UK Biobank GWAS Studies: Cohorts and Bibliometric Analysis
Isobel Beasley
Last updated: 2025-08-06
Checks: 6 1
Knit directory:
genomics_ancest_disease_dispar/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220216) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 925266a. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: data/gwas_catalog/
Untracked files:
Untracked: analysis/collapse_cohorts.Rmd
Untracked: data/.DS_Store
Untracked: renv/
Unstaged changes:
Modified: .Rprofile
Modified: analysis/icite_rcr.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
knitr::opts_chunk$set(echo = TRUE, message = FALSE, warning = FALSE)
library(data.table)
library(dplyr)
library(ggplot2)1 Overview / introduction:
I’m interested in identifying which studies use the same cohort/samples in the GWAS catalogue. Why? To produce ancestry proportion information that controls for the same samples (particularly big biobanks) being reused many times.
Within the same research article, this will likely be easier to achieve as: - Labelling of the cohort is likely to be consistent within the same research article (for e.g. one article will likely refer to UKBB samples as UKBB always, and not sometimes UK Biobank) - Many papers study the same cohort across diseases. Thus, this kind of cohort reuse can be identified by noticing that these papers have the same or similar sample sizes across diseases - Many papers use the same cohort/s, and then study particular subsamples and the overall cohort. This kind of sample reuse cohort can be identified when it involves, e.g. ancestry groups, as the overall study sample demographics are provided and can be matched to the study subsamples - (?) I believe the GWAS catalog somewhat reduces / identifys cohort overlap in the same study when producing ancestry metadata for the whole catalog
More recently, the GWAS catalog has allowed authors to supply cohort label information (e.g. UK Biobank). However, this doesn’t make it necessarily easier to compare across studies as: - Not all studies provide cohort information/label (especially studies before this policy was implemented) - Studies inconsistency label the same cohort (e.g. UK Biobank is listed as UKBB, UKB etc.) - Many studies use multiple cohorts
