TRAIT_URI)
)
# Capecitabine-induced hand-foot syndrome in breast or colorectal cancer
gwas_study_info =
gwas_study_info |>
mutate(
MAPPED_TRAIT =
ifelse(grepl("Capecitabine-induced hand-foot syndrome in breast or colorectal cancer", `DISEASE/TRAIT`),
"breast cancer, colorectal cancer, hand-foot syndrome",
MAPPED_TRAIT
),
MAPPED_TRAIT_URI =
ifelse(grepl("Capecitabine-induced hand-foot syndrome in breast or colorectal cancer", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/MONDO_0007254, http://purl.obolibrary.org/obo/MONDO_0005575, http://purl.obolibrary.org/obo/MONDO_0700048",
MAPPED_TRAIT_URI
)
)
# GCST001789
# replace MAPPED_TRAIT from "bronchopulmonary dysplasia"
# to "blood high density lipoprotein particle diameter"
# and MAPPED_TRAIT_URI to "http://purl.obolibrary.org/obo/CMO_0002692"
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(STUDY_ACCESSION == "GCST001789" &
MAPPED_TRAIT == "bronchopulmonary dysplasia",
"blood high density lipoprotein particle diameter",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(STUDY_ACCESSION == "GCST001789" &
MAPPED_TRAIT_URI == "http://purl.obolibrary.org/obo/MONDO_0019091",
"http://purl.obolibrary.org/obo/CMO_0002692",
MAPPED_TRAIT_URI)
)
# Periodontal disease related phenotype
# set trait to periodontal disorder
# rather than periodontal measurement
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(grepl("Periodontal disease related phenotype", `DISEASE/TRAIT`),
str_replace_all(pattern = "periodontal measurement",
replacement = "periodontal disorder",
MAPPED_TRAIT
),
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(grepl("Periodontal disease related phenotype", `DISEASE/TRAIT`),
str_replace_all(pattern = "http://www.ebi.ac.uk/efo/EFO_0007780",
replacement = "http://purl.obolibrary.org/obo/MONDO_0002635",
MAPPED_TRAIT_URI
),
MAPPED_TRAIT_URI)
)1.4.2 Add and correct MAPPED_BACKGROUND_TRAIT terms
# for MAPPED_TRAIT contains "sars-cov-2"
# set background trait to covid-19
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(grepl("sars-cov-2", MAPPED_TRAIT),
"covid-19",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(grepl("sars-cov-2", MAPPED_TRAIT),
"http://purl.obolibrary.org/obo/MONDO_0100096",
MAPPED_BACKGROUND_TRAIT_URI)
)
# for pubmed id: 32247823
# set background trait to non-alcoholic steatohepatitis
# and background trait uri to EFO_1001249
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(PUBMED_ID == 32247823,
"non-alcoholic steatohepatitis",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(PUBMED_ID == 32247823,
"http://www.ebi.ac.uk/efo/EFO_1001249",
MAPPED_BACKGROUND_TRAIT_URI)
)
# Exploratory eye movement dysfunction in schizophrenia
# set background trait to schizophrenia
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(grepl("Exploratory eye movement dysfunction in schizophrenia", `DISEASE/TRAIT`),
"schizophrenia",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(grepl("Exploratory eye movement dysfunction in schizophrenia", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/MONDO_0005090",
MAPPED_BACKGROUND_TRAIT_URI)
)
# for pubmed_id: 21107309
# set background trait to schizophrenia
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(PUBMED_ID == 21107309,
"schizophrenia",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(PUBMED_ID == 21107309,
"http://purl.obolibrary.org/obo/MONDO_0005090",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if `DISEASE/TRAIT` contains Adverse response to chemotherapy in breast cancer
# set MAPPED_BACKGROUND_TRAIT to breast cancer
# http://purl.obolibrary.org/obo/MONDO_0007254
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(grepl("Adverse response to chemotherapy in breast cancer", `DISEASE/TRAIT`),
"breast cancer",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(grepl("Adverse response to chemotherapy in breast cancer", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/MONDO_0007254",
MAPPED_BACKGROUND_TRAIT_URI)
)
# for pubmed id: 30188897
# and DISEASE/TRAIT contains "miscarriages"
# add spontaneous abortion, http://www.ebi.ac.uk/efo/EFO_1001255
# as MAPPED_BACKGROUND_TRAIT
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(PUBMED_ID == 30188897 &
grepl("miscarriage", `DISEASE/TRAIT`),
"spontaneous abortion",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(PUBMED_ID == 30188897 &
grepl("miscarriage", `DISEASE/TRAIT`),
"http://www.ebi.ac.uk/efo/EFO_1001255",
MAPPED_BACKGROUND_TRAIT_URI)
)
# for pubmed id: 30188897
# and DISEASE/TRAIT contains "stillbirth"
# add stillbirth, http://purl.obolibrary.org/obo/NCIT_C49151
# as MAPPED_BACKGROUND_TRAIT
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(PUBMED_ID == 30188897 &
grepl("stillbirth", `DISEASE/TRAIT`),
"stillbirth",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(PUBMED_ID == 30188897 &
grepl("stillbirth", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/NCIT_C49151",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "remission"
# and "Hepatitis C" is in DISEASE/TRAIT, then
# set MAPPED_BACKGROUND_TRAIT to hepatitis C virus infection
# and MAPPED_BACKGROUND_TRAIT_URI to: http://purl.obolibrary.org/obo/MONDO_0005231
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "remission" &
grepl("Hepatitis C", `DISEASE/TRAIT`, ignore.case = T),
"hepatitis C virus infection",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "hepatitis C virus infection",
"http://purl.obolibrary.org/obo/MONDO_0005231",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "progression free survival"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains epithelial ovarian cancer
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "progression free survival" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("epithelial ovarian cancer", `DISEASE/TRAIT`, ignore.case = T),
"ovarian cancer",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "ovarian cancer" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://purl.obolibrary.org/obo/MONDO_0008170",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "progression free survival"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains colorectal cancer
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "progression free survival" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("colorectal cancer|colon cancer", `DISEASE/TRAIT`, ignore.case = T),
"colorectal cancer",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "colorectal cancer" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://purl.obolibrary.org/obo/MONDO_0005575",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "progression free survival"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains leukemia
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "progression free survival" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("leukemia", `DISEASE/TRAIT`, ignore.case = T),
"leukemia",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "leukemia" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://www.ebi.ac.uk/efo/EFO_0000565",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "progression free survival"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains glioma
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "progression free survival" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("glioma", `DISEASE/TRAIT`, ignore.case = T),
"glioma",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "glioma" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://www.ebi.ac.uk/efo/EFO_0005543",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "progression free survival"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains bladder cancer
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "progression free survival" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("bladder cancer", `DISEASE/TRAIT`, ignore.case = T),
"urinary bladder carcinoma",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "urinary bladder carcinoma" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://purl.obolibrary.org/obo/MONDO_0004986",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "progression free survival"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains lung adenocarcinoma
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "progression free survival" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("lung adenocarcinoma", `DISEASE/TRAIT`, ignore.case = T),
"lung adenocarcinoma",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "lung adenocarcinoma" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://www.ebi.ac.uk/efo/EFO_0000571",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "survival time"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains prostate cancer
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "survival time" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("prostate cancer", `DISEASE/TRAIT`, ignore.case = T),
"prostate cancer",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "prostate cancer" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://purl.obolibrary.org/obo/DOID_10283",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "survival time"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains breast cancer
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "survival time" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("breast cancer", `DISEASE/TRAIT`, ignore.case = T),
"breast cancer",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "breast cancer" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://purl.obolibrary.org/obo/MONDO_0007254",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "survival time"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains colorectal cancer
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "survival time" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("colorectal cancer|colon cancer|rectal cancer", `DISEASE/TRAIT`, ignore.case = T),
"colorectal cancer",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "colorectal cancer" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://purl.obolibrary.org/obo/MONDO_0005575",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "survival time"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains lung cancer
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "survival time" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("lung cancer", `DISEASE/TRAIT`, ignore.case = T),
"lung cancer",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "lung cancer" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://purl.obolibrary.org/obo/MONDO_0008903",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "illness severity status"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains hand, foot, and mouth disease
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "illness severity status" &
MAPPED_BACKGROUND_TRAIT == "" &
grepl("hand, foot, and mouth disease", `DISEASE/TRAIT`, ignore.case = T),
"hand, foot, and mouth disease",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_BACKGROUND_TRAIT == "hand, foot, and mouth disease" &
MAPPED_BACKGROUND_TRAIT_URI == "",
"http://www.ebi.ac.uk/efo/EFO_0007294",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "response to covid-19 vaccine"
# set MAPPED_BACKGROUND_TRAIT to covid-19
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "response to covid-19 vaccine",
"covid-19",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_TRAIT == "response to covid-19 vaccine",
"http://purl.obolibrary.org/obo/MONDO_0100096",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "response to vaccine"
# and DISEASE/TRAIT contains "smallpox"
# set MAPPED_BACKGROUND_TRAIT to smallpox
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "response to vaccine" &
grepl("smallpox", `DISEASE/TRAIT`),
"smallpox",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_TRAIT == "response to vaccine" &
grepl("smallpox", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/DOID_8736",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "response to vaccine"
# and DISEASE/TRAIT contains "influenza"
# set MAPPED_BACKGROUND_TRAIT to influenza
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "response to vaccine" &
grepl("influenza", `DISEASE/TRAIT`),
"influenza",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_TRAIT == "response to vaccine" &
grepl("influenza", `DISEASE/TRAIT`),
"http://www.ebi.ac.uk/efo/EFO_0007328",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if. MAPPED_TRAIT == "response to vaccine"
# and DISEASE/TRAIT contains "hepatitis B"
# set MAPPED_BACKGROUND_TRAIT to hepatitis B
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "response to vaccine" &
grepl("hepatitis B|Hepatitis B", `DISEASE/TRAIT`),
"hepatitis b",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_TRAIT == "response to vaccine" &
grepl("hepatitis B|Hepatitis B", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/DOID_2043",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "response to covid-19 vaccine"
# and MAPPED_BACKGROUND_TRAIT == ""
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "response to covid-19 vaccine" &
(is.na(MAPPED_BACKGROUND_TRAIT) | MAPPED_BACKGROUND_TRAIT == ""),
"covid-19",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_TRAIT == "response to covid-19 vaccine" &
(is.na(MAPPED_BACKGROUND_TRAIT_URI) | MAPPED_BACKGROUND_TRAIT_URI == ""),
"http://purl.obolibrary.org/obo/MONDO_0100096",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "response to vaccine"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains "Immune response to smallpox"
# set MAPPED_BACKGROUND_TRAIT to smallpox
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "response to vaccine" &
(is.na(MAPPED_BACKGROUND_TRAIT) | MAPPED_BACKGROUND_TRAIT == "") &
grepl("Immune response to smallpox", `DISEASE/TRAIT`),
"smallpox",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_TRAIT == "response to vaccine" &
(is.na(MAPPED_BACKGROUND_TRAIT_URI) | MAPPED_BACKGROUND_TRAIT_URI == "") &
grepl("Immune response to smallpox", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/OMIT_0013787",
MAPPED_BACKGROUND_TRAIT_URI)
)
# if MAPPED_TRAIT == "response to vaccine"
# and MAPPED_BACKGROUND_TRAIT == ""
# and DISEASE/TRAIT contains "Immune response to measles vaccine"
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(MAPPED_TRAIT == "response to vaccine" &
(is.na(MAPPED_BACKGROUND_TRAIT) | MAPPED_BACKGROUND_TRAIT == "") &
grepl("Immune response to measles vaccine", `DISEASE/TRAIT`),
"measles",
MAPPED_BACKGROUND_TRAIT)
) |>
mutate(MAPPED_BACKGROUND_TRAIT_URI = ifelse(MAPPED_TRAIT == "response to vaccine" &
(is.na(MAPPED_BACKGROUND_TRAIT_URI) | MAPPED_BACKGROUND_TRAIT_URI == "") &
grepl("Immune response to measles vaccine", `DISEASE/TRAIT`),
"http://purl.obolibrary.org/obo/DOID_8622",
MAPPED_BACKGROUND_TRAIT_URI)
)1.5 Replacing * for , inside a MAPPED_TERM
To ensure splitting MAPPED_TERM column by commas would split distinct traits
