Juicer Gene Expression
Ittai Eres
2018-09-14
Last updated: 2019-05-05
Checks: 6 0
Knit directory: HiCiPSC/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190311) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: analysis/.DS_Store
Ignored: code/.DS_Store
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: Rplot.jpeg
Untracked: Rplot001.jpeg
Untracked: Rplot002.jpeg
Untracked: Rplot003.jpeg
Untracked: Rplot004.jpeg
Untracked: Rplot005.jpeg
Untracked: Rplot006.jpeg
Untracked: Rplot007.jpeg
Untracked: Rplot008.jpeg
Untracked: Rplot009.jpeg
Untracked: Rplot010.jpeg
Untracked: Rplot011.jpeg
Untracked: Rplot012.jpeg
Untracked: Rplot013.jpeg
Untracked: Rplot014.jpeg
Untracked: Rplot015.jpeg
Untracked: Rplot016.jpeg
Untracked: Rplot017.jpeg
Untracked: Rplot018.jpeg
Untracked: Rplot019.jpeg
Untracked: Rplot020.jpeg
Untracked: Rplot021.jpeg
Untracked: Rplot022.jpeg
Untracked: Rplot023.jpeg
Untracked: Rplot024.jpeg
Untracked: Rplot025.jpeg
Untracked: Rplot026.jpeg
Untracked: Rplot027.jpeg
Untracked: Rplot028.jpeg
Untracked: Rplot029.jpeg
Untracked: Rplot030.jpeg
Untracked: Rplot031.jpeg
Untracked: Rplot032.jpeg
Untracked: Rplot033.jpeg
Untracked: Rplot034.jpeg
Untracked: Rplot035.jpeg
Untracked: Rplot036.jpeg
Untracked: Rplot037.jpeg
Untracked: Rplot038.jpeg
Untracked: Rplot039.jpeg
Untracked: Rplot040.jpeg
Untracked: Rplot041.jpeg
Untracked: Rplot042.jpeg
Untracked: Rplot043.jpeg
Untracked: Rplot044.jpeg
Untracked: Rplot045.jpeg
Untracked: Rplot046.jpeg
Untracked: Rplot047.jpeg
Untracked: Rplot048.jpeg
Untracked: Rplot049.jpeg
Untracked: Rplot050.jpeg
Untracked: Rplot051.jpeg
Untracked: Rplot052.jpeg
Untracked: Rplot053.jpeg
Untracked: Rplot054.jpeg
Untracked: Rplot055.jpeg
Untracked: Rplot056.jpeg
Untracked: Rplot057.jpeg
Untracked: Rplot058.jpeg
Untracked: Rplot059.jpeg
Untracked: Rplot060.jpeg
Untracked: Rplot061.jpeg
Untracked: Rplot062.jpeg
Untracked: Rplot063.jpeg
Untracked: Rplot064.jpeg
Untracked: Rplot065.jpeg
Untracked: Rplot066.jpeg
Untracked: Rplot067.jpeg
Untracked: Rplot068.jpeg
Untracked: Rplot069.jpeg
Untracked: Rplot070.jpeg
Untracked: Rplot071.jpeg
Untracked: Rplot072.jpeg
Untracked: Rplot073.jpeg
Untracked: Rplot074.jpeg
Untracked: Rplot075.jpeg
Untracked: Rplot076.jpeg
Untracked: Rplot077.jpeg
Untracked: Rplot078.jpeg
Untracked: Rplot079.jpeg
Untracked: Rplot080.jpeg
Untracked: Rplot081.jpeg
Untracked: Rplot082.jpeg
Untracked: Rplot083.jpeg
Untracked: Rplot084.jpeg
Untracked: Rplot085.jpeg
Untracked: Rplot086.jpeg
Untracked: Rplot087.jpeg
Untracked: Rplot088.jpeg
Untracked: Rplot089.jpeg
Untracked: Rplot090.jpeg
Untracked: Rplot091.jpeg
Untracked: Rplot092.jpeg
Untracked: Rplot093.jpeg
Untracked: Rplot094.jpeg
Untracked: Rplot095.jpeg
