Collapse / filtering traits
Isobel Beasley
2025-08-21
Last updated: 2025-08-23
Checks: 7 0
Knit directory:
genomics_ancest_disease_dispar/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220216) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9a9803b. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: data/gwas_catalog/
Ignored: output/gwas_study_info_cohort_corrected.csv
Ignored: output/gwas_study_info_trait_corrected.csv
Untracked files:
Untracked: .DS_Store
Untracked: code/custom_plotting.R
Untracked: data/.DS_Store
Untracked: gwas_time.pdf
Untracked: renv/
Unstaged changes:
Modified: .Rprofile
Modified: analysis/disease_inves_by_ancest.Rmd
Modified: code/collapse_diseases.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/collapse_traits.Rmd) and
HTML (docs/collapse_traits.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9a9803b | IJbeasley | 2025-08-23 | Update correcting traits |
| html | 605013e | IJbeasley | 2025-08-21 | Build site. |
| Rmd | d4ec61e | IJbeasley | 2025-08-21 | Adding investigation harmonizing mapped traits |
1 Set up
knitr::opts_chunk$set(echo = TRUE,
message = FALSE,
warning = FALSE
)
library(data.table)
library(dplyr)
library(ggplot2)
library(stringr)1.1 Load data
gwas_study_info <- fread(here::here("output/gwas_study_info_cohort_corrected.csv"))2 Number of studies per mapped trait
# number of studies per mapped trait
n_studies_trait = gwas_study_info |>
dplyr::group_by(MAPPED_TRAIT, MAPPED_TRAIT_URI) |>
dplyr::summarise(n_studies = dplyr::n()) |>
dplyr::arrange(desc(n_studies))
n_studies_trait |>
head()# A tibble: 6 × 3
# Groups: MAPPED_TRAIT [6]
MAPPED_TRAIT MAPPED_TRAIT_URI n_studies
<chr> <chr> <int>
1 neuroimaging measurement http://www.ebi.ac.uk/efo/EFO_0004346 9773
2 protein measurement http://www.ebi.ac.uk/efo/EFO_0004747 8748
3 blood protein amount http://purl.obolibrary.org/obo/OBA_VT000… 5336
4 metabolite measurement http://www.ebi.ac.uk/efo/EFO_0004725 4559
5 blood metabolite level http://purl.obolibrary.org/obo/OBA_20500… 2738
6 gut microbiome measurement http://www.ebi.ac.uk/efo/EFO_0007874 2363n_studies_trait |>
head() |>
ggplot(aes(y = reorder(MAPPED_TRAIT,
n_studies),
x = n_studies)
) +
geom_col() +
theme_bw() +
labs(y = "Trait", x = "Number of studies")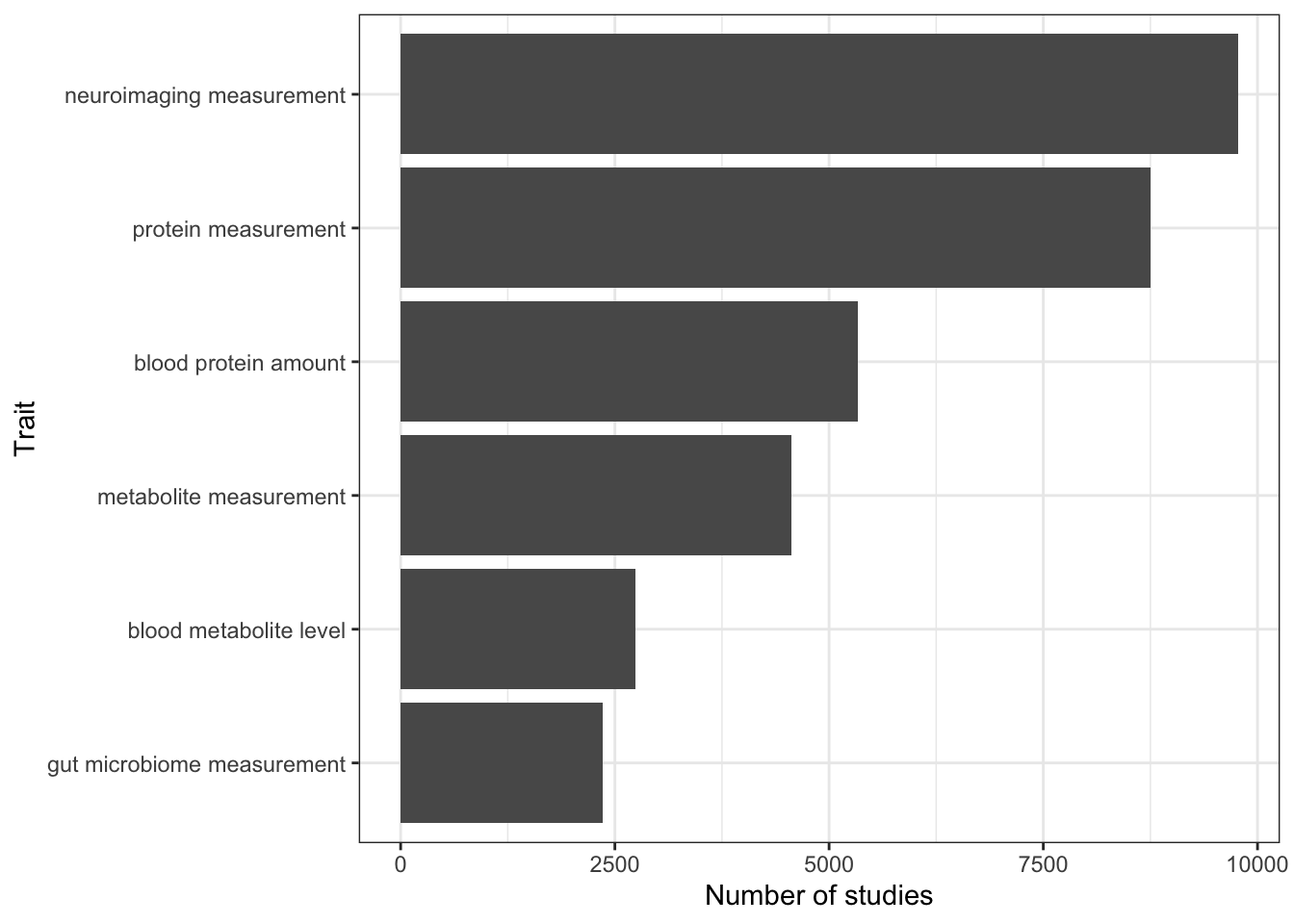
3 Unmapped / badly mapped traits:
# number of traits only observed once in the catalog
# likely badly mapped traits:
n_studies_trait |>
dplyr::filter(n_studies == 1) |>
nrow()[1] 11592unique_mapped_traits = n_studies_trait |>
dplyr::filter(n_studies == 1) |>
dplyr::pull(MAPPED_TRAIT)
gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits) |>
group_by(PUBMED_ID) |>
summarise(n_unmapped_per_study = n()) |>
arrange(desc(n_unmapped_per_study)) |>
head(n = 10)# A tibble: 10 × 2
PUBMED_ID n_unmapped_per_study
<int> <int>
1 35870639 3411
2 38412862 2821
3 39789286 612
4 29875488 388
5 39528826 384
6 33634981 197
7 24816252 173
8 36482414 121
9 26068415 99
10 37253714 953.1 PUBMED_ID 35870639
pubmed_id = 35870639
# this study tests
# 6,790 proteins or protein complexes
print(paste0("How many studies for pubmed id: ", pubmed_id))[1] "How many studies for pubmed id: 35870639"gwas_study_info |>
filter(PUBMED_ID == pubmed_id) |>
nrow()[1] 6790# not all are unmapped
print(paste0("How many studies for pubmed id: ", pubmed_id,
"have unmapped/not well mapped traits"
)
)[1] "How many studies for pubmed id: 35870639have unmapped/not well mapped traits"gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
nrow()[1] 3411not_well_mapped_traits =
gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
pull(MAPPED_TRAIT)
print("Of these unmapped studies, how many contain phrase in blood")[1] "Of these unmapped studies, how many contain phrase in blood"grepl("in blood serum", not_well_mapped_traits) |> sum()[1] 3408print("level of or amount of")[1] "level of or amount of"grepl("level of|amount of", not_well_mapped_traits) |> sum()[1] 3408# all unmapped
# map to blood protein amount
# serum proteome - i.e. blood proteome
gwas_study_info =
gwas_study_info |>
dplyr::rows_update(tibble(PUBMED_ID = pubmed_id,
MAPPED_TRAIT = "blood protein amount",
MAPPED_TRAIT_URI = "http://purl.obolibrary.org/obo/OBA_VT0005416"),
unmatched = "ignore")3.2 PUBMED_ID: 39789286
pubmed_id = 39789286
print(paste0("How many studies for pubmed id: ", pubmed_id))[1] "How many studies for pubmed id: 39789286"gwas_study_info |>
filter(PUBMED_ID == pubmed_id) |>
nrow()[1] 3049# not all are unmapped
print(paste0("How many studies for pubmed id: ", pubmed_id, "have unmapped/not well mapped traits"))[1] "How many studies for pubmed id: 39789286have unmapped/not well mapped traits"gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