# in MAPPED_BACKGROUND_TRAIT, replace commas with "*" in:
# migraine without aura, susceptibility to, 4
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_BACKGROUND_TRAIT = ifelse(grepl("migraine without aura, susceptibility to, 4", MAPPED_BACKGROUND_TRAIT),
stringr::str_replace_all(MAPPED_BACKGROUND_TRAIT,
pattern = "migraine without aura, susceptibility to, 4",
"migraine without aura* susceptibility to* 4"),
MAPPED_BACKGROUND_TRAIT)
)
# in MAPPED_TRAIT, replace commas with "*" in:
# migraine without aura, susceptibility to, 4
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(grepl("migraine without aura, susceptibility to, 4", MAPPED_TRAIT),
stringr::str_replace_all(MAPPED_TRAIT,
pattern = "migraine without aura, susceptibility to, 4",
"migraine without aura* susceptibility to* 4"),
MAPPED_TRAIT)
)
# Other MAPPED_TRAIT fixes
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = case_when(
# osteoarthritis, hip ... http://www.ebi.ac.uk/efo/EFO_1000786
grepl("EFO_1000786", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "osteoarthritis, hip",
"osteoarthritis* hip"),
# osteoarthritis, hand ... http://www.ebi.ac.uk/efo/EFO_1000789
grepl("EFO_1000789", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "osteoarthritis, hand",
"osteoarthritis* hand"
),
# osteoarthritis, knee ... http://www.ebi.ac.uk/efo/EFO_0004616
grepl("EFO_0004616", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "osteoarthritis, knee",
"osteoarthritis* knee"),
# osteoarthritis, spine ... http://www.ebi.ac.uk/efo/EFO_1000787
grepl("EFO_1000787", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "osteoarthritis, spine",
"osteoarthritis* spine"),
# Hepatitis, Alcoholic, http://www.ebi.ac.uk/efo/EFO_1001345
grepl("EFO_1001345", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "hepatitis, alcoholic",
"hepatitis* alcoholic"),
# psoriasis 14, pustular http://purl.obolibrary.org/obo/MONDO_0013626
grepl("MONDO_0013626", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "psoriasis 14, pustular",
"psoriasis 14* pustular"),
# hypertension, pregnancy-induced http://purl.obolibrary.org/obo/MONDO_0024664
grepl("MONDO_0024664", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "hypertension, pregnancy-induced",
"hypertension* pregnancy-induced"),
# renal agenesis, unilateral http://purl.obolibrary.org/obo/MONDO_0019636
grepl("MONDO_0019636", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "renal agenesis, unilateral",
"renal agenesis* unilateral"),
# Cholecystitis, Acute http://www.ebi.ac.uk/efo/EFO_1001289
grepl("EFO_1001289", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "cholecystitis, acute",
"cholecystitis* acute"),
# Genital neoplasm, female http://www.ebi.ac.uk/efo/EFO_1001331
grepl("EFO_1001331", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "genital neoplasm, female",
"genital neoplasm* female"),
# Anemia, Hemolytic, Autoimmune http://www.ebi.ac.uk/efo/EFO_1001264
grepl("EFO_1001264", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "anemia, hemolytic, autoimmune",
"anemia* hemolytic* autoimmune"),
grepl("EFO_1002020", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "polyarticular juvenile idiopathic arthritis, rheumatoid factor negative",
"polyarticular juvenile idiopathic arthritis* rheumatoid factor negative"),
# http://www.ebi.ac.uk/efo/EFO_0007294, hand, foot and mouth disease,
grepl("EFO_0007294", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "hand, foot and mouth disease",
"hand* foot and mouth disease"),
# neural tube defects, susceptibility to, http://purl.obolibrary.org/obo/MONDO_0020705
grepl("MONDO_0020705", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "neural tube defects, susceptibility to",
"neural tube defects* susceptibility to"),
# self-reported traits
grepl("EFO_0009803|EFO_0009822|EFO_0009803|EFO_0009817|EFO_0009822|EFO_0009819|EFO_0009823|EFO_0009824", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = ", self-reported$",
"* self-reported"),
# Hodgkins lymphoma, mixed cellularity http://www.ebi.ac.uk/efo/EFO_1002031
grepl("EFO_1002031", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "hodgkins lymphoma, mixed cellularity",
"hodgkins lymphoma* mixed cellularity"),
# encephalopathy, acute, infection-induced, http://purl.obolibrary.org/obo/MONDO_0000166
grepl("MONDO_0000166", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "encephalopathy, acute, infection-induced",
"encephalopathy* acute* infection-induced"),
# Diarrhea, Infantile http://www.ebi.ac.uk/efo/EFO_1001306
grepl("EFO_1001306", MAPPED_TRAIT_URI) ~ stringr::str_replace_all(MAPPED_TRAIT,
pattern = "diarrhea, infantile",
"diarrhea* infantile"),
TRUE ~ MAPPED_TRAIT
)
)
# in MAPPED_TRAIT, replace commas with "*" in:
# chromosome, telomeric region length
# fractures, ununited
# osteoarthritis, knee ... http://www.ebi.ac.uk/efo/EFO_0004616
# localized superficial swelling, mass, or lump
# cys-gly, oxidized measurement
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(grepl("chromosome, telomeric region length", MAPPED_TRAIT),
stringr::str_replace_all(MAPPED_TRAIT,
pattern = "chromosome, telomeric region length",
"chromosome* telomeric region length"),
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT = ifelse(grepl("fractures, ununited", MAPPED_TRAIT),
stringr::str_replace_all(MAPPED_TRAIT,
pattern = "fractures, ununited",
"fractures* ununited"),
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT = # osteoarthritis, knee ... http://www.ebi.ac.uk/efo/EFO_0004616
ifelse(grepl("EFO_0004616", MAPPED_TRAIT_URI),
stringr::str_replace_all(MAPPED_TRAIT,
pattern = "osteoarthritis, knee",
"osteoarthritis* knee"),
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT = ifelse(grepl("localized superficial swelling, mass, or lump", MAPPED_TRAIT),
stringr::str_replace_all(MAPPED_TRAIT,
pattern = "localized superficial swelling, mass, or lump",
"localized superficial swelling* mass* or lump"),
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT = ifelse(grepl("cys-gly, oxidized measurement", MAPPED_TRAIT),
stringr::str_replace_all(MAPPED_TRAIT,
pattern = "cys-gly, oxidized measurement",
"cys-gly* oxidized measurement"),
MAPPED_TRAIT)
)1.5.1 Fix by the number of commas
# fixing weird terms- where comma is in the term
# count number of separating commas in
# MAPPED_TRAIT, MAPPED_TRAIT_URI, MAPPED_BACKGROUND_TRAIT, MAPPED_BACKGROUND_TRAIT_URI
gwas_study_info =
gwas_study_info |>
mutate(n_commas_trait = str_count(MAPPED_TRAIT, ", (?![^()]*\\))"),
#", "),
n_commas_trait_uri = str_count(MAPPED_TRAIT_URI, ", (?![^()]*\\))"),
n_commas_bg_trait = str_count(MAPPED_BACKGROUND_TRAIT, ", (?![^()]*\\))"),
n_commas_bg_trait_uri = str_count(MAPPED_BACKGROUND_TRAIT_URI, ", (?![^()]*\\))")
# ", ")
) |>
# select(contains("n_commas"),
# MAPPED_TRAIT, MAPPED_TRAIT_URI,
# MAPPED_BACKGROUND_TRAIT, MAPPED_BACKGROUND_TRAIT_URI
# ) |>
distinct()
# if n_commas_trait >= 1, n_commas_trait_uri == 0,
# replace comma in MAPPED_TRAIT with "*"
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = ifelse(n_commas_trait >= 1 &
n_commas_trait_uri == 0,
stringr::str_replace_all(MAPPED_TRAIT,
pattern = ", ",
"* "),
MAPPED_TRAIT)
)
# now that's been correct, recalculate number of commas
gwas_study_info =
gwas_study_info |>
mutate(n_commas_trait = str_count(MAPPED_TRAIT, ", (?![^()]*\\))"),
#", "),
n_commas_trait_uri = str_count(MAPPED_TRAIT_URI, ", (?![^()]*\\))"),
n_commas_bg_trait = str_count(MAPPED_BACKGROUND_TRAIT, ", (?![^()]*\\))"),
n_commas_bg_trait_uri = str_count(MAPPED_BACKGROUND_TRAIT_URI, ", (?![^()]*\\))")
# ", ")
)
# check the number of commas in MAPPED_TRAIT is always equal to number of commas in MAPPED_TRAIT_URI
# and the number of commas in MAPPED_BACKGROUND_TRAIT is not equal to number of commas in MAPPED_BACKGROUND_TRAIT_URI
gwas_study_info =
gwas_study_info |>
mutate(match_comma_trait = ifelse(n_commas_trait != n_commas_trait_uri,
FALSE,
TRUE),
match_comma_bg_trait = ifelse(n_commas_bg_trait != n_commas_bg_trait_uri,
FALSE,
TRUE)
)
gwas_study_info |>
filter(match_comma_trait == FALSE |
match_comma_bg_trait == FALSE)Empty data.table (0 rows and 32 cols): DATE_ADDED_TO_CATALOG,PUBMED_ID,FIRST_AUTHOR,DATE,JOURNAL,LINK...# yay! all match now2 Overlap ontology terms and GWAS traits
2.1 Create data.frame of GWAS traits with one trait per row
gwas_study_info =
gwas_study_info |>
select(
`DISEASE/TRAIT`,
PUBMED_ID,
YEAR,
STUDY,
STUDY_ACCESSION,
contains("MAPPED")
)
# now split by commas to get each MAPPED_TRAIT on an individual row
gwas_study_info <-
gwas_study_info |>
tidyr::separate_longer_delim(cols = c("MAPPED_TRAIT",
"MAPPED_TRAIT_URI"
),
delim = stringr::regex(", (?![^()]*\\))")
) |>
tidyr::separate_longer_delim(cols = c("MAPPED_BACKGROUND_TRAIT",
"MAPPED_BACKGROUND_TRAIT_URI"
),
delim = stringr::regex(", (?![^()]*\\))")
) |>
distinct()
# now replace '*' back to commas
gwas_study_info =
gwas_study_info |>
mutate(MAPPED_TRAIT = stringr::str_replace_all(MAPPED_TRAIT,
pattern = "\\* ",
", "),
MAPPED_BACKGROUND_TRAIT = stringr::str_replace_all(MAPPED_BACKGROUND_TRAIT,
pattern = "\\* ",
", ")
)
gwas_study_info <-
gwas_study_info |>
mutate(MAPPED_TRAIT = stringr::str_trim(tolower(MAPPED_TRAIT))) |>
mutate(MAPPED_BACKGROUND_TRAIT = stringr::str_trim(tolower(MAPPED_BACKGROUND_TRAIT))) all_gwas_terms = gwas_study_info$MAPPED_TRAIT
all_gwas_terms = stringr::str_trim(tolower(all_gwas_terms))
all_gwas_terms = unique(all_gwas_terms)
print("Number of unique GWAS traits")[1] "Number of unique GWAS traits"length(all_gwas_terms)[1] 199752.2 Disease Overlap (How many GWAS traits fall within disease or disorder terms?)
2.2.1 Combine disease terms
efo_descendants <- readLines(here::here("output/trait_ontology/efo_0000408_descendants.txt"))
mondo_descendants <- readLines(here::here("output/trait_ontology/mondo_0700096_descendants.txt"))
ncit_descendants <- readLines(here::here("output/trait_ontology/ncit_C2991_descendants.txt"))
orphanet_descendants <- readLines(here::here("output/trait_ontology/orphanet_557493_descendants.txt"))
age_of_onset_descendants <- readLines(here::here("output/trait_ontology/oba_2020000_descendants.txt"))
disease_measurement_terms <- readLines(here::here("output/trait_ontology/efo_0001444_disease_measurement_terms.txt"))
disease_typos = c("Alzheimer disease",
"late-onset Alzheimers disease",
"Chagas cardiomyopathy",
"Parkinson disease",
"Iron deficiency anemia"
)
biomarker_terms <- c("cardiovascular disease biomarker measurement",
"cancer biomarker measurement",
"diabetes mellitus biomarker",
"osteoarthritis biomarker measurement",
"liver disease biomarker",
"alzheimer's disease biomarker measurement",
"iron deficiency anemia (disorder)"
)
other_disorders <- c(
"Allergic disease",
"Churg-Strauss syndrome",
"Iridocyclitis",
"Phlebitis",
"pregnancy induced alloimmunization",
"somnambulism",
"suicide",
"attempted suicide",
"suicide behaviour",
"suicide ideation measurement",
"suicide behaviour measurement",
"Lewy body dementia",
"Lewy body attribute",
"non-Hodgkins lymphoma",
"Ischemic Stroke",
"Lung disease",
"Respiratory System Disease",
"Alzheimer disease, APOE carrier status",
"Genital neoplasm, female",
"HIV-associated neurocognitive disorder",
"encephalopathy acute infection-induced",
"anomalous atrioventricular excitation (disorder)",
"scleritis and episcleritis (disorder)",
"atopic march",
"infection",
"neural tube defects, susceptibility to",
"migraine without aura, susceptibility to, 4",
"hiv mother to child transmission",
"hemolysis",
"chromosomal aberration",
"dna methylation",
"gata1 gene mutation",
"atropy",
"premature birth",
"growth delay",
"reduced left ventricular ejection fraction",
"hepatitis b",
"vascular brain injury measurement",
"borderline personality disorder symptom",
"miscarriage",
"emphysema pattern measurement",
"emphysema imaging measurement",
"persistent staphylococcus aureus carrier status",
"intermittent staphylococcus aureus carrier status",
"influenza a severity measurement",
"pneumonia severity measurement",
"hsv2 virologic severity measurement",
"opioid overdose severity measurement",
"nausea and vomiting of pregnancy severity measurement",
"myopic maculopathy severity measurement",
"hepatitis C virus infection"
)
disease_status_terms <- c(
"benign",
"remission",
"disease recurrence",
"complicated disease course",
"disease prognosis measurement",
"mild disease course",
"disease free survival",
"progression free survival",
"survival time",
"overall survival",
"illness severity status"
)
family_disease_terms <- c("family history of breast cancer",
"family history of cancer",
"family history of prostate cancer",
"family history of upper gastrointestinal cancer",
"family history of uterine fibroids")
disease_terms = c(mondo_descendants,
efo_descendants,
ncit_descendants,
orphanet_descendants,
age_of_onset_descendants,
disease_measurement_terms,
family_disease_terms,
disease_typos,
biomarker_terms,
disease_status_terms,
other_disorders) |>
unique()
disease_terms = stringr::str_trim(tolower(disease_terms))
disease_terms = unique(disease_terms)
print("Number of ontology terms found related to disease or disorder")[1] "Number of ontology terms found related to disease or disorder"length(disease_terms)[1] 451462.2.2 Find disease terms in GWAS traits
# Find GWAS traits that fall within disease or disorder terms
#simple_disease_terms = all_gwas_terms[all_gwas_terms %in% disease_terms]
disease_gwas <- all_gwas_terms[all_gwas_terms %in% disease_terms]
not_disease_terms = all_gwas_terms[!all_gwas_terms %in% disease_gwas]
print("Number of GWAS traits under disease or disorder terms")[1] "Number of GWAS traits under disease or disorder terms"length(disease_gwas)[1] 1978print("Percentage of GWAS traits under disease or disorder terms")[1] "Percentage of GWAS traits under disease or disorder terms"round(100 * (length(disease_gwas)) / length(all_gwas_terms),
digits = 1)[1] 9.9print("Percentage of GWAS traits not under disease or disorder terms")[1] "Percentage of GWAS traits not under disease or disorder terms"round(100 * length(not_disease_terms) / length(all_gwas_terms),
digits = 1)[1] 90.1not_accounted_for = not_disease_terms 2.3 Phenotype abnormality overlap
pheno_abnorm <- readLines(here::here("output/trait_ontology/hp_0000118_descendants.txt"))
pheno_abnorm = stringr::str_trim(tolower(pheno_abnorm))
pheno_abnorm = unique(pheno_abnorm)
pheno_abnorm <- c("abnormal pap smear",
"abnormal result of function studies",
"abnormal result of diagnostic imaging",
pheno_abnorm)
pheno_abnorm_gwas <- not_accounted_for[not_accounted_for %in% pheno_abnorm]
print("Percentage of GWAS traits under phenotype abnormality terms")[1] "Percentage of GWAS traits under phenotype abnormality terms"round(100 * length(pheno_abnorm_gwas) / length(all_gwas_terms),
digits = 1)[1] 1.9not_accounted_for = not_accounted_for[!not_accounted_for %in% pheno_abnorm_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 88.2print("Number of GWAS traits not accounted for by so far")[1] "Number of GWAS traits not accounted for by so far"length(not_accounted_for)[1] 176242.4 Measurement Overlap (how many GWAS traits fall within measurement terms?)