Untracked: Rplot096.jpeg
Untracked: Rplot097.jpeg
Untracked: Rplot098.jpeg
Untracked: Rplot099.jpeg
Untracked: Rplot100.jpeg
Untracked: Rplot101.jpeg
Untracked: Rplot102.jpeg
Untracked: Rplot103.jpeg
Untracked: Rplot104.jpeg
Untracked: Rplot105.jpeg
Untracked: Rplot106.jpeg
Untracked: Rplot107.jpeg
Untracked: Rplot108.jpeg
Untracked: Rplot109.jpeg
Untracked: Rplot110.jpeg
Untracked: Rplot111.jpeg
Untracked: Rplot112.jpeg
Untracked: Rplot113.jpeg
Untracked: Rplot114.jpeg
Untracked: Rplot115.jpeg
Untracked: Rplot116.jpeg
Untracked: Rplot117.jpeg
Untracked: Rplot118.jpeg
Untracked: Rplot119.jpeg
Untracked: Rplot120.jpeg
Untracked: Rplot121.jpeg
Untracked: Rplot122.jpeg
Untracked: Rplot123.jpeg
Untracked: Rplot124.jpeg
Untracked: Rplot125.jpeg
Untracked: Rplot126.jpeg
Untracked: Rplot127.jpeg
Untracked: Rplot128.jpeg
Untracked: Rplot129.jpeg
Untracked: Rplot130.jpeg
Untracked: Rplot131.jpeg
Untracked: Rplot132.jpeg
Untracked: Rplot133.jpeg
Untracked: Rplot134.jpeg
Untracked: Rplot135.jpeg
Untracked: Rplot136.jpeg
Untracked: Rplot137.jpeg
Untracked: Rplot138.jpeg
Untracked: Rplot139.jpeg
Untracked: Rplot140.jpeg
Untracked: Rplot141.jpeg
Untracked: Rplot142.jpeg
Untracked: Rplot143.jpeg
Untracked: Rplot144.jpeg
Untracked: Rplot145.jpeg
Untracked: Rplot146.jpeg
Untracked: Rplot147.jpeg
Untracked: Rplot148.jpeg
Untracked: Rplot149.jpeg
Untracked: Rplot150.jpeg
Untracked: Rplot151.jpeg
Untracked: Rplot152.jpeg
Untracked: Rplot153.jpeg
Untracked: Rplot154.jpeg
Untracked: Rplot155.jpeg
Untracked: Rplot156.jpeg
Untracked: Rplot157.jpeg
Untracked: Rplot158.jpeg
Untracked: Rplot159.jpeg
Untracked: Rplot160.jpeg
Untracked: Rplot161.jpeg
Untracked: Rplot162.jpeg
Untracked: Rplot163.jpeg
Untracked: Rplot164.jpeg
Untracked: Rplot165.jpeg
Untracked: Rplot166.jpeg
Untracked: Rplot167.jpeg
Untracked: Rplot168.jpeg
Untracked: Rplot169.jpeg
Untracked: Rplot170.jpeg
Untracked: Rplot171.jpeg
Untracked: Rplot172.jpeg
Untracked: Rplot173.jpeg
Untracked: Rplot174.jpeg
Untracked: Rplot175.jpeg
Untracked: Rplot176.jpeg
Untracked: Rplot177.jpeg
Untracked: Rplot178.jpeg
Untracked: Rplot179.jpeg
Untracked: Rplot180.jpeg
Untracked: Rplot181.jpeg
Untracked: Rplot182.jpeg
Untracked: Rplot183.jpeg
Untracked: Rplot184.jpeg
Untracked: Rplot185.jpeg
Untracked: Rplot186.jpeg
Untracked: Rplot187.jpeg
Untracked: Rplot188.jpeg
Untracked: Rplot189.jpeg
Untracked: Rplot190.jpeg
Untracked: Rplot191.jpeg
Untracked: Rplot192.jpeg
Untracked: Rplot193.jpeg
Untracked: Rplot194.jpeg
Untracked: Rplot195.jpeg
Untracked: Rplot196.jpeg
Untracked: Rplot197.jpeg
Untracked: Rplot198.jpeg
Untracked: Rplot199.jpeg
Untracked: Rplot200.jpeg
Untracked: Rplot201.jpeg
Untracked: Rplot202.jpeg
Untracked: Rplot203.jpeg