nrow()[1] 612# 2,821 ratios between protein levels
# i.e. each study is a association study between ratio between two proteins
# ? put under blood protein amount (even though it is a ratio between amounts not an amount...)
gwas_study_info =
gwas_study_info |>
dplyr::rows_update(tibble(PUBMED_ID = pubmed_id,
MAPPED_TRAIT = "blood protein amount",
MAPPED_TRAIT_URI = "http://purl.obolibrary.org/obo/OBA_VT0005416"),
unmatched = "ignore")3.3 PUBMED_ID: 39789286
pubmed_id = 39789286
print(paste0("How many studies for pubmed id: ", pubmed_id))[1] "How many studies for pubmed id: 39789286"gwas_study_info |>
filter(PUBMED_ID == pubmed_id) |>
nrow()[1] 3049# not all are unmapped
print(paste0("How many studies for pubmed id: ", pubmed_id, "have unmapped/not well mapped traits"))[1] "How many studies for pubmed id: 39789286have unmapped/not well mapped traits"gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
nrow()[1] 0not_well_mapped_traits =
gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
pull(MAPPED_TRAIT)
print("Of these unmapped studies, how many contain phrase in blood")[1] "Of these unmapped studies, how many contain phrase in blood"grepl("in blood", not_well_mapped_traits) |> sum()[1] 0print("level of or amount of")[1] "level of or amount of"grepl("level of|amount of", not_well_mapped_traits) |> sum()[1] 0print("All unmapped studies are measurements in blood")[1] "All unmapped studies are measurements in blood"gwas_study_info = gwas_study_info |>
rowwise() |>
mutate(MAPPED_TRAIT = ifelse(PUBMED_ID == pubmed_id && MAPPED_TRAIT %in% unique_mapped_traits,
"blood protein amount",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(PUBMED_ID == pubmed_id && MAPPED_TRAIT %in% unique_mapped_traits,
"http://purl.obolibrary.org/obo/OBA_VT0005416",
MAPPED_TRAIT_URI)
) |>
ungroup()3.4 PUBMED_ID: 29875488
pubmed_id = 29875488
print(paste0("How many studies for pubmed id: ", pubmed_id))[1] "How many studies for pubmed id: 29875488"gwas_study_info |>
filter(PUBMED_ID == pubmed_id) |>
nrow()[1] 3283# not all are unmapped
print(paste0("How many studies for pubmed id: ", pubmed_id, "have unmapped/not well mapped traits"))[1] "How many studies for pubmed id: 29875488have unmapped/not well mapped traits"gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
nrow()[1] 388not_well_mapped_traits =
gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
pull(MAPPED_TRAIT)
print("Of these unmapped studies, how many contain phrase in blood")[1] "Of these unmapped studies, how many contain phrase in blood"grepl("in blood", not_well_mapped_traits) |> sum()[1] 0print("level of or amount of")[1] "level of or amount of"grepl("level of|amount of|measurement", not_well_mapped_traits) |> sum()[1] 388grepl("measurement", not_well_mapped_traits) |> sum()[1] 388# study title is: Genomic atlas of the human plasma proteome.
# assume therefore, all these values are protein blood measurements
gwas_study_info = gwas_study_info |>
rowwise() |>
mutate(MAPPED_TRAIT = ifelse(PUBMED_ID == pubmed_id && MAPPED_TRAIT %in% unique_mapped_traits,
"blood protein amount",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(PUBMED_ID == pubmed_id && MAPPED_TRAIT %in% unique_mapped_traits,
"http://purl.obolibrary.org/obo/OBA_VT0005416",
MAPPED_TRAIT_URI)
) |>
ungroup()# PUBMED_ID: 39528826
# suspect:
# metabolite measurement http://www.ebi.ac.uk/efo/EFO_0004725
# Title:
# Publication: Genetic architecture of cerebrospinal fluid and brain metabolite levels and the genetic colocalization of metabolites with human traits.# PUBMED+ID: 33634981
# suspect:
# metabolite measurement http://www.ebi.ac.uk/efo/EFO_0004725
# Genome-wide association study of metabolites in patients with coronary artery disease identified novel metabolite quantitative trait loci.# 24816252