2.4.1 Combine measurement
measurement <- readLines(here::here("output/trait_ontology/efo_0001444_descendants.txt"))
total_choles <- readLines(here::here("output/trait_ontology/efo_0004574_descendants.txt"))
measurement <- c(total_choles,
measurement)
measurement <- unique(measurement)
measurement <- c("cerebrospinal fluid composition attribute",
"blood protein amount",
"fatty acid measurement",
"obsolete_3,3',5-triiodo-l-thyronine measurement",
"1-(1-enyl-stearoyl)-2-linoleoyl-gpe (p-18:0/18:2), measurement",
"microtubule-associated protein tau",
measurement)
measurement = stringr::str_trim(tolower(measurement))
measurement = unique(measurement)2.4.2 BMI / weight terms / body fat terms
bmi_weight_terms <- grep("bmi|body mass index|weight|bmi", measurement, value = T)
bmi_weight_terms <- grep("fetal|birth|gestational", bmi_weight_terms, value = T, invert = T)
bmi_weight_terms <- c(bmi_weight_terms,
"body composition measurement",
"body fat percentage",
"body fat distribution")
measurement <- measurement[!(measurement %in% bmi_weight_terms)]
bmi_weight_gwas = not_accounted_for[not_accounted_for %in% bmi_weight_terms]
print("Number of GWAS traits under BMI / weight / body fat terms")[1] "Number of GWAS traits under BMI / weight / body fat terms"length(bmi_weight_gwas)[1] 23print("Percentage of GWAS traits under BMI / weight / body fat terms")[1] "Percentage of GWAS traits under BMI / weight / body fat terms"round(100 * length(bmi_weight_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.1not_accounted_for = not_accounted_for[!not_accounted_for %in% bmi_weight_gwas]
print("Percentage of GWAS traits not accounted for by disease, disorder or BMI / weight / body fat terms")[1] "Percentage of GWAS traits not accounted for by disease, disorder or BMI / weight / body fat terms"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 88.12.4.3 Lipid / cholesterol measurement terms
lipid_cholesterol_terms <- grep("cholesterol|lipid|triglyceride|ldl|hdl|apolipoprotein",
measurement,
value = T)
measurement <- measurement[!(measurement %in% lipid_cholesterol_terms)]
lipid_cholesterol_gwas = not_accounted_for[not_accounted_for %in% lipid_cholesterol_terms]
print("Number of GWAS traits under lipid / cholesterol terms")[1] "Number of GWAS traits under lipid / cholesterol terms"length(lipid_cholesterol_gwas)[1] 253print("Percentage of GWAS traits under lipid / cholesterol terms")[1] "Percentage of GWAS traits under lipid / cholesterol terms"round(100 * length(lipid_cholesterol_gwas) / length(all_gwas_terms),
digits = 1)[1] 1.3not_accounted_for = not_accounted_for[!not_accounted_for %in% lipid_cholesterol_gwas]
print("Percentage of GWAS traits not accounted for by disease, disorder or lipid / cholesterol terms")[1] "Percentage of GWAS traits not accounted for by disease, disorder or lipid / cholesterol terms"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 86.82.4.4 Brain measurement terms
brain_measurement_terms <- grep("brain|volume",
measurement,
value = T)
brain_measurement_terms <- grep("bone|muscle|reticulocyte|erythrocyte|expiratory|platelet|urinary|thyroid|pancreas|kidney|spleen|liver|ventricular|blood",
brain_measurement_terms,
value = T,
invert = T)2.4.5 Blood pressure measurement terms
blood_pressure_terms <- grep("blood pressure",
measurement,
value = T)
measurement <- measurement[!(measurement %in% blood_pressure_terms)]
blood_pressure_gwas = not_accounted_for[not_accounted_for %in% blood_pressure_terms]
print("Number of GWAS traits under blood pressure terms")[1] "Number of GWAS traits under blood pressure terms"length(blood_pressure_gwas)[1] 7print("Percentage of GWAS traits under blood pressure terms")[1] "Percentage of GWAS traits under blood pressure terms"round(100 * length(blood_pressure_gwas) / length(all_gwas_terms),
digits = 1)[1] 0not_accounted_for = not_accounted_for[!not_accounted_for %in% blood_pressure_gwas]
print("Percentage of GWAS traits not accounted for by disease, disorder or blood pressure terms")[1] "Percentage of GWAS traits not accounted for by disease, disorder or blood pressure terms"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 86.82.4.6 Find seropositivity terms in GWAS traits
seropositivity_terms <- grep("seropositivity|antibody", measurement, value = T)
seropositivity_terms <- c(seropositivity_terms,
"foot-and-mouth disease virus seropositivity",
"bacillus phage virus seropositivity")
measurement <- measurement[!(measurement %in% seropositivity_terms)]
seropositivity_gwas = not_accounted_for[not_accounted_for %in% seropositivity_terms]
print("Number of GWAS traits under seropositivity terms")[1] "Number of GWAS traits under seropositivity terms"length(seropositivity_gwas)[1] 143print("Percentage of GWAS traits under seropositivity terms")[1] "Percentage of GWAS traits under seropositivity terms"round(100 * length(seropositivity_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.7not_accounted_for = not_accounted_for[!not_accounted_for %in% seropositivity_gwas]
print("Percentage of GWAS traits not accounted for by disease, disorder or seropositivity terms")[1] "Percentage of GWAS traits not accounted for by disease, disorder or seropositivity terms"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 86.12.4.7 General measurement terms
behavior_measurement <- c(
"smoking",
"alcohol consumption",
"alcoholic beverage consumption",
"alcohol exposure",
"behavior",
"farm exposure",
"tobacco",
"cannabis",
"physical activity",
"cognitive function",
"pack-years",
"coffee",
"opioid",
"environment",
"exercise"
)
behavior_measurement< grep(paste0(behavior_measurement,
collapse = "|"),
measurement,
value = T
) [1] TRUE TRUE TRUE TRUE TRUE FALSE TRUE FALSE FALSE TRUE FALSE TRUE
[13] TRUE TRUE TRUE FALSE TRUE TRUE TRUE FALSE FALSE FALSE FALSE FALSE
[25] FALSE FALSE FALSE FALSE TRUE TRUE FALSE TRUE TRUEmeasurement <- measurement[!(measurement %in% behavior_measurement)]
measurement <- grep("emphysema|eye colour|lifestyle",
measurement,
value = T,
invert = T)
measurement_gwas <- not_accounted_for[not_accounted_for %in% measurement]
print("Number of GWAS traits under measurement terms")[1] "Number of GWAS traits under measurement terms"length(measurement_gwas)[1] 16704print("Percentage of GWAS traits under measurement terms")[1] "Percentage of GWAS traits under measurement terms"round(100 * length(measurement_gwas) / length(all_gwas_terms),
digits = 1)[1] 83.6not_accounted_for = not_accounted_for[!not_accounted_for %in% measurement_gwas]
print("Percentage of GWAS traits not accounted for by disease, disorder or measurement terms")[1] "Percentage of GWAS traits not accounted for by disease, disorder or measurement terms"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 2.5print("Number of GWAS traits not accounted for by disease, disorder or measurement terms")[1] "Number of GWAS traits not accounted for by disease, disorder or measurement terms"length(not_accounted_for)[1] 4942.5 Response to stimulus
2.5.1 Combine response terms
go_response = readLines(here::here("output/trait_ontology/go_0050896_descendants.txt"))
efo_response <- readLines(here::here("output/trait_ontology/efo_go_0050896_descendants.txt"))
response <- c(go_response,
efo_response,
"response to stimulus")
response <- unique(response)
response = stringr::str_trim(tolower(response))
response = unique(response)response_gwas <- not_accounted_for[not_accounted_for %in% response]
#additional_response <- not_accounted_for[not_accounted_for %in% response]
#response_gwas = c(response_gwas, additional_response) |> unique()
print("Percentage of GWAS traits under response terms")[1] "Percentage of GWAS traits under response terms"round(100 * length(response_gwas) / length(all_gwas_terms),
digits = 1)[1] 1.2not_accounted_for = not_accounted_for[!not_accounted_for %in% response_gwas]
print("Percentage of GWAS traits not accounted for by disease, measurement or response terms")[1] "Percentage of GWAS traits not accounted for by disease, measurement or response terms"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 1.3print("Number of GWAS traits not accounted for by disease, measurement or response terms")[1] "Number of GWAS traits not accounted for by disease, measurement or response terms"length(not_accounted_for)[1] 2542.6 Mental process
mental <- readLines(here::here("output/trait_ontology/efo_0004323_descendants.txt"))
mental = stringr::str_trim(tolower(mental))
mental <- unique(mental)
mental <- c(mental,
"memory performance",
"visual memory process attribute",
"verbal memory measurement",
"executive function measurement",
"cognitive function measurement"
)
mental_gwas = not_accounted_for[not_accounted_for %in% mental]
print("Percentage of GWAS traits under mental process terms")[1] "Percentage of GWAS traits under mental process terms"round(100 * length(mental_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.1not_accounted_for = not_accounted_for[!not_accounted_for %in% mental_gwas]
print("Percentage of GWAS traits not accounted for thus far")[1] "Percentage of GWAS traits not accounted for thus far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 1.2print("Number of GWAS traits not accounted for thus far")[1] "Number of GWAS traits not accounted for thus far"length(not_accounted_for)[1] 2362.7 Behavior
behavior <- readLines(here::here("output/trait_ontology/go_0007610_descendants.txt"))
behavior = stringr::str_trim(tolower(behavior))
behavior <- unique(behavior)
behavior <- c(behavior,
behavior_measurement,
"physical activity")
behavior_gwas = not_accounted_for[not_accounted_for %in% behavior]
print("Percentage of GWAS traits under behavouir terms")[1] "Percentage of GWAS traits under behavouir terms"round(100 * length(behavior_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.1not_accounted_for = not_accounted_for[!not_accounted_for %in% behavior_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 1.1print("Number of GWAS traits not accounted for so far")[1] "Number of GWAS traits not accounted for so far"length(not_accounted_for)[1] 2142.8 Injury
injury <- readLines(here::here("output/trait_ontology/efo_0000546_descendants.txt"))
injury = stringr::str_trim(tolower(injury))
injury <- c(injury,
"fall")
injury_gwas = not_accounted_for[not_accounted_for %in% injury]
print("Percentage of GWAS traits under injury terms")[1] "Percentage of GWAS traits under injury terms"round(100 * length(injury_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.1not_accounted_for = not_accounted_for[!not_accounted_for %in% injury_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 1print("Number of GWAS traits not accounted for so far")[1] "Number of GWAS traits not accounted for so far"length(not_accounted_for)[1] 1932.9 Phenotype
phenotype <- readLines(here::here("output/trait_ontology/efo_0000651_descendants.txt"))
phenotype = stringr::str_trim(tolower(phenotype))
phenotype <- unique(c(phenotype,
"aging",
"biological sex",
"comparative body size at age 10, self-reported",
"complex trait",
"eye colour measurement",
"strand of hair color",
"high altitude adaptation",
"multiple gestation",
"normal",
"personality trait",
"skin pigmentation",
"personality",
"growth delay",
"sensory perception of taste",
"sensory perception of bitter taste",
"sensory perception of sweet taste",
"sensory perception of smell",
"sensory perception of sound",
"size",
"skin aging",
"sexual dimorphism",
"voice quality trait")
)
phenotype_gwas = not_accounted_for[not_accounted_for %in% phenotype]
print("Percentage of GWAS traits under phenotype terms")[1] "Percentage of GWAS traits under phenotype terms"round(100 * length(phenotype_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.4not_accounted_for = not_accounted_for[!not_accounted_for %in% phenotype_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 0.6print("Number of GWAS traits not accounted for so far")[1] "Number of GWAS traits not accounted for so far"length(not_accounted_for)[1] 1232.10 Medical procedure
medical_procedure <- readLines(here::here("output/trait_ontology/efo_0002571_descendants.txt"))
surgical_procedure <- readLines(here::here("output/trait_ontology/maxo_0000004_descendants.txt"))
clinical_history <- c("clinical history",
"encounter with health service",
"encounter with health service for adjustment and management of implanted device",
"encounter with health service related to reproduction")