Untracked: Rplot204.jpeg
Untracked: Rplot205.jpeg
Untracked: Rplot206.jpeg
Untracked: Rplot207.jpeg
Untracked: Rplot208.jpeg
Untracked: Rplot209.jpeg
Untracked: Rplot210.jpeg
Untracked: Rplot211.jpeg
Untracked: Rplot212.jpeg
Untracked: Rplot213.jpeg
Untracked: Rplot214.jpeg
Untracked: Rplot215.jpeg
Untracked: Rplot216.jpeg
Untracked: Rplot217.jpeg
Untracked: Rplot218.jpeg
Untracked: Rplot219.jpeg
Untracked: Rplot220.jpeg
Untracked: Rplot221.jpeg
Untracked: Rplot222.jpeg
Untracked: Rplot223.jpeg
Untracked: Rplot224.jpeg
Untracked: Rplot225.jpeg
Untracked: Rplot226.jpeg
Untracked: Rplot227.jpeg
Untracked: Rplot228.jpeg
Untracked: Rplot229.jpeg
Untracked: Rplot230.jpeg
Untracked: Rplot231.jpeg
Untracked: Rplot232.jpeg
Untracked: Rplot233.jpeg
Untracked: Rplot234.jpeg
Untracked: Rplot235.jpeg
Untracked: Rplot236.jpeg
Untracked: Rplot237.jpeg
Untracked: Rplot238.jpeg
Untracked: Rplot239.jpeg
Untracked: Rplot240.jpeg
Untracked: Rplot241.jpeg
Untracked: Rplot242.jpeg
Untracked: Rplot243.jpeg
Untracked: Rplot244.jpeg
Untracked: Rplot245.jpeg
Untracked: Rplot246.jpeg
Untracked: Rplot247.jpeg
Untracked: Rplot248.jpeg
Untracked: Rplot249.jpeg
Untracked: Rplot250.jpeg
Untracked: Rplot251.jpeg
Untracked: Rplot252.jpeg
Untracked: Rplot253.jpeg
Untracked: Rplot254.jpeg
Untracked: Rplot255.jpeg
Untracked: Rplot256.jpeg
Untracked: Rplot257.jpeg
Untracked: Rplot258.jpeg
Untracked: Rplot259.jpeg
Untracked: Rplot260.jpeg
Untracked: Rplot261.jpeg
Untracked: Rplot262.jpeg
Untracked: Rplot263.jpeg
Untracked: Rplot264.jpeg
Untracked: Rplot265.jpeg
Untracked: Rplot266.jpeg
Untracked: Rplot267.jpeg
Untracked: Rplot268.jpeg
Untracked: Rplot269.jpeg
Untracked: Rplot270.jpeg
Untracked: Rplot271.jpeg
Untracked: Rplot272.jpeg
Untracked: Rplot273.jpeg
Untracked: Rplot274.jpeg
Untracked: Rplot275.jpeg
Untracked: Rplot276.jpeg
Untracked: Rplot277.jpeg
Untracked: Rplot278.jpeg
Untracked: Rplot279.jpeg
Untracked: Rplot280.jpeg
Untracked: Rplot281.jpeg
Untracked: Rplot282.jpeg
Untracked: Rplot283.jpeg
Untracked: Rplot284.jpeg
Untracked: Rplot285.jpeg
Untracked: Rplot286.jpeg
Untracked: Rplot287.jpeg
Untracked: Rplot288.jpeg
Untracked: Rplot289.jpeg
Untracked: Rplot290.jpeg
Untracked: Rplot291.jpeg
Untracked: Rplot292.jpeg
Untracked: Rplot293.jpeg
Untracked: Rplot294.jpeg
Untracked: Rplot295.jpeg
Untracked: Rplot296.jpeg
Untracked: Rplot297.jpeg
Untracked: Rplot298.jpeg
Untracked: Rplot299.jpeg
Untracked: Rplot300.jpeg
Untracked: Rplot301.jpeg
Untracked: Rplot302.jpeg
Untracked: Rplot303.jpeg
Untracked: Rplot304.jpeg
Untracked: S2A.jpeg
Untracked: S2B.jpeg
Untracked: code/mediate.test.regressing.R
Untracked: data/Chimp_orthoexon_extended_info.txt
Untracked: data/Human_orthoexon_extended_info.txt