pubmed_id = 24816252
# suspect:
# metabolite measurement http://www.ebi.ac.uk/efo/EFO_0004725
# An atlas of genetic influences on human blood metabolites.
print(paste0("How many studies for pubmed id: ", pubmed_id))[1] "How many studies for pubmed id: 24816252"gwas_study_info |>
filter(PUBMED_ID == pubmed_id) |>
nrow()[1] 533# not all are unmapped
print(paste0("How many studies for pubmed id: ", pubmed_id, "have unmapped/not well mapped traits"))[1] "How many studies for pubmed id: 24816252have unmapped/not well mapped traits"gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
nrow()[1] 173not_well_mapped_traits =
gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits,
PUBMED_ID == pubmed_id) |>
pull(MAPPED_TRAIT)
print("Of these unmapped studies, how many contain phrase in blood")[1] "Of these unmapped studies, how many contain phrase in blood"grepl("in blood", not_well_mapped_traits) |> sum()[1] 0print("level of or amount of")[1] "level of or amount of"grepl("level of|amount of|measurement|ratio", not_well_mapped_traits) |> sum()[1] 173gwas_study_info = gwas_study_info |>
rowwise() |>
mutate(MAPPED_TRAIT = ifelse(PUBMED_ID == pubmed_id && MAPPED_TRAIT %in% unique_mapped_traits,
"blood protein amount",
MAPPED_TRAIT)
) |>
mutate(MAPPED_TRAIT_URI = ifelse(PUBMED_ID == pubmed_id && MAPPED_TRAIT %in% unique_mapped_traits,
"http://purl.obolibrary.org/obo/OBA_VT0005416",
MAPPED_TRAIT_URI)
) |>
ungroup()# 36482414
# Publication: Comprehensive characterization of putative genetic influences on plasma metabolome in a pediatric cohort.gwas_study_info |>
dplyr::filter(MAPPED_TRAIT %in% unique_mapped_traits) |>
group_by(PUBMED_ID, STUDY, COHORT) |>
summarise(n_unmapped_per_study = n()) |>
arrange(desc(n_unmapped_per_study))# A tibble: 1,548 × 4
# Groups: PUBMED_ID, STUDY [1,512]
PUBMED_ID STUDY COHORT n_unmapped_per_study
<int> <chr> <chr> <int>
1 38412862 Genetic associations with ratios betwe… "UKBB" 2821
2 39528826 Genetic architecture of cerebrospinal … "Knig… 381
3 33634981 Genome-wide association study of metab… "" 197
4 36482414 Comprehensive characterization of puta… "othe… 121
5 37253714 Whole-Genome Sequencing Analysis of Hu… "ARIC… 95
6 37794183 Rare variant associations with plasma … "UKBB" 89
7 34737426 A generalized linear mixed model assoc… "UKBB" 81
8 26068415 Genome-wide association study identifi… "EGCU… 76
9 35347128 Genome-wide association studies of met… "METS… 55
10 39024449 Diversity and scale: Genetic architect… "MVP" 51
# ℹ 1,538 more rows4 Saving:
data.table::fwrite(gwas_study_info,
here::here("output/gwas_study_info_trait_corrected.csv"),
sep = ",")
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 15.6.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/Los_Angeles
tzcode source: internal
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] stringr_1.5.1 ggplot2_3.5.2 dplyr_1.1.4 data.table_1.17.8
[5] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] gtable_0.3.6 jsonlite_2.0.0 compiler_4.3.1 renv_1.0.3
[5] promises_1.3.3 tidyselect_1.2.1 Rcpp_1.1.0 git2r_0.36.2
[9] callr_3.7.6 later_1.4.2 jquerylib_0.1.4 scales_1.4.0
[13] yaml_2.3.10 fastmap_1.2.0 here_1.0.1 R6_2.6.1
[17] labeling_0.4.3 generics_0.1.4 knitr_1.50 tibble_3.3.0
[21] rprojroot_2.1.0 RColorBrewer_1.1-3 bslib_0.9.0 pillar_1.11.0
[25] rlang_1.1.6 utf8_1.2.6 cachem_1.1.0 stringi_1.8.7
[29] httpuv_1.6.16 xfun_0.52 getPass_0.2-4 fs_1.6.6
[33] sass_0.4.10 cli_3.6.5 withr_3.0.2 magrittr_2.0.3
[37] ps_1.9.1 grid_4.3.1 digest_0.6.37 processx_3.8.6
[41] rstudioapi_0.17.1 lifecycle_1.0.4 vctrs_0.6.5 evaluate_1.0.4
[45] glue_1.8.0 farver_2.1.2 whisker_0.4.1 rmarkdown_2.29
[49] httr_1.4.7 tools_4.3.1 pkgconfig_2.0.3 htmltools_0.5.8.1