medical_procedure = stringr::str_trim(tolower(medical_procedure))
medical_procedure = unique(c(medical_procedure,
surgical_procedure,
clinical_history,
"braces",
"vaccination",
"hormone replacement therapy",
"cognitive behavioural therapy",
"organ extraction",
"gastric bypass",
"medical procedure",
"number of treatments or medications taken, self-reported",
"treatment",
"test result",
"hospitalisation",
"clinical treatment")
)
medical_procedure_gwas = not_accounted_for[not_accounted_for %in% medical_procedure]
print("Percentage of GWAS traits under medical procedure terms")[1] "Percentage of GWAS traits under medical procedure terms"round(100 * length(medical_procedure_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.3not_accounted_for = not_accounted_for[!not_accounted_for %in% medical_procedure_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 0.4print("Number of GWAS traits not accounted for so far")[1] "Number of GWAS traits not accounted for so far"length(not_accounted_for)[1] 702.11 Environmental factors
enviro_factors <- c(
"diet measurement",
"economic and social preference",
"educational attainment",
"encounter with health service related to socioeconomic and psychosocial circumstances" ,
"energy intake",
"environmental factor",
"family relationship",
"household income",
"income",
"lifestyle measurement",
"risk factor",
"self reported educational attainment",
"social deprivation",
"social risk factor",
"socioeconomic status",
"townsend deprivation index"
)
enviro_factors_gwas = not_accounted_for[not_accounted_for %in% enviro_factors]
print("Percentage of GWAS traits under environmental factor terms")[1] "Percentage of GWAS traits under environmental factor terms"round(100 * length(enviro_factors_gwas) / length(all_gwas_terms),
digits = 1)[1] 0.1not_accounted_for = not_accounted_for[!not_accounted_for %in% enviro_factors_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 0.3print("Number of GWAS traits not accounted for so far")[1] "Number of GWAS traits not accounted for so far"length(not_accounted_for)[1] 572.12 Other
2.12.1 Biological process
bio_process <- c("pregnancy",
"puberty",
"menopause",
"ovulation",
"positive regulation of ovulation")
bio_process = stringr::str_trim(tolower(bio_process))
bio_process_gwas = not_accounted_for[not_accounted_for %in% bio_process]
print("Percentage of GWAS traits under biological process terms")[1] "Percentage of GWAS traits under biological process terms"round(100 * length(bio_process_gwas) / length(all_gwas_terms),
digits = 1)[1] 0not_accounted_for = not_accounted_for[!not_accounted_for %in% bio_process_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 0.3print("Number of GWAS traits not accounted for so far")[1] "Number of GWAS traits not accounted for so far"length(not_accounted_for)[1] 522.12.2 Traditional medicine constitutional types
tm_constitution <- c("yu-zhi constitution type",
"sasang constitutional medicine",
"sasang constitutional medicine type",
"hepatonia constitution type",
"pulmotonia constitution type",
"tae-yang",
"tae-eum",
"so-eum",
"so-yang"
)
tm_constitution = stringr::str_trim(tolower(tm_constitution))
tm_constitution_gwas = not_accounted_for[not_accounted_for %in% tm_constitution]
print("Percentage of GWAS traits under traditional medicine constitutional type terms")[1] "Percentage of GWAS traits under traditional medicine constitutional type terms"round(100 * length(tm_constitution_gwas) / length(all_gwas_terms),
digits = 1)[1] 0not_accounted_for = not_accounted_for[!not_accounted_for %in% tm_constitution_gwas]
print("Percentage of GWAS traits not accounted for so far")[1] "Percentage of GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(all_gwas_terms),
digits = 1)[1] 0.2print("Number of GWAS traits not accounted for so far")[1] "Number of GWAS traits not accounted for so far"length(not_accounted_for)[1] 432.12.3 Ancestry-info
ancestry_gwas <- "latin or admixed american ancestry"
not_accounted_for = not_accounted_for[!not_accounted_for %in% ancestry_gwas]2.12.4 Cell lines
cell_line_gwas <- "gm11992"
not_accounted_for = not_accounted_for[!not_accounted_for %in% cell_line_gwas]3 Add Categories to GWAS Info
3.1 Add disease & phenotypic abnormality terms to GWAS study info dataset
disease_or_disorder <- c(disease_gwas,
pheno_abnorm_gwas,
seropositivity_gwas
)
disease_progress_measure <-
gwas_study_info |>
filter(MAPPED_TRAIT == "disease prognosis measurement") |>
pull(STUDY_ACCESSION) |>
unique()
# for all GWAS Catalog studies with trait, "disease prognosis measurement"
# the actual disease is captured / recorded in trait, so we can remove disease prognosis measurement and safely capture all disease studies
gwas_study_info |>
filter(STUDY_ACCESSION %in% disease_progress_measure) |>
select(STUDY_ACCESSION, MAPPED_TRAIT, MAPPED_BACKGROUND_TRAIT) STUDY_ACCESSION MAPPED_TRAIT MAPPED_BACKGROUND_TRAIT
1 GCST004053 disease prognosis measurement crohn's disease
2 GCST008222 urinary bladder carcinoma
3 GCST008222 disease prognosis measurement
4 GCST90014045 non-small cell lung carcinoma smoking status measurement
5 GCST90014045 disease prognosis measurement smoking status measurement
6 GCST009878 disease prognosis measurement crohn's disease# similarly for:
# disease free survival
# complicated disease course
# mild disease course
# remission
# progression free survival
# illness severity status
unneeded_disease_progress_terms <-
c("disease prognosis measurement",
"complicated disease course",
"mild disease course",
"remission",
"disease free survival",
"survival time",
"overall survival",
"progression free survival",
"illness severity status"
)
# accessory eyelid
# dna methylation
# tube feeding
# widow's peak
# gata1 gene mutation
other_not_disease_terms <-
c(
"anti-drug antibody measurement",
"accessory eyelid",
"dna methylation",
"tube feeding",
"widow's peak",
"gata1 gene mutation"
)
disease_or_disorder <-
disease_or_disorder[!(disease_or_disorder %in%
c(unneeded_disease_progress_terms,
other_not_disease_terms
)
)]
gwas_study_info <-
gwas_study_info |>
#dplyr::rowwise() |>
dplyr::mutate(
disease_terms =
ifelse(MAPPED_TRAIT %in% disease_or_disorder,
MAPPED_TRAIT,
NA)
)3.2 Map GWAS traits to high-level categories
gwas_study_info =
gwas_study_info |>
dplyr::mutate(MAPPED_TRAIT_CATEGORY = dplyr::case_when(is.na(MAPPED_TRAIT) ~ NA,
MAPPED_TRAIT == "" ~ NA,
tolower(MAPPED_TRAIT) %in% disease_or_disorder ~ "Disease/Disorder",
#tolower(MAPPED_TRAIT) %in% pheno_abnorm_gwas ~ "Phenotypic Abnormality",
tolower(MAPPED_TRAIT) %in% seropositivity_gwas ~ "Seropositivity",
tolower(MAPPED_TRAIT) %in% bmi_weight_gwas ~ "BMI/Weight/Body Fat Measurement",
tolower(MAPPED_TRAIT) %in% lipid_cholesterol_gwas ~ "Lipid/Cholesterol Measurement",
tolower(MAPPED_TRAIT) %in% brain_measurement_terms ~ "Brain Measurement",
tolower(MAPPED_TRAIT) %in% blood_pressure_gwas ~ "Blood Pressure Measurement",
tolower(MAPPED_TRAIT) %in% measurement_gwas ~ "Measurement",
tolower(MAPPED_TRAIT) %in% response_gwas ~ "Response",
tolower(MAPPED_TRAIT) %in% mental_gwas ~ "Mental Process",
tolower(MAPPED_TRAIT) %in% behavior_gwas ~ "Behavior",
tolower(MAPPED_TRAIT) %in% injury_gwas ~ "Injury",
tolower(MAPPED_TRAIT) %in% phenotype_gwas ~ "Phenotype",
tolower(MAPPED_TRAIT) %in% medical_procedure_gwas ~ "Medical Procedure",
tolower(MAPPED_TRAIT) %in% enviro_factors_gwas ~ "Environmental Factor",
TRUE ~ "Other"
)
)4 Background traits
gwas_background <- gwas_study_info$MAPPED_BACKGROUND_TRAIT
gwas_background = stringr::str_trim(tolower(gwas_background))
gwas_background <- unique(gwas_background)
gwas_background <- gwas_background[gwas_background != ""]
print("Number of unique background GWAS traits")[1] "Number of unique background GWAS traits"length(gwas_background)[1] 2894.1 Overlap with disease/disorder traits
disease_gwas = gwas_background[gwas_background %in% disease_terms]
print("Number of background GWAS traits under disease or disorder terms")[1] "Number of background GWAS traits under disease or disorder terms"length(disease_gwas)[1] 198print("Percentage of background GWAS traits under disease or disorder terms")[1] "Percentage of background GWAS traits under disease or disorder terms"round(100 * length(disease_gwas) / length(gwas_background),
digits = 1)[1] 68.5not_accounted_for = gwas_background[!gwas_background %in% disease_gwas]
print("Percentage of background GWAS traits not under disease or disorder terms")[1] "Percentage of background GWAS traits not under disease or disorder terms"round(100 * length(not_accounted_for) / length(gwas_background),
digits = 1)[1] 31.54.2 Phenotype abnormality overlap
pheno_abnorm_gwas = pheno_abnorm_gwas
print("Percentage of background GWAS traits under phenotype abnormality terms")[1] "Percentage of background GWAS traits under phenotype abnormality terms"round(100 * length(pheno_abnorm_gwas) / length(gwas_background),
digits = 1)[1] 129.1not_accounted_for = not_accounted_for[!not_accounted_for %in% pheno_abnorm_gwas]
print("Percentage of background GWAS traits not accounted for so far")[1] "Percentage of background GWAS traits not accounted for so far"round(100 * length(not_accounted_for) / length(gwas_background),
digits = 1)[1] 29.4print("Number of background GWAS traits not accounted for so far")[1] "Number of background GWAS traits not accounted for so far"length(not_accounted_for)[1] 854.3 Add disease & phenotypic abnormality terms to GWAS study info dataset
disease_or_disorder <- c(disease_gwas,
pheno_abnorm_gwas
)
gwas_study_info <-
gwas_study_info |>
#rowwise() |>
dplyr::mutate(
background_disease_terms =
ifelse(MAPPED_BACKGROUND_TRAIT %in% disease_or_disorder,
MAPPED_BACKGROUND_TRAIT,
NA)
) |>
ungroup()4.4 Measurement traits
measurement_gwas = measurement[measurement %in% not_accounted_for]
not_accounted_for = not_accounted_for[!not_accounted_for %in% measurement_gwas]
print("Percentage of background GWAS traits not accounted for by disease, disorder or measurement terms")[1] "Percentage of background GWAS traits not accounted for by disease, disorder or measurement terms"round(100 * length(not_accounted_for) / length(gwas_background),
digits = 1)[1] 13.1print("Number of background GWAS traits not accounted for by disease, disorder or measurement terms")[1] "Number of background GWAS traits not accounted for by disease, disorder or measurement terms"length(not_accounted_for)[1] 384.5 Response traits
response_gwas = response[response %in% not_accounted_for]
not_accounted_for = not_accounted_for[!not_accounted_for %in% response_gwas]
print("Percentage of background GWAS traits under response terms")[1] "Percentage of background GWAS traits under response terms"round(100 * length(response_gwas) / length(gwas_background),
digits = 1)[1] 2.4print("Number of background GWAS traits under response terms")[1] "Number of background GWAS traits under response terms"length(response_gwas)[1] 7print("Number of background GWAS traits not accounted for by disease, measurement or response terms")[1] "Number of background GWAS traits not accounted for by disease, measurement or response terms"length(not_accounted_for)[1] 31print("Percentage of background GWAS traits not accounted for by disease, measurement or response terms")[1] "Percentage of background GWAS traits not accounted for by disease, measurement or response terms"round(100 * length(not_accounted_for) / length(gwas_background),
digits = 1)[1] 10.74.6 Medical procedure traits
medical_procedure_gwas = medical_procedure[medical_procedure %in% not_accounted_for]
not_accounted_for = not_accounted_for[!not_accounted_for %in% medical_procedure_gwas]
print("Percentage of background GWAS traits not accounted for by disease, measurement, response or medical procedure terms")[1] "Percentage of background GWAS traits not accounted for by disease, measurement, response or medical procedure terms"round(100 * length(not_accounted_for) / length(gwas_background),
digits = 1)[1] 6.9print("Number of background GWAS traits not accounted for by disease, measurement, response or medical procedure terms")[1] "Number of background GWAS traits not accounted for by disease, measurement, response or medical procedure terms"length(not_accounted_for)[1] 204.7 Background trait categories
gwas_study_info =
gwas_study_info |>
dplyr::mutate(BACKGROUND_TRAIT_CATEGORY =
dplyr::case_when(