I believe I will be able to identify when the same samples are used across different research articles by matching sample sizes (e.g., does their sample description match the cohort of the UK Biobank), citation patterns (e.g., cite the UK Biobank dataset papers), etc. This will likely require considering publication date, as the sample sizes of biobanks/cohorts and the canonical dataset papers to cite will change over time.
This page aims to investigate how feasible this analysis will be.
Specifically, for sample re-use in the same study: - Can I easily categorise it into: subsample use / testing, & re-use across disease? - Are their papers which don’t match either of these categories?
For sample re-use across study: - How much missingness is there in cohort labelling? - Can I identify the UK Biobank dataset papers?
1.1 1. Load / pre-process GWAS Catalog data
# Load GWAS Catalog studies
gwas_study_info <- fread("data/gwas_catalog/gwas-catalog-v1.0.3.1-studies-r2025-07-21.tsv",
sep = "\t", quote = "")
# Standardize column names (remove spaces)
gwas_study_info <- gwas_study_info |>
rename_all(~gsub(" ", "_", .x))1.2 2. Cohort Summary
# Number of unique cohorts
length(unique(gwas_study_info$COHORT))[1] 1239# Studies per cohort
studies_per_cohort = gwas_study_info |>
group_by(COHORT) |>
summarise(n_studies = n()) |>
arrange(desc(n_studies))
studies_per_cohort |>
ggplot(aes(x=n_studies)) +
geom_histogram() +
theme_bw() +
labs(title = "Distribution of the number of studies per cohort label")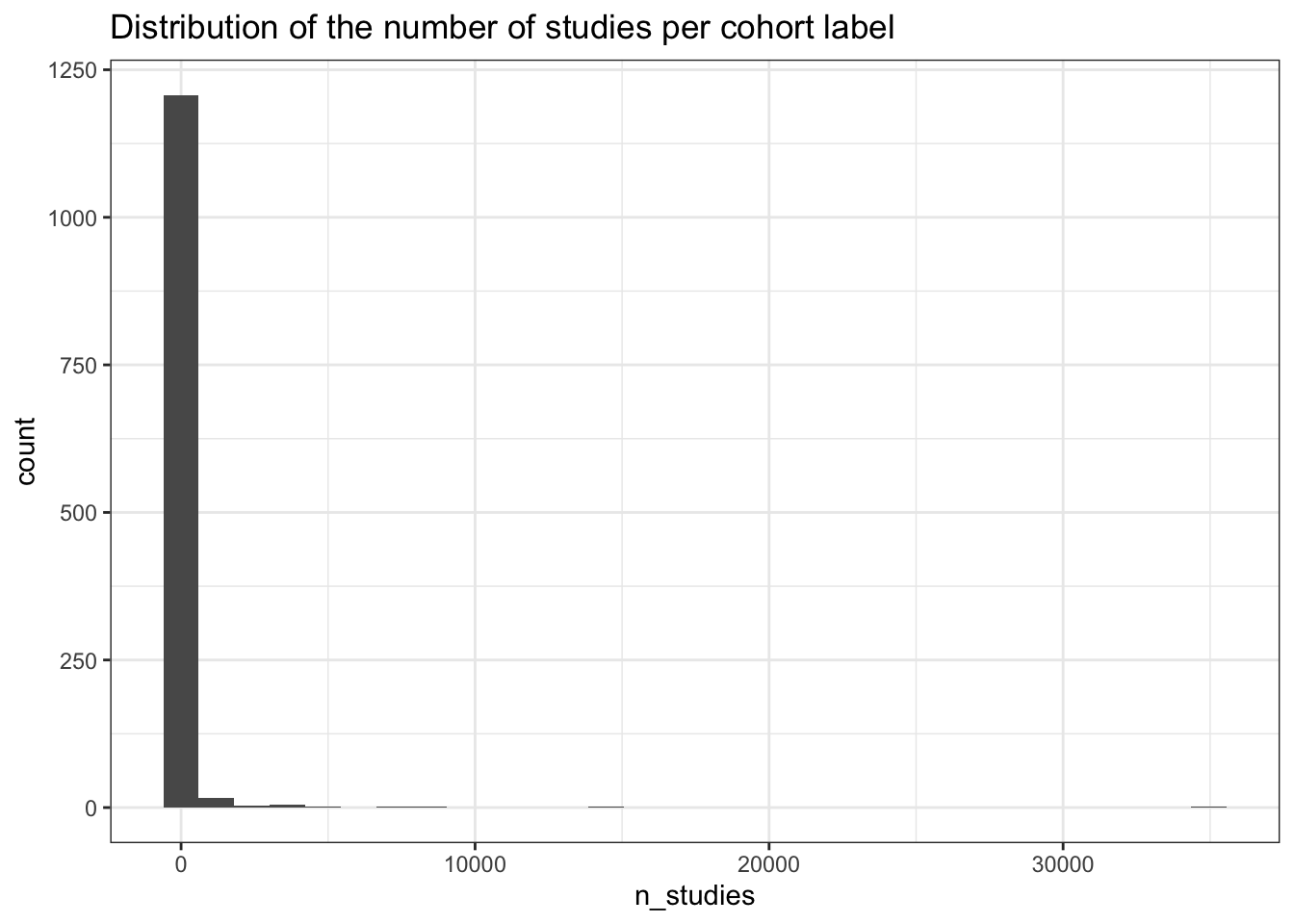
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 15.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/Los_Angeles
tzcode source: internal
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] ggplot2_3.5.2 dplyr_1.1.4 data.table_1.17.8 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] gtable_0.3.6 jsonlite_2.0.0 compiler_4.3.1 renv_1.0.3
[5] promises_1.3.3 tidyselect_1.2.1 Rcpp_1.1.0 stringr_1.5.1
[9] git2r_0.36.2 callr_3.7.6 later_1.4.2 jquerylib_0.1.4
[13] scales_1.4.0 yaml_2.3.10 fastmap_1.2.0 R6_2.6.1
[17] labeling_0.4.3 generics_0.1.4 knitr_1.50 tibble_3.3.0
[21] rprojroot_2.1.0 RColorBrewer_1.1-3 bslib_0.9.0 pillar_1.11.0
[25] rlang_1.1.6 cachem_1.1.0 stringi_1.8.7 httpuv_1.6.16
[29] xfun_0.52 getPass_0.2-4 fs_1.6.6 sass_0.4.10
[33] cli_3.6.5 withr_3.0.2 magrittr_2.0.3 ps_1.9.1
[37] grid_4.3.1 digest_0.6.37 processx_3.8.6 rstudioapi_0.17.1
[41] lifecycle_1.0.4 vctrs_0.6.5 evaluate_1.0.4 glue_1.8.0
[45] farver_2.1.2 whisker_0.4.1 rmarkdown_2.29 httr_1.4.7
[49] tools_4.3.1 pkgconfig_2.0.3 htmltools_0.5.8.1