Untracked: data/Meta_data.txt
Untracked: data/TADs/
Untracked: data/chimp_lengths.txt
Untracked: data/counts_iPSC.txt
Untracked: data/epigenetic_enrichments/
Untracked: data/final.10kb.homer.df
Untracked: data/final.juicer.10kb.KR
Untracked: data/final.juicer.10kb.VC
Untracked: data/hic_gene_overlap/
Untracked: data/human_lengths.txt
Untracked: data/old_mediation_permutations/
Untracked: output/DC_regions.txt
Untracked: output/IEE.RPKM.RDS
Untracked: output/IEE_voom_object.RDS
Untracked: output/data.4.filtered.lm.QC
Untracked: output/data.4.fixed.init.LM
Untracked: output/data.4.fixed.init.QC
Untracked: output/data.4.init.LM
Untracked: output/data.4.init.QC
Untracked: output/data.4.lm.QC
Untracked: output/full.data.10.init.LM
Untracked: output/full.data.10.init.QC
Untracked: output/full.data.10.lm.QC
Untracked: output/full.data.annotations.RDS
Untracked: output/gene.hic.filt.KR.RDS
Untracked: output/gene.hic.filt.RDS
Untracked: output/gene.hic.filt.VC.RDS
Untracked: output/juicer.IEE.RPKM.RDS
Untracked: output/juicer.IEE_voom_object.RDS
Untracked: output/juicer.filt.KR
Untracked: output/juicer.filt.KR.final
Untracked: output/juicer.filt.KR.lm
Untracked: output/juicer.filt.VC
Untracked: output/juicer.filt.VC.final
Untracked: output/juicer.filt.VC.lm
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2419813 | Ittai Eres | 2019-05-05 | Update all files. |
| html | ff886b1 | Ittai Eres | 2019-04-30 | Build site. |
| html | b7d82fc | Ittai Eres | 2019-04-30 | Build site. |
| Rmd | 8380e8b | Ittai Eres | 2019-04-30 | Add variety of juicer analyses into website files. |
###Preparing the gene expression data Here I read in a dataframe of counts summarized at the gene-level, then do some pre-processing and normalization to obtain a voom object to later run linear modeling analyses on. This is identical code to what was done in the case with HOMER, since this gene expression analysis is completely orthogonal to the Hi-C normalization and significance calling schemes employed.
#Read in counts data, create DGEList object out of them and convert to log counts per million (CPM). Also read in metadata.
setwd("/Users/ittaieres/HiCiPSC")
counts <- fread("data/counts_iPSC.txt", header=TRUE, data.table=FALSE, stringsAsFactors = FALSE, na.strings=c("NA",""))
colnames(counts) <- c("genes", "C-3649", "G-3624", "H-3651", "D-40300", "F-28834", "B-28126", "E-28815", "A-21792")
rownames(counts) <- counts$genes
counts <- counts[,-1]
dge <- DGEList(counts, genes=rownames(counts))
#Now, convert counts into RPKM to account for gene length differences between species. First load in and re-organize metadata, then the gene lengths for both species, and then the function to convert counts to RPKM.