MAPPED_BACKGROUND_TRAIT == "" ~ NA,
is.na(MAPPED_BACKGROUND_TRAIT) ~ NA,
stringr::str_trim(tolower(MAPPED_BACKGROUND_TRAIT)) %in% disease_or_disorder ~ "Disease/Disorder",
#stringr::str_trim(tolower(MAPPED_BACKGROUND_TRAIT)) %in% pheno_abnorm_gwas ~ "Phenotypic Abnormality",
stringr::str_trim(tolower(MAPPED_BACKGROUND_TRAIT)) %in% measurement_gwas ~ "Measurement",
stringr::str_trim(tolower(MAPPED_BACKGROUND_TRAIT)) %in% response_gwas ~ "Response",
stringr::str_trim(tolower(MAPPED_BACKGROUND_TRAIT)) %in% medical_procedure_gwas ~ "Medical Procedure",
TRUE ~ "Other")
)5 Summary of number of disease studies (and studies of each kind of trait)
gwas_study_info |>
group_by(MAPPED_TRAIT_CATEGORY, BACKGROUND_TRAIT_CATEGORY) |>
summarise(n_studies = n()) |>
arrange(desc(n_studies))# A tibble: 51 × 3
# Groups: MAPPED_TRAIT_CATEGORY [15]
MAPPED_TRAIT_CATEGORY BACKGROUND_TRAIT_CATEGORY n_studies
<chr> <chr> <int>
1 Measurement <NA> 91964
2 Disease/Disorder <NA> 27794
3 Measurement Disease/Disorder 18824
4 Brain Measurement <NA> 7143
5 Lipid/Cholesterol Measurement <NA> 4150
6 Medical Procedure <NA> 1266
7 Response <NA> 987
8 Disease/Disorder Disease/Disorder 821
9 Response Disease/Disorder 790
10 Other <NA> 747
# ℹ 41 more rowsgwas_study_info =
gwas_study_info |>
#dplyr::rowwise() |>
dplyr::mutate(DISEASE_STUDY =
case_when(MAPPED_TRAIT_CATEGORY == "Disease/Disorder" |
MAPPED_TRAIT_CATEGORY == "Phenotypic Abnormality" |
MAPPED_TRAIT_CATEGORY == "Seropositivity" |
BACKGROUND_TRAIT_CATEGORY == "Disease/Disorder" |
BACKGROUND_TRAIT_CATEGORY == "Phenotypic Abnormality" ~ T,
T ~ F )
) |>
dplyr::ungroup()
print("Number of studies of each kind of trait")[1] "Number of studies of each kind of trait"gwas_study_info |>
group_by(DISEASE_STUDY,
MAPPED_TRAIT_CATEGORY,
BACKGROUND_TRAIT_CATEGORY) |>
summarise(n = n()) # A tibble: 51 × 4
# Groups: DISEASE_STUDY, MAPPED_TRAIT_CATEGORY [27]
DISEASE_STUDY MAPPED_TRAIT_CATEGORY BACKGROUND_TRAIT_CATEGORY n
<lgl> <chr> <chr> <int>
1 FALSE BMI/Weight/Body Fat Measurement Measurement 7
2 FALSE BMI/Weight/Body Fat Measurement Other 6
3 FALSE BMI/Weight/Body Fat Measurement <NA> 726
4 FALSE Behavior Measurement 2
5 FALSE Behavior <NA> 249
6 FALSE Blood Pressure Measurement <NA> 531
7 FALSE Brain Measurement Measurement 1
8 FALSE Brain Measurement Response 6
9 FALSE Brain Measurement <NA> 7143
10 FALSE Environmental Factor <NA> 179
# ℹ 41 more rows# Number of papers with at least one disease study
print("Number of papers with at least one disease study")[1] "Number of papers with at least one disease study"gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY == T)) |>
group_by(DISEASE_STUDY) |>
summarise(n = n())# A tibble: 2 × 2
DISEASE_STUDY n
<lgl> <int>
1 FALSE 2716
2 TRUE 4610# ~ 60% of papers have at least one disease study
gwas_study_info |>
group_by(PUBMED_ID, YEAR) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY == T)) |>
group_by(YEAR) |>
summarise(n_disease_studies = sum(DISEASE_STUDY == T),
n_total_studies = n()) |>
mutate(percentage_disease_studies = 100 * n_disease_studies / n_total_studies) |>
ggplot(aes(x= YEAR,
y= percentage_disease_studies)) +
geom_line() +
geom_point() +
labs(title = "Percentage of papers with at least one disease GWAS over time",
x = "Year",
y = "Percentage of papers with at least one disease GWAS") +
theme_bw() +
lims(y = c(0,100))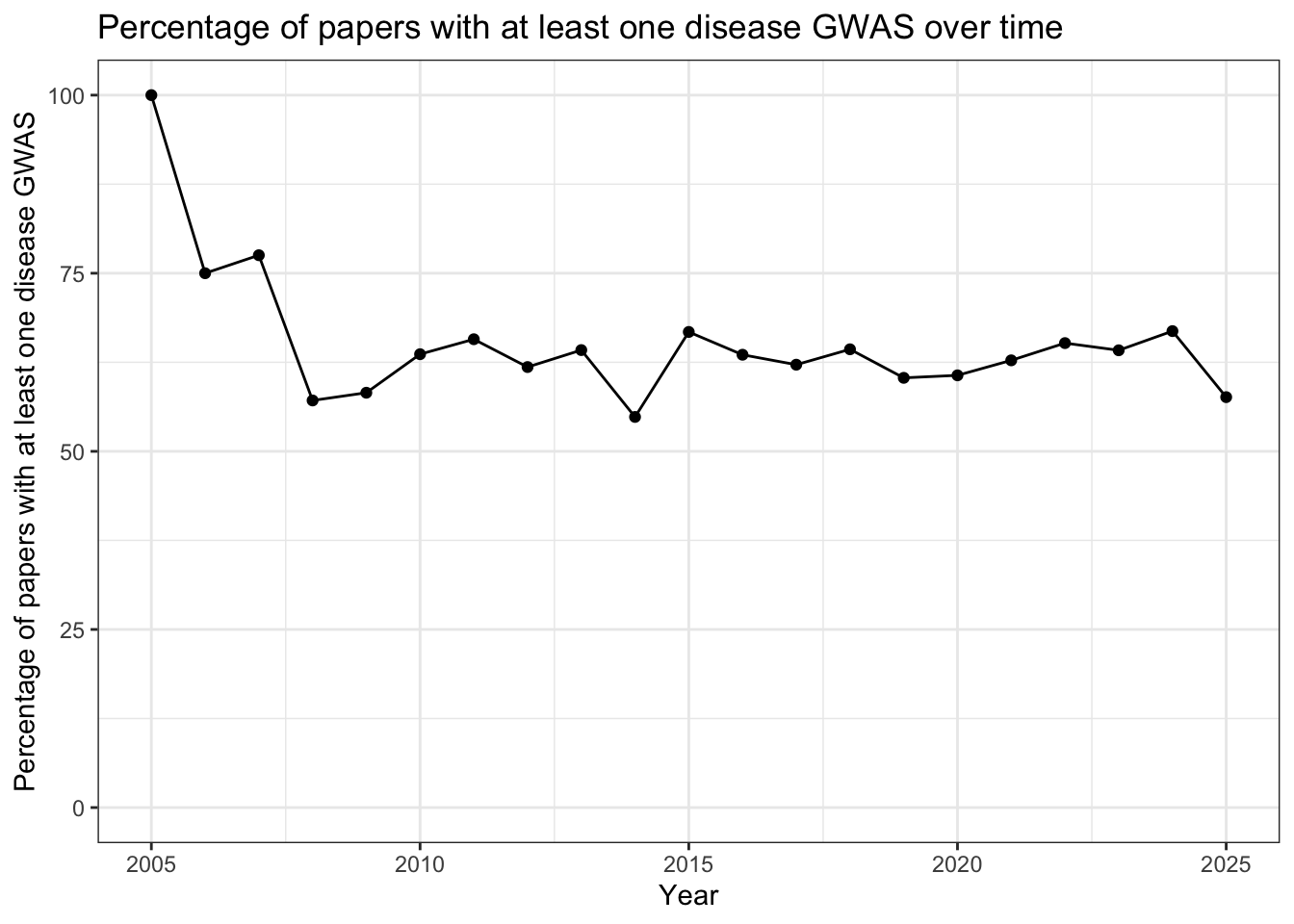
| Version | Author | Date |
|---|---|---|
| 1d36e53 | IJbeasley | 2026-03-20 |
6 Check for disease terms in traits of studies that don’t have a disease term mapped to either the trait or background trait - to see if there are any disease studies that we missed
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Number of papers with no disease study")[1] "Number of papers with no disease study"length(not_disease_pubmeds)[1] 27166.1 Cancer
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("breast cancer", `DISEASE/TRAIT`, ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`, STUDY) |>
distinct() PUBMED_ID
1 28763429
2 28763429
3 28763429
4 23518928
5 21245432
6 22180457
7 34648354
8 28240269
9 29875488
10 39528825
11 39528825
12 39528825
13 36168886
DISEASE/TRAIT
1 Change in LVEF in response to paclitaxel in HER2+ breast cancer
2 Change in LVEF in response to paclitaxel and trastuzumab in HER2+ breast cancer
3 Change in LVEF in response to paclitaxel and in response to paclitaxel and trastuzumab in HER+ breast cancer
4 Estradiol plasma levels (breast cancer)
5 Lapatinib-induced hepatotoxicity in breast cancer
6 Response to tamoxifen in breast cancer
7 Breast cancer anti-estrogen resistance protein 3 levels
8 Breast cancer anti-estrogen resistance protein 3 levels
9 Breast cancer anti-estrogen resistance protein 3 levels (BCAR3.12634.79.3)
10 Breast cancer anti-estrogen resistance protein 3:Src Homology domain levels
11 Breast cancer metastasis-suppressor 1-like protein levels
12 Breast cancer anti-estrogen resistance protein 3:Guanine Nucleotide Exchange Factor Domain levels
13 Breast cancer anti-estrogen resistance protein 3 levels
STUDY
1 Genome-wide association study of cardiotoxicity in the NCCTG N9831 (Alliance) adjuvant trastuzumab trial.
2 Genome-wide association study of cardiotoxicity in the NCCTG N9831 (Alliance) adjuvant trastuzumab trial.
3 Genome-wide association study of cardiotoxicity in the NCCTG N9831 (Alliance) adjuvant trastuzumab trial.
4 TSPYL5 SNPs: association with plasma estradiol concentrations and aromatase expression.
5 HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer.
6 A genome-wide association study identifies locus at 10q22 associated with clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients in Japanese.
7 Mapping the proteo-genomic convergence of human diseases.
8 Connecting genetic risk to disease end points through the human blood plasma proteome.
9 Genomic atlas of the human plasma proteome.
10 Proteogenomic analysis of human cerebrospinal fluid identifies neurologically relevant regulation and implicates causal proteins for Alzheimer's disease.
11 Proteogenomic analysis of human cerebrospinal fluid identifies neurologically relevant regulation and implicates causal proteins for Alzheimer's disease.
12 Proteogenomic analysis of human cerebrospinal fluid identifies neurologically relevant regulation and implicates causal proteins for Alzheimer's disease.
13 Differences and commonalities in the genetic architecture of protein quantitative trait loci in European and Arab populations.# for these studies, add breast cancer to the disease terms column, and disease/disorder to the trait category column, and then we can re-run the summary of number of studies of each kind
add_missed_breast_cancer <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("breast cancer", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "breast carcinoma",
DISEASE_STUDY = T)
# colorectal cancer
add_missed_colorectal_cancer <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("colorectal cancer", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "colorectal cancer",
DISEASE_STUDY = T)
# colon cancer
add_missed_colon_cancer <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("colon cancer", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "colorectal adenocarcinoma",
DISEASE_STUDY = T)
# small-cell lung cancer
add_small_cell_lung_carcinoma <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("small-cell lung cancer", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "small cell lung carcinoma",
DISEASE_STUDY = T)
# lung cancer
add_lung_cancer <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("lung cancer", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "lung carcinoma",
DISEASE_STUDY = T)
# head and neck cancer
add_head_neck_cancer <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("head and neck cancer", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "head and neck malignant neoplasia",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
rbind(add_missed_breast_cancer,
add_missed_colorectal_cancer,
add_small_cell_lung_carcinoma,
add_lung_cancer,
add_head_neck_cancer
)
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Papers mentioning cancer in DISEASE/TRAIT that are still not categorized as disease studies after adding missed cancer studies")[1] "Papers mentioning cancer in DISEASE/TRAIT that are still not categorized as disease studies after adding missed cancer studies"gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("cancer",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 35144566 Serum cancer antigen 50 (CA 50) levels
2 35144566 Serum cancer antigen 125 (CA 125) levels
3 35144566 Serum cancer antigen 15.3 levels
4 35144566 Serum cancer antigen 19.9 levels
5 31666285 Serum cancer antigen 15.3 levels
6 31666285 Serum cancer antigen 19.9 levels
7 31666285 Serum cancer antigen 125 (CA 125) levels
8 33563976 Gut microbiota presence (Enterobacter_cancerogenus)
9 37794183 Cancer/testis antigen 1 levels
10 37794183 Receptor-binding cancer antigen expressed on SiSo cells levels
11 37794183 Serologically defined colon cancer antigen 8 levels
12 33067605 Ovarian cancer-related tumor marker CA 125 levelsgwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("cancer",
STUDY,
ignore.case = T)) |>
select(PUBMED_ID, STUDY) |>
distinct() |>
head() PUBMED_ID
1 28471803
2 27488534
3 26053186
4 26414677
5 28173075
6 21460395
STUDY
1 Search for genetic factor association with cancer-free prostate-specific antigen level elevation on the basis of a genome-wide association study in the Korean population.
2 Novel Association of Genetic Markers Affecting CYP2A6 Activity and Lung Cancer Risk.
3 Mercapturic Acids Derived from the Toxicants Acrolein and Crotonaldehyde in the Urine of Cigarette Smokers from Five Ethnic Groups with Differing Risks for Lung Cancer.
4 Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair.
5 Metformin pharmacogenomics: a genome-wide association study to identify genetic and epigenetic biomarkers involved in metformin anticancer response using human lymphoblastoid cell lines.
6 A genome-wide association study identifies a locus on chromosome 14q21 as a predictor of leukocyte telomere length and as a marker of susceptibility for bladder cancer.print("Number of papers with no disease study after adding missed cancer studies")[1] "Number of papers with no disease study after adding missed cancer studies"length(not_disease_pubmeds)[1] 27036.2 Diabetes
# type 2 diabetes
add_missed_type_2_diabetes <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("type 2 diabetes|\\bT2D\\b", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "type 2 diabetes mellitus",
DISEASE_STUDY = T)
# type 1 diabetes
add_missed_type_1_diabetes <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("type 1 diabetes", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "type 1 diabetes mellitus",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
rbind(add_missed_type_2_diabetes,
add_missed_type_1_diabetes
)
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Papers mentioning diabetes in DISEASE/TRAIT that are still not categorized as disease studies after adding missed diabetes studies")[1] "Papers mentioning diabetes in DISEASE/TRAIT that are still not categorized as disease studies after adding missed diabetes studies"gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("diabetes",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 31015401 Medication use (drugs used in diabetes)gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("\\bdiabetes",
STUDY,
ignore.case = T)) |>
select(PUBMED_ID, STUDY) |>
distinct() |>
head() PUBMED_ID
1 30575882
2 26902266
3 28490609
4 27898682
5 28898252
6 20081858
STUDY
1 Association of Genetic Variants Related to Gluteofemoral vs Abdominal Fat Distribution With Type 2 Diabetes, Coronary Disease, and Cardiovascular Risk Factors.
2 Genome wide association study of uric acid in Indian population and interaction of identified variants with Type 2 diabetes.
3 A Genome-Wide Association Study of IVGTT-Based Measures of First-Phase Insulin Secretion Refines the Underlying Physiology of Type 2 Diabetes Variants.
4 Genetic Predisposition to an Impaired Metabolism of the Branched-Chain Amino Acids and Risk of Type 2 Diabetes: A Mendelian Randomisation Analysis.
5 Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis.
6 New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk.# diabetes add
add_missed_diabetes <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("diabetes", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "diabetes mellitus",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
add_missed_diabetes
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Number of papers with no disease study after adding missed diabetes studies")[1] "Number of papers with no disease study after adding missed diabetes studies"length(not_disease_pubmeds)[1] 26976.3 COPD, and other chronic lung/airway diseases
add_missed_copd <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("chronic osbtructive pulmonary disease|COPD|chronic obstructive pulmonary disease", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "chronic obstructive pulmonary disease",
DISEASE_STUDY = T)
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("asthma", `DISEASE/TRAIT`, ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 21991891 Asthma treatment response
2 23541324 Pulmonary function in asthmatics
3 24486069 Asthma (corticosteroid response)add_missed_asthma <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("asthma", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "asthma",
DISEASE_STUDY = T)
# interstitial lung disease
add_missed_ild <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("interstitial lung disease", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "interstitial lung disease",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
rbind(add_missed_copd,
add_missed_asthma,
add_missed_ild
)
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Papers mentioning COPD in DISEASE/TRAIT that are still not categorized as disease studies after adding missed COPD studies")[1] "Papers mentioning COPD in DISEASE/TRAIT that are still not categorized as disease studies after adding missed COPD studies"gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("COPD|chronic obstructive pulmonary disease|lung|asthma",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID
1 26635082
2 26635082
3 26635082
4 24023788
5 24023788
6 24023788
7 24023788
8 25044411
9 24929828
10 28166213
11 28166213
12 28166213
13 26423011
14 26423011
15 26423011
16 26423011
17 26423011
18 26423011
19 26423011
20 26423011
21 31902109
22 24387323
23 25713168
24 29095316
25 29095316
26 29095316
27 29095316
28 29095316
29 31453325
30 31453325
31 31453325
32 31453325
33 31453325
34 31453325
35 36638096
36 36638096
37 36914875
38 36914875
39 36914875
40 33755393
41 34128465
42 30804560
43 30804560
44 30049742
45 34782693
46 34782693
47 30175238
48 30175238
49 30175238
50 31846791
51 30061609
52 30061609
53 30061609
54 33766948
55 33766948
56 33766948
57 33766948
58 33766948
59 33766948
60 33766948
61 33766948
62 33766948
63 33766948
64 34226706
65 34226706
DISEASE/TRAIT
1 Lung function (FEV1/FVC)
2 Lung function (FEV1)
3 Lung function (FVC)
4 Lung function (FEV1)
5 Lung function (FEV1/FVC)
6 Lung function (FVC)
7 Lung function (forced expiratory flow during mid-portion (25% and 75%) of forced vital capacity)
8 Lung function (forced expiratory volume in 1 second)
9 Lung function (forced vital capacity)
10 Lung function (FEV1)
11 Lung function (FVC)
12 Lung function (FEV1/FVC)
13 Lung function in heavy smokers (low FEV1 vs high FEV1)
14 Lung function in never smokers (high FEV1 vs average FEV1)
15 Lung function in heavy smokers (high FEV1 vs average FEV1)
16 Lung function in never smokers (low FEV1 vs average FEV1)
17 Lung function in heavy smokers (low FEV1 vs average FEV1)
18 Lung function (low FEV1 vs high FEV1) x smoking interaction
19 Lung function (low FEV1 vs high FEV1)
20 Lung function in never smokers (low FEV1 vs high FEV1)
21 Lung function
22 Lung function
23 Gene methylation in lung tissue
24 Lung function (maximal voluntary ventilation)
25 Lung function (forced expiratory volume in 1 second)
26 Lung function (forced expiratory flow during mid-portion (25% and 75%) of forced vital capacity)
27 Lung function (forced vital capacity)
28 Lung function (FEV1/FVC)
29 Lung function (forced vital capacity) variance
30 Lung function (FEV1) variance
31 Lung function (FEV1/FVC) variance
32 Lung function (forced vital capacity)
33 Lung function (FEV1)
34 Lung function (FEV1/FVC)
35 Childhood lung function (FEV1/FVC z score) in low-income urban environment
36 Childhood lung function (FEV1 percent predicted) in low-income urban environment
37 Lung function (forced vital capacity)
38 Lung function (FEV1/FVC)
39 Lung function (FEV1)
40 Tacrolimus trough level in transplant patients (heart, kidney, lung, and liver)
41 Lung volume
42 Lung function (FVC)
43 Lung function (FEV1/FVC)
44 Diffusing capacity of the lung for carbon monoxide traits
45 Lung function (forced expiratory volume in 1 second)
46 Lung function (FEV1/FVC)
47 Lung function (FVC)
48 Lung function (FEV1/FVC)
49 Lung function (FEV1)
50 Lung function x fine particulate matter exposure levels interaction
51 Lung function (FEV1)
52 Lung function (FEV1/FVC)
53 Lung function (FVC)
54 Lung function (FEV1)
55 Lung function (FVC)
56 Lung function (FEV1/FVC)
57 Lung function (FEV1) and body mass index
58 Lung function (FEV1) and waist to hip ratio adjusted for BMI
59 Lung function (FEV1) and waist circumference adjusted for BMI
60 Lung function (FVC) and waist to hip ratio adjusted for BMI
61 Lung function (FVC) and waist circumference adjusted for BMI
62 Lung function (FEV1/FVC) and body mass index
63 Lung function (FVC) and body mass index
64 Lung function (FEV1/FVC)
65 Lung function (FVC)gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("COPD|chronic obstructive pulmonary disease|lung|asthma",
STUDY,
ignore.case = T)) |>
select(PUBMED_ID, STUDY) |>
distinct() |>
head() PUBMED_ID
1 26635082
2 27488534
3 26053186
4 26183928
5 28738859
6 21946350
STUDY
1 Sixteen new lung function signals identified through 1000 Genomes Project reference panel imputation.
2 Novel Association of Genetic Markers Affecting CYP2A6 Activity and Lung Cancer Risk.
3 Mercapturic Acids Derived from the Toxicants Acrolein and Crotonaldehyde in the Urine of Cigarette Smokers from Five Ethnic Groups with Differing Risks for Lung Cancer.
4 Implication of a Chromosome 15q15.2 Locus in Regulating UBR1 and Predisposing Smokers to MGMT Methylation in Lung.
5 Genes and pathways underlying susceptibility to impaired lung function in the context of environmental tobacco smoke exposure.
6 Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function.print("Number of papers with no disease study after adding missed chronic lung/airway studies")[1] "Number of papers with no disease study after adding missed chronic lung/airway studies"length(not_disease_pubmeds)[1] 26926.4 Psychiatric/Neurological
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("bipolar disorder", `DISEASE/TRAIT`, ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`, STUDY) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 21961650 Response to lithium treatment in bipolar disorder
2 19448189 Response to lithium treatment in bipolar disorder
STUDY
1 Evidence for association of an ACCN1 gene variant with response to lithium treatment in Sardinian patients with bipolar disorder.
2 A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder.# bipolar disorder
add_missed_bipolar_disorder <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("bipolar disorder", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "bipolar disorder",
DISEASE_STUDY = T)
# major depressive disorder
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("major depressive disorder", `DISEASE/TRAIT`, ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`, STUDY) |>
distinct() PUBMED_ID
1 22041458
2 24528284
DISEASE/TRAIT
1 Response to anti-depressant treatment in major depressive disorder
2 Response to serotonin reuptake inhibitors in major depressive disorder (plasma drug and metabolite levels)
STUDY
1 Pharmacogenomic study of side-effects for antidepressant treatment options in STAR*D.
2 Citalopram and escitalopram plasma drug and metabolite concentrations: genome-wide associations.add_missed_major_depressive_disorder <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("major depressive disorder", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "major depressive disorder",
DISEASE_STUDY = T)
# ADHD
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("attention deficit hyperactivity disorder|ADHD|attention-deficit/hyperactivity disorder",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`, STUDY) |>
distinct() PUBMED_ID
1 21130132
DISEASE/TRAIT
1 Response to methylphenidate treatment in attention-deficit/hyperactivity disorder (blood pressure)
STUDY
1 Genome-wide association study of blood pressure response to methylphenidate treatment of attention-deficit/hyperactivity disorder.add_missed_adhd <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("attention deficit hyperactivity disorder|ADHD|attention-deficit/hyperactivity disorder",
`DISEASE/TRAIT`,
ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "attention deficit hyperactivity disorder",
DISEASE_STUDY = T)
# schizophrenia
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("schizophrenia", `DISEASE/TRAIT`, ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`, STUDY) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 26856250 Early response to risperidone in schizophrenia
2 25963331 Endophenotypes for schizophrenia in healthy individuals
STUDY
1 The GRM7 gene, early response to risperidone, and schizophrenia: a genome-wide association study and a confirmatory pharmacogenetic analysis.
2 Common genetic variation and schizophrenia polygenic risk influence neurocognitive performance in young adulthood.add_missed_schizophrenia <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("schizophrenia", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "schizophrenia",
DISEASE_STUDY = T)
# Alzheimer's disease
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("Alzheimer", `DISEASE/TRAIT`, ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`, STUDY) |>
distinct() PUBMED_ID
1 30584014
2 31095298
DISEASE/TRAIT
1 Voxel-wise structural brain imaging measurements in Alzheimer’s disease
2 Voxel-wise structural brain imaging measurements in Alzheimer’s disease
STUDY
1 Spatial correlations exploitation based on nonlocal voxel-wise GWAS for biomarker detection of AD.
2 Incorporating spatial-anatomical similarity into the VGWAS framework for AD biomarker detection.add_missed_alzheimers <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("Alzheimer", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "Alzheimer's disease",
DISEASE_STUDY = T)
# dementia
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("dementia",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 34224794 Global cognition (Clinical Dementia Rating Scale Sum of Boxes)
2 35250029 Plasma p-tau181 levels in non-dementia individualsgwas_study_info <-
rows_append(gwas_study_info,
rbind(add_missed_bipolar_disorder,
add_missed_major_depressive_disorder,
add_missed_adhd,
add_missed_schizophrenia,
add_missed_alzheimers
)
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Papers mentioning psychiatric or neurological diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed psychiatric/neurological disease studies")[1] "Papers mentioning psychiatric or neurological diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed psychiatric/neurological disease studies"gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("bipolar disorder|major depressive disorder|attention deficit hyperactivity disorder|ADHD|schizophrenia|Alzheimer|dementia",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 34224794 Global cognition (Clinical Dementia Rating Scale Sum of Boxes)
2 35250029 Plasma p-tau181 levels in non-dementia individualsgwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("bipolar disorder|major depressive disorder|attention deficit hyperactivity disorder|ADHD|schizophrenia|Alzheimer|dementia",
STUDY,
ignore.case = T)) |>
select(PUBMED_ID, STUDY) |>
distinct() PUBMED_ID
1 29527006
2 29907492
3 31497858
4 31413141
5 25993607
6 29130521
7 26268530
8 24342994
9 29187730
10 28577822
11 19846067
12 19680635
13 23471985
14 20215924
15 22907730
16 25562672
17 24152035
18 22584459
19 18521091
20 21810643
21 28247064
22 31708768
23 32062564
24 32066700
25 32310165
26 32066663
27 31996736
28 29752348
29 30954325
30 31628463
31 31596458
32 31689377
33 31755389
34 36764567
35 32844198
36 32427856
37 32450446
38 37634885
39 37539664
40 33640202
41 34224794
42 35023831
43 35028426
44 36585402
45 37208024
46 32568366
47 30649180
48 30941828
49 33134509
50 36066633
51 35386118
52 40111762
53 31204042
54 32804141
55 29936532
56 30150663
57 38172904
58 30319691
59 34785643
STUDY
1 Genome-wide analyses of self-reported empathy: correlations with autism, schizophrenia, and anorexia nervosa.
2 Polygenic risk score, genome-wide association, and gene set analyses of cognitive domain deficits in schizophrenia.
3 Sex differences in the genetic predictors of Alzheimer's pathology.
4 The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer's disease risk.
5 Meta-analysis of Genome-wide Association Studies for Neuroticism, and the Polygenic Association With Major Depressive Disorder.
6 A variant in PPP4R3A protects against alzheimer-related metabolic decline.
7 GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer's disease implicates microglial activation gene IL1RAP.
8 Molecular genetic evidence for overlap between general cognitive ability and risk for schizophrenia: a report from the Cognitive Genomics consorTium (COGENT).
9 Genome-wide analysis in UK Biobank identifies four loci associated with mood instability and genetic correlation with major depressive disorder, anxiety disorder and schizophrenia.
10 Genome-wide association study of language performance in Alzheimer's disease.
11 A genomewide association study of citalopram response in major depressive disorder.
12 Genome-wide association study of antipsychotic-induced parkinsonism severity among schizophrenia patients.
13 Genome-wide scan of healthy human connectome discovers SPON1 gene variant influencing dementia severity.
14 A genome-wide association study of amygdala activation in youths with and without bipolar disorder.
15 Pharmacogenomics of selective serotonin reuptake inhibitor treatment for major depressive disorder: genome-wide associations and functional genomics.
16 Seasonality shows evidence for polygenic architecture and genetic correlation with schizophrenia and bipolar disorder.
17 Variants in the 1q21 risk region are associated with a visual endophenotype of autism and schizophrenia.
18 Possible association of CUX1 gene polymorphisms with antidepressant response in major depressive disorder.
19 Whole genome association study identifies polymorphisms associated with QT prolongation during iloperidone treatment of schizophrenia.
20 Association of genetic variants on 15q12 with cortical thickness and cognition in schizophrenia.
21 Genome-wide association study identifies four novel loci associated with Alzheimer's endophenotypes and disease modifiers.
22 Genome-Wide Association Studies for Cerebrospinal Fluid Soluble TREM2 in Alzheimer's Disease.
23 Genome-wide association study of white matter hyperintensity volume in elderly persons without dementia.
24 Genome-wide study of immune biomarkers in cerebrospinal fluid and serum from patients with bipolar disorder and controls.
25 Identification of Novel Genes Associated with Cortical Thickness in Alzheimer's Disease: Systems Biology Approach to Neuroimaging Endophenotype.
26 Genome-wide association study of dietary intake in the UK biobank study and its associations with schizophrenia and other traits.
27 Genome-Wide Association Study of Brain Connectivity Changes for Alzheimer's Disease.
28 Genetic Study of White Matter Integrity in UK Biobank (N=8448) and the Overlap With Stroke, Depression, and Dementia.
29 Population-based genome-wide association study of cognitive decline in older adults free of dementia: identification of a novel locus for the attention domain.
30 Associations with metabolites in Chinese suggest new metabolic roles in Alzheimer's and Parkinson's diseases.
31 Genome-wide Association of Endophenotypes for Schizophrenia From the Consortium on the Genetics of Schizophrenia (COGS) Study.
32 Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study.
33 Genome-wide Network-assisted Association and Enrichment Study of Amyloid Imaging Phenotype in Alzheimer's Disease.
34 Metabolomic Investigation of Major Depressive Disorder Identifies a Potentially Causal Association With Polyunsaturated Fatty Acids.
35 Genetic variants and functional pathways associated with resilience to Alzheimer's disease.
36 CDH6 and HAGH protein levels in plasma associate with Alzheimer's disease in APOE ε4 carriers.
37 Genome-wide interaction analysis of pathological hallmarks in Alzheimer's disease.
38 Brain-wide genome-wide colocalization study for integrating genetics, transcriptomics and brain morphometry in Alzheimer's disease.
39 Genome-Wide Meta-Analysis of Cerebrospinal Fluid Biomarkers in Alzheimer's Disease and Parkinson's Disease Cohorts.
40 Genome-wide association study identifies susceptibility loci of brain atrophy to NFIA and ST18 in Alzheimer's disease.
41 Genome-wide association study identified INSC gene associated with Trail Making Test Part A and Alzheimer's disease related cognitive phenotypes.
42 GWAS and ExWAS of blood Mitochondrial DNA copy number identifies 71 loci and highlights a potential causal role in dementia.
43 The genetic and epigenetic profile of serum S100β in the Lothian Birth Cohort 1936 and its relationship to Alzheimer's disease.
44 Visual masking deficits in schizophrenia: a view into the genetics of the disease through an endophenotype.
45 Genetic architecture of plasma Alzheimer disease biomarkers.
46 Association Between Common Variants in RBFOX1, an RNA-Binding Protein, and Brain Amyloidosis in Early and Preclinical Alzheimer Disease.
47 Association of a Schizophrenia-Risk Nonsynonymous Variant With Putamen Volume in Adolescents: A Voxelwise and Genome-Wide Association Study.