meta_data <- fread("data/Meta_data.txt", sep="\t",stringsAsFactors = FALSE,header=T,na.strings=c("NA",""))
meta_data$fullID <- c("C-3649", "H-3651", "B-28126", "D-40300", "G-3624", "A-21792", "E-28815", "F-28834")
ord <- data.frame(fullID=colnames(counts)) #Pull order of samples from expression object
left_join(ord, meta_data, by="fullID") -> group_ref #left join meta data to this to make sure sample IDs correctWarning: Column `fullID` joining factor and character vector, coercing into
character vector#Read in human and chimp gene lengths for the RPKM function:
human_lengths<- fread("data/human_lengths.txt", sep="\t",stringsAsFactors = FALSE,header=T,na.strings=c("NA",""))
chimp_lengths<- fread("data/chimp_lengths.txt", sep="\t",stringsAsFactors = FALSE,header=T,na.strings=c("NA",""))
#The function for RPKM conversion.
vRPKM <- function(expr.obj,chimp_lengths,human_lengths,meta4) {
if (is.null(expr.obj$E)) {
meta4%>%filter(SP=="C" & fullID %in% colnames(counts))->chimp_meta
meta4%>%filter(SP=="H" & fullID %in% colnames(counts))->human_meta
#using RPKM function:
#Put genes in correct order:
expr.obj$genes %>%select(Geneid=genes)%>%
left_join(.,chimp_lengths,by="Geneid")%>%select(Geneid,ch.length)->chlength
expr.obj$genes %>%select(Geneid=genes)%>%
left_join(.,human_lengths,by="Geneid")%>%select(Geneid,hu.length)->hulength
#Chimp RPKM
expr.obj$genes$Length<-(chlength$ch.length)
RPKMc=rpkm(expr.obj,normalized.lib.sizes=TRUE, log=TRUE)
RPKMc[,colnames(RPKMc) %in% chimp_meta$fullID]->rpkm_chimp
#Human RPKM
expr.obj$genes$Length<-hulength$hu.length
RPKMh=rpkm(expr.obj,normalized.lib.sizes=TRUE, log=TRUE)
RPKMh[,colnames(RPKMh) %in% human_meta$fullID]->rpkm_human
cbind(rpkm_chimp,rpkm_human)->allrpkm
expr.obj$E <- allrpkm
return(expr.obj)
}
else {
#Pull out gene order from voom object and add in gene lengths from feature counts file
#Put genes in correct order:
expr.obj$genes %>%select(Geneid=genes)%>%
left_join(.,chimp_lengths,by="Geneid")%>%select(Geneid,ch.length)->chlength
expr.obj$genes %>%select(Geneid=genes)%>%
left_join(.,human_lengths,by="Geneid")%>%select(Geneid,hu.length)->hulength
#Filter meta data to be able to separate human and chimp
meta4%>%filter(SP=="C")->chimp_meta
meta4%>%filter(SP=="H")->human_meta
#Pull out the expression data in cpm to convert to RPKM
expr.obj$E->forRPKM
forRPKM[,colnames(forRPKM) %in% chimp_meta$fullID]->rpkm_chimp
forRPKM[,colnames(forRPKM) %in% human_meta$fullID]->rpkm_human
#Make log2 in KB:
row.names(chlength)=chlength$Geneid
chlength %>% select(-Geneid)->chlength
as.matrix(chlength)->chlength
row.names(hulength)=hulength$Geneid
hulength %>% select(-Geneid)->hulength
as.matrix(hulength)->hulength
log2(hulength/1000)->l2hulength
log2(chlength/1000)->l2chlength
#Subtract out log2 kb:
sweep(rpkm_chimp, 1,l2chlength,"-")->chimp_rpkm
sweep(rpkm_human, 1,l2hulength,"-")->human_rpkm
colnames(forRPKM)->column_order
cbind(chimp_rpkm,human_rpkm)->vRPKMS
#Put RPKMS back into the VOOM object:
expr.obj$E <- (vRPKMS[,colnames(vRPKMS) %in% column_order])
return(expr.obj)
}
}
dge <- vRPKM(dge, chimp_lengths, human_lengths, group_ref) #Normalize via log2 RPKM.
#A typical low-expression filtering step: use default prior count adding (0.25), and filtering out anything that has