48 Common genetic variants have associations with human cortical brain regions and risk of schizophrenia.
49 Matrix metalloproteinase-degraded type I collagen is associated with APOE/TOMM40 variants and preclinical dementia.
50 Genome-wide meta-analysis for Alzheimer's disease cerebrospinal fluid biomarkers.
51 Genome-Wide Association Study of Alzheimer's Disease Brain Imaging Biomarkers and Neuropsychological Phenotypes in the European Medical Information Framework for Alzheimer's Disease Multimodal Biomarker Discovery Dataset.
52 Novel modelling approaches to elucidate the genetic architecture of resilience to Alzheimer's disease.
53 Genome-wide association study identifies Alzheimer's risk variant in MS4A6A influencing cerebrospinal fluid sTREM2 levels.
54 Genome-Wide Association Study of Brain Alzheimer's Disease-Related Metabolic Decline as Measured by [18F] FDG-PET Imaging.
55 Association Between Population Density and Genetic Risk for Schizophrenia.
56 GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia.
57 Proteo-genomics of soluble TREM2 in cerebrospinal fluid provides novel insights and identifies novel modulators for Alzheimer's disease.
58 Genome-Wide Association and Mechanistic Studies Indicate That Immune Response Contributes to Alzheimer's Disease Development.
59 A missense variant in SHARPIN mediates Alzheimer's disease-specific brain damages.print("Number of papers with no disease study after adding missed psychiatric/neurological disease studies")[1] "Number of papers with no disease study after adding missed psychiatric/neurological disease studies"length(not_disease_pubmeds)[1] 26836.5 Pubmed ID 35760791
gwas_study_info |>
filter(PUBMED_ID =="35760791") |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID
1 35760791
2 35760791
3 35760791
4 35760791
5 35760791
DISEASE/TRAIT
1 Cryptic phenotype that captures hereditary hemorrhagic telangiectasia severity
2 Cryptic phenotype that captures Marfan syndrome severity
3 Cryptic phenotype that captures Alport syndrome severity
4 Cryptic phenotype that captures autosomal dominant polycystic kidney disease severity
5 Cryptic phenotype that captures alpha-1-antitrypsin deficiency severityadd_missed_hereditary_hemorrhagic_telangiectasia <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("hereditary hemorrhagic telangiectasia",
`DISEASE/TRAIT`,
ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "hereditary hemorrhagic telangiectasia",
DISEASE_STUDY = T)
add_missed_marfan_syndrome <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("marfan syndrome", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "marfan syndrome",
DISEASE_STUDY = T)
add_missed_alport_syndrome <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("alport syndrome", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "alport syndrome",
DISEASE_STUDY = T)
# autosomal dominant polycystic kidney disease
add_missed_adpkd <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("autosomal dominant polycystic kidney disease|ADPKD", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "autosomal dominant polycystic kidney disease",
DISEASE_STUDY = T)
# alpha-1-antitrypsin deficiency
add_missed_a1at_deficiency <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("alpha-1-antitrypsin deficiency", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "alpha-1-antitrypsin deficiency",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
rbind(
add_missed_hereditary_hemorrhagic_telangiectasia,
add_missed_marfan_syndrome,
add_missed_alport_syndrome,
add_missed_adpkd,
add_missed_a1at_deficiency
)
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Number of papers with no disease study after adding missed hereditary hemorrhagic telangiectasia study")[1] "Number of papers with no disease study after adding missed hereditary hemorrhagic telangiectasia study"length(not_disease_pubmeds)[1] 26826.6 Infectious disease
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("HIV|human immunodeficiency virus|AIDS|acquired immunodeficiency syndrome",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID
1 17641165
2 24554482
3 24554482
4 24554482
5 37532928
DISEASE/TRAIT
1 HIV-1 viral setpoint
2 Response to anti-retroviral therapy (ddI/d4T) in HIV-1 infection (Grade 1 peripheral neuropathy)
3 Response to anti-retroviral therapy (ddI/d4T) in HIV-1 infection (Grade 3 peripheral neuropathy)
4 Response to anti-retroviral therapy (ddI/d4T) in HIV-1 infection (Grade 2 peripheral neuropathy)
5 HIV setpoint viral loadadd_hiv_1_infection <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("HIV-1|human immunodeficiency virus 1",
`DISEASE/TRAIT`,
ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "hiv-1 infection",
DISEASE_STUDY = T)
# hepatitis c virus infection
add_hep_c_virus_infection <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("hepatitis c virus infection",
`DISEASE/TRAIT`,
ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "hepatitis c virus infection",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
rbind(add_hiv_1_infection,
add_hep_c_virus_infection
)
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Papers mentioning infectious diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed infectious disease studies")[1] "Papers mentioning infectious diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed infectious disease studies"gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("HIV|human immunodeficiency virus|AIDS|acquired immunodeficiency syndrome|hepatitis c virus infection",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 37532928 HIV setpoint viral loadgwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("HIV|human immunodeficiency virus|AIDS|acquired immunodeficiency syndrome|hepatitis c virus infection",
STUDY,
ignore.case = T)) |>
select(PUBMED_ID, STUDY) |>
distinct() |>
head() PUBMED_ID
1 21507922
2 20045101
3 37532928
4 35888748
STUDY
1 Duffy-null-associated low neutrophil counts influence HIV-1 susceptibility in high-risk South African black women.
2 Quantitative trait loci for CD4:CD8 lymphocyte ratio are associated with risk of type 1 diabetes and HIV-1 immune control.
3 Africa-specific human genetic variation near CHD1L associates with HIV-1 load.
4 Crosstalk between Host Genome and Metabolome among People with HIV in South Africa.print("Number of papers with no disease study after adding missed infectious disease studies")[1] "Number of papers with no disease study after adding missed infectious disease studies"length(not_disease_pubmeds)[1] 26806.7 Autoimmune/Inflammatory:
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("inflammatory bowel disease|IBD",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 20014019 Response to anti-TNF alpha therapy in inflammatory bowel disease# inflammatory bowel disease
add_missed_ibd <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("inflammatory bowel disease|IBD", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "inflammatory bowel disease",
DISEASE_STUDY = T)
# rheumatoid arthritis
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("in rheumatoid arthritis\\b", `DISEASE/TRAIT`, ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 28512992 Bone erosion in rheumatoid arthritis
2 21061259 Response to anti-TNF therapy in rheumatoid arthritis
3 24583629 Response to methotrexate in rheumatoid arthritisadd_missed_ra <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("in rheumatoid arthritis\\b", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "rheumatoid arthritis",
DISEASE_STUDY = T)
# juvenile idiopathic arthritis
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("juvenile idiopathic arthritis",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 24709693 Response to methotrexate in juvenile idiopathic arthritisadd_missed_jia <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("juvenile idiopathic arthritis", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "juvenile idiopathic arthritis",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
rbind(add_missed_ibd,
add_missed_ra,
add_missed_jia
)
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Papers mentioning autoimmune/inflammatory diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed autoimmune/inflammatory disease studies")[1] "Papers mentioning autoimmune/inflammatory diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed autoimmune/inflammatory disease studies"gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("inflammatory bowel disease|IBD|rheumatoid arthritis|juvenile idiopathic arthritis",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 28861588 Disease topic 24 (Rheumatoid arthritis-plus)gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("inflammatory bowel disease|IBD|rheumatoid arthritis|juvenile idiopathic arthritis",
STUDY,
ignore.case = T)) |>
select(PUBMED_ID, STUDY) |>
distinct() PUBMED_ID
1 18615156
2 19287509
3 37127109
4 34780722
STUDY
1 Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis.
2 Genome-wide association study of determinants of anti-cyclic citrullinated peptide antibody titer in adults with rheumatoid arthritis.
3 Metabolic signature of healthy lifestyle and risk of rheumatoid arthritis: observational and Mendelian randomization study.
4 Integrative analysis of the Inflammatory Bowel Disease serum metabolome improves our understanding of genetic etiology and points to novel putative therapeutic targets.print("Number of papers with no disease study after adding missed autoimmune/inflammatory disease studies")[1] "Number of papers with no disease study after adding missed autoimmune/inflammatory disease studies"length(not_disease_pubmeds)[1] 26756.8 Cardiovascular disease
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("cardiovascular disease|coronary artery disease|myocardial infarction|heart attack|heart disease",
`DISEASE/TRAIT`,
ignore.case = T)
) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID
1 21943158
2 22029572
3 29748315
4 29703846
DISEASE/TRAIT
1 Cardiovascular disease risk factors
2 Cardiovascular disease risk factors
3 Plasma proprotein convertase subtilisin/kexin type 9 levels in stable coronary artery disease
4 Coronary heart disease events during statin therapyadd_missed_coronary_artery_disease <-
gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("stable coronary artery disease", `DISEASE/TRAIT`, ignore.case = T)) |>
mutate(MAPPED_TRAIT_CATEGORY = "Disease/Disorder",
disease_terms = "coronary artery disease",
DISEASE_STUDY = T)
gwas_study_info <-
rows_append(gwas_study_info,
add_missed_coronary_artery_disease
)
not_disease_pubmeds <- gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY)) |>
filter(DISEASE_STUDY == F) |>
pull(PUBMED_ID) |>
unique()
print("Papers mentioning cardiovascular diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed cardiovascular disease studies")[1] "Papers mentioning cardiovascular diseases in DISEASE/TRAIT that are still not categorized as disease studies after adding missed cardiovascular disease studies"gwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("cardiovascular disease|coronary artery disease|myocardial infarction|heart attack|heart disease",
`DISEASE/TRAIT`,
ignore.case = T)) |>
select(PUBMED_ID, `DISEASE/TRAIT`) |>
distinct() PUBMED_ID DISEASE/TRAIT
1 21943158 Cardiovascular disease risk factors
2 22029572 Cardiovascular disease risk factors
3 29703846 Coronary heart disease events during statin therapygwas_study_info |>
filter(PUBMED_ID %in% not_disease_pubmeds) |>
filter(grepl("cardiovascular disease|coronary artery disease|myocardial infarction|heart attack|heart disease",
STUDY,
ignore.case = T)) |>
select(PUBMED_ID, STUDY) |>
distinct() PUBMED_ID
1 31551469
2 31169883
3 29563342
4 26822151
5 29212897
6 20529992
7 19567438
8 22703881
9 22029572
10 23824729
11 20864672
12 18179892
13 20884846
14 18193043
15 19198610
16 19060911
17 22068335
18 21757653
19 22003152
20 23969696
21 20031564
22 32154731
23 32203549
24 28369058
25 32876488
26 31597446
27 36974753
28 36869765
29 36918541
30 34233476
31 33287642
32 32649856
33 32805626
34 37188768
35 39048560
36 26540294
37 35668104
38 35692035
39 38437179
40 33339817
41 30111768
42 31070104
43 38507016
44 34706549
45 24507774
46 31070471
47 31070453
48 31584380
49 33937362
50 33910371
51 38180560
52 30866520
53 30685440
54 29728394
55 33469137
56 30305239
57 33356394
58 35884923
59 36578646
STUDY
1 Genetic architecture of human plasma lipidome and its link to cardiovascular disease.
2 Genetic association study of eight steroid hormones and implications for sexual dimorphism of coronary artery disease.
3 Untargeted metabolomics identifies trimethyllysine, a TMAO-producing nutrient precursor, as a predictor of incident cardiovascular disease risk.
4 Genome-wide association study and targeted metabolomics identifies sex-specific association of CPS1 with coronary artery disease.
5 Genetic Variants Contributing to Circulating Matrix Metalloproteinase 8 Levels and Their Association With Cardiovascular Diseases: A Genome-Wide Analysis.
6 Genetic regulation of serum phytosterol levels and risk of coronary artery disease.
7 Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease.
8 Genetic associations for activated partial thromboplastin time and prothrombin time, their gene expression profiles, and risk of coronary artery disease.
9 Recent methods for polygenic analysis of genome-wide data implicate an important effect of common variants on cardiovascular disease risk.
10 Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease.
11 Genetic variants influencing circulating lipid levels and risk of coronary artery disease.
12 Genome-wide association study identifies genes for biomarkers of cardiovascular disease: serum urate and dyslipidemia.
13 Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors.
14 Newly identified loci that influence lipid concentrations and risk of coronary artery disease.
15 Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction.
16 Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts.
17 Common genetic variation in the 3'-BCL11B gene desert is associated with carotid-femoral pulse wave velocity and excess cardiovascular disease risk: the AortaGen Consortium.
18 Assessment of genetic determinants of the association of γ' fibrinogen in relation to cardiovascular disease.
19 Eight genetic loci associated with variation in lipoprotein-associated phospholipase A2 mass and activity and coronary heart disease: meta-analysis of genome-wide association studies from five community-based studies.
20 Multiethnic meta-analysis of genome-wide association studies in >100 000 subjects identifies 23 fibrinogen-associated Loci but no strong evidence of a causal association between circulating fibrinogen and cardiovascular disease.
21 Polymorphism in the CETP gene region, HDL cholesterol, and risk of future myocardial infarction: Genomewide analysis among 18 245 initially healthy women from the Women's Genome Health Study.
22 Polygenic Hyperlipidemias and Coronary Artery Disease Risk.
23 Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis.
24 Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease.
25 The Genetics of Circulating Resistin Level, A Biomarker for Cardiovascular Diseases, Is Informed by Mendelian Randomization and the Unique Characteristics of African Genomes.
26 Novel Genetic Locus Influencing Retinal Venular Tortuosity Is Also Associated With Risk of Coronary Artery Disease.
27 Unprocessed Red Meat and Processed Meat Consumption, Plasma Metabolome, and Risk of Ischemic Heart Disease: A Prospective Cohort Study of UK Biobank.
28 VEGF-D plasma levels and VEGFD genetic variants are independently associated with outcomes in patients with cardiovascular disease.
29 Genetic architecture of spatial electrical biomarkers for cardiac arrhythmia and relationship with cardiovascular disease.
30 Genome-Wide Association Study Identifies a Functional <i>SIDT2</i> Variant Associated With HDL-C (High-Density Lipoprotein Cholesterol) Levels and Premature Coronary Artery Disease.
31 Associations of Observational and Genetically Determined Caffeine Intake With Coronary Artery Disease and Diabetes Mellitus.
32 A Platelet Function Modulator of Thrombin Activation Is Causally Linked to Cardiovascular Disease and Affects PAR4 Receptor Signaling.
33 Effects of tumour necrosis factor on cardiovascular disease and cancer: A two-sample Mendelian randomization study.
34 Fine-mapping of retinal vascular complexity loci identifies Notch regulation as a shared mechanism with myocardial infarction outcomes.
35 Causal relevance of different blood pressure traits on risk of cardiovascular diseases: GWAS and Mendelian randomisation in 100,000 Chinese adults.
36 Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis.
37 Comprehensive genetic analysis of the human lipidome identifies loci associated with lipid homeostasis with links to coronary artery disease.
38 Role of circulating polyunsaturated fatty acids on cardiovascular diseases risk: analysis using Mendelian randomization and fatty acid genetic association data from over 114,000 UK Biobank participants.
39 Sex-specific and polygenic effects underlying resting heart rate and associated risk of cardiovascular disease.
40 Loss-of-function genomic variants highlight potential therapeutic targets for cardiovascular disease.
41 Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease.
42 Genetic Determinants of Circulating Glycine Levels and Risk of Coronary Artery Disease.
43 Genetic evidence for T-wave area from 12-lead electrocardiograms to monitor cardiovascular diseases in patients taking diabetes medications.
44 Soluble Urokinase Plasminogen Activator Receptor: Genetic Variation and Cardiovascular Disease Risk in Blacks.
45 Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks.
46 Group IIA Secretory Phospholipase A2 and Incident Cardiovascular Disease.
47 Genetic Association of Finger Photoplethysmography-Derived Arterial Stiffness Index With Blood Pressure and Coronary Artery Disease.
48 The Assessment of Interleukin-18 on the Risk of Coronary Heart Disease.
49 Sortilin as a Biomarker for Cardiovascular Disease Revisited.
50 Soluble CD14 Levels in the Jackson Heart Study: Associations With Cardiovascular Disease Risk and Genetic Variants.
51 Genome-wide association and Mendelian randomization analysis provide insights into the shared genetic architecture between high-dimensional electrocardiographic features and ischemic heart disease.
52 Circulating Chemerin Levels, but not the RARRES2 Polymorphisms, Predict the Long-Term Outcome of Angiographically Confirmed Coronary Artery Disease.
53 LDL triglycerides, hepatic lipase activity, and coronary artery disease: An epidemiologic and Mendelian randomization study.
54 CETP (Cholesteryl Ester Transfer Protein) Concentration: A Genome-Wide Association Study Followed by Mendelian Randomization on Coronary Artery Disease.
55 Variation in the SERPINA6/SERPINA1 locus alters morning plasma cortisol, hepatic corticosteroid binding globulin expression, gene expression in peripheral tissues, and risk of cardiovascular disease.
56 Genetic contributors to serum uric acid levels in Mexicans and their effect on premature coronary artery disease.
57 Urate, Blood Pressure, and Cardiovascular Disease: Evidence From Mendelian Randomization and Meta-Analysis of Clinical Trials.
58 Dyslipidaemia-Genotype Interactions with Nutrient Intake and Cerebro-Cardiovascular Disease.
59 Circulating serum amyloid A levels but not SAA1 variants predict long-term outcomes of angiographically confirmed coronary artery disease.print("Number of papers with no disease study after adding missed cardiovascular disease studies")[1] "Number of papers with no disease study after adding missed cardiovascular disease studies"length(not_disease_pubmeds)[1] 26747 Creating disease labels column of just disease or phenotype abnormality terms for each study - so that we can see what diseases are being studied
7.1 Make disease label column - combining disease terms from both mapped trait and background trait
combined_disease_terms = function(MAPPED_TRAIT_1, MAPPED_TRAIT_2){
#MAPPED_TRAIT_1 = stringr::str_split(MAPPED_TRAIT_1, ", ") |> unlist()
#MAPPED_TRAIT_2 = stringr::str_split(MAPPED_TRAIT_2, ", ") |> unlist()
all_mapped_disease_terms =
c(MAPPED_TRAIT_1, MAPPED_TRAIT_2) |>
unique()
combined_mapped_disease_terms = str_flatten(all_mapped_disease_terms,
collapse = "; ",
na.rm = T
)
return(combined_mapped_disease_terms)
}
gwas_study_info <-
gwas_study_info |>
tidyr::unite(col = "all_disease_terms",
c("disease_terms", "background_disease_terms"),
remove = F,
sep = "; ",
na.rm = TRUE)
#dplyr::rowwise() |>
# dplyr::mutate(all_disease_terms =
# case_when(is.na(background_disease_terms) & is.na(disease_terms) ~ NA,
# is.na(background_disease_terms) & !is.na(disease_terms) ~ disease_terms,
# !is.na(background_disease_terms) & is.na(disease_terms) ~ background_disease_terms,
# !is.na(background_disease_terms) & !is.na(disease_terms) ~
# combined_disease_terms(disease_terms,
# background_disease_terms,
# ))
#
# ) |>
# dplyr::ungroup()
# correct commas
gwas_study_info =
gwas_study_info |>
mutate(
all_disease_terms =
str_replace(
all_disease_terms,
"^osteoarthritis,\\s*(.+)$",
"osteoarthritis of \\1"
)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "hodgkins lymphoma, mixed cellularity",
replacement = "hodgkins lymphoma",
string = all_disease_terms)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "hypertension, pregnancy-induced",
replacement = "pregnancy-induced hypertension",
string = all_disease_terms)
) |>
mutate(all_disease_terms =
str_replace_all(
pattern = "renal agenesis, unilateral",
replacement = "unilateral renal agenesis",
string = all_disease_terms)
) |>
mutate(all_disease_terms =
str_replace_all(
pattern = "diarrhea, infantile",
replacement = "infantile diarrhea",
string = all_disease_terms)
) |>
mutate(all_disease_terms =
str_replace_all(
pattern = "fractures, ununited",
replacement = "ununited fractures",
string = all_disease_terms)
) |>
mutate(all_disease_terms =
str_replace_all(
pattern = "cholecystitis, acute",
replacement = "acute cholecystitis",
string = all_disease_terms)
) |>
mutate(all_disease_terms =
str_replace_all(
pattern = "hepatitis, alcoholic",
replacement = "alcoholic hepatitis",
string = all_disease_terms)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "encephalopathy, acute, infection-induced",
replacement = "infectious encephalitis",
string = all_disease_terms
)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "genital neoplasm, female",
replacement = "female genital neoplasm",
string = all_disease_terms
)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "psoriasis 14, pustular",
replacement = "pustular psoriasis 14",
string = all_disease_terms
)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "hand, foot and mouth disease",
replacement = "hand foot and mouth disease",
string = all_disease_terms
)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "anemia, hemolytic, autoimmune",
replacement = "autoimmune hemolytic anemia",
string = all_disease_terms
)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "polyarticular juvenile idiopathic arthritis, rheumatoid factor negative",
replacement = "rheumatoid factor-negative polyarticular juvenile idiopathic arthritis",
string = all_disease_terms
)
)
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "neural tube defects, susceptibility to",
replacement = "neural tube defects",
string = all_disease_terms
))
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "migraine without aura, susceptibility to, 4",
replacement = "migraine without aura",
string = all_disease_terms
))
gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms =
str_replace_all(
pattern = "genital neoplasm, female",
replacement = "female genital neoplasm",
string = all_disease_terms
))
print("Number of studies with bad commas")[1] "Number of studies with bad commas"gwas_study_info |> filter(grepl(",", all_disease_terms)) [1] DISEASE/TRAIT PUBMED_ID
[3] YEAR STUDY
[5] STUDY_ACCESSION MAPPED_TRAIT
[7] MAPPED_TRAIT_URI MAPPED_BACKGROUND_TRAIT
[9] MAPPED_BACKGROUND_TRAIT_URI all_disease_terms
[11] disease_terms MAPPED_TRAIT_CATEGORY
[13] background_disease_terms BACKGROUND_TRAIT_CATEGORY
[15] DISEASE_STUDY
<0 rows> (or 0-length row.names)gwas_study_info =
gwas_study_info |>
mutate(all_disease_terms = str_replace_all(
pattern = "; ",
replacement = ", ",
string = all_disease_terms
))7.2 Minor fixes of trait categorisation and returning traits
# What studies are disease studies but have no collected disease terms?
gwas_study_info |>
filter(DISEASE_STUDY == T) |>
filter(all_disease_terms == "") |>
select(PUBMED_ID) |>
distinct() |>
nrow()[1] 0gwas_study_info |>
filter(DISEASE_STUDY == T) |>
filter(all_disease_terms == "") |>
select(MAPPED_TRAIT, MAPPED_TRAIT_CATEGORY) |>
distinct() |>
head()[1] MAPPED_TRAIT MAPPED_TRAIT_CATEGORY
<0 rows> (or 0-length row.names)7.2.1 Fix bug where MAPPED_TRAIT/BACKGROUND_MAPPED_TRAIT is empty string but TRAIT_CATEGORY is listed as disease/phenotypic abnormality
gwas_study_info = gwas_study_info |>
#rowwise() |>
mutate(MAPPED_TRAIT_CATEGORY = ifelse(MAPPED_TRAIT == "",
"Other",
MAPPED_TRAIT_CATEGORY)) |>
mutate(BACKGROUND_TRAIT_CATEGORY = ifelse(MAPPED_BACKGROUND_TRAIT == "",
"Other",
BACKGROUND_TRAIT_CATEGORY)) 7.2.2 Recalculate disease study flag (is disease study or not?)
now that I have corrected any mistakes in categorization and added some missing disease terms
gwas_study_info =
gwas_study_info |>
#dplyr::rowwise() |>
dplyr::mutate(DISEASE_STUDY =
ifelse(MAPPED_TRAIT_CATEGORY == "Disease/Disorder" |
MAPPED_TRAIT_CATEGORY == "Phenotypic Abnormality" |
BACKGROUND_TRAIT_CATEGORY == "Disease/Disorder" |
BACKGROUND_TRAIT_CATEGORY == "Phenotypic Abnormality",
T, F )
) |>
dplyr::ungroup()
gwas_study_info |>
filter(DISEASE_STUDY == T) |>
group_by(MAPPED_TRAIT_CATEGORY,
BACKGROUND_TRAIT_CATEGORY) |>
summarise(n = n()) |>
arrange(desc(n))# A tibble: 18 × 3
# Groups: MAPPED_TRAIT_CATEGORY [14]
MAPPED_TRAIT_CATEGORY BACKGROUND_TRAIT_CATEGORY n
<chr> <chr> <int>
1 Disease/Disorder Other 27919
2 Measurement Disease/Disorder 18824
3 Disease/Disorder Disease/Disorder 821
4 Response Disease/Disorder 790
5 Lipid/Cholesterol Measurement Disease/Disorder 401
6 Disease/Disorder Measurement 93
7 Other Disease/Disorder 45
8 Seropositivity Disease/Disorder 39
9 BMI/Weight/Body Fat Measurement Disease/Disorder 33
10 Disease/Disorder Medical Procedure 23
11 Blood Pressure Measurement Disease/Disorder 20
12 Behavior Disease/Disorder 15
13 Phenotype Disease/Disorder 11
14 Brain Measurement Disease/Disorder 8
15 Medical Procedure Disease/Disorder 8
16 Mental Process Disease/Disorder 7
17 Disease/Disorder Response 5
18 Injury Disease/Disorder 2# gwas_study_info |>
# filter(DISEASE_STUDY == T) |>
# filter(all_disease_terms == "" | is.na(all_disease_terms)) |>
# select(MAPPED_TRAIT, MAPPED_TRAIT_CATEGORY) |>
# distinct() |>
# nrow()
print("Number of studies of each kind of trait")[1] "Number of studies of each kind of trait"gwas_study_info |>
group_by(DISEASE_STUDY,
MAPPED_TRAIT_CATEGORY,
BACKGROUND_TRAIT_CATEGORY) |>
summarise(n = n()) |>
arrange(desc(n))# A tibble: 47 × 4
# Groups: DISEASE_STUDY, MAPPED_TRAIT_CATEGORY [27]
DISEASE_STUDY MAPPED_TRAIT_CATEGORY BACKGROUND_TRAIT_CATEGORY n
<lgl> <chr> <chr> <int>
1 FALSE Measurement Other 92058
2 TRUE Disease/Disorder Other 27919
3 TRUE Measurement Disease/Disorder 18824
4 FALSE Brain Measurement Other 7143
5 FALSE Lipid/Cholesterol Measurement Other 4165
6 FALSE Medical Procedure Other 1266
7 FALSE Response Other 987
8 TRUE Disease/Disorder Disease/Disorder 821
9 TRUE Response Disease/Disorder 790
10 FALSE Other Other 747
# ℹ 37 more rowsprint("Number of papers with at least one disease study")[1] "Number of papers with at least one disease study"gwas_study_info |>
group_by(PUBMED_ID) |>
summarise(DISEASE_STUDY = any(DISEASE_STUDY == T)) |>
group_by(DISEASE_STUDY) |>
summarise(n = n())# A tibble: 2 × 2
DISEASE_STUDY n
<lgl> <int>
1 FALSE 2674
2 TRUE 46528 Saving:
data.table::fwrite(gwas_study_info,
here::here("output/gwas_cat/gwas_study_info_trait_cat.csv"),
sep = ",")
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 26.3.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/Los_Angeles
tzcode source: internal
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] stringr_1.6.0 ggplot2_3.5.2 dplyr_1.1.4 data.table_1.17.8
[5] workflowr_1.7.2
loaded via a namespace (and not attached):
[1] utf8_1.2.6 sass_0.4.10 generics_0.1.4
[4] tidyr_1.3.1 renv_1.1.8 stringi_1.8.7
[7] digest_0.6.37 magrittr_2.0.4 evaluate_1.0.5
[10] grid_4.3.1 timechange_0.3.0 RColorBrewer_1.1-3
[13] fastmap_1.2.0 rprojroot_2.1.0 jsonlite_2.0.0
[16] processx_3.8.6 whisker_0.4.1 ps_1.9.1
[19] promises_1.3.3 BiocManager_1.30.26 httr_1.4.7
[22] purrr_1.1.0 scales_1.4.0 jquerylib_0.1.4
[25] cli_3.6.5 rlang_1.1.6 withr_3.0.2
[28] cachem_1.1.0 yaml_2.3.10 tools_4.3.1
[31] httpuv_1.6.16 here_1.0.1 vctrs_0.6.5
[34] R6_2.6.1 lifecycle_1.0.4 lubridate_1.9.4
[37] git2r_0.36.2 fs_1.6.6 pkgconfig_2.0.3
[40] callr_3.7.6 pillar_1.11.1 bslib_0.9.0
[43] later_1.4.4 gtable_0.3.6 glue_1.8.0
[46] Rcpp_1.1.0 xfun_0.55 tibble_3.3.0
[49] tidyselect_1.2.1 rstudioapi_0.17.1 knitr_1.50
[52] farver_2.1.2 htmltools_0.5.8.1 labeling_0.4.3
[55] rmarkdown_2.30 compiler_4.3.1 getPass_0.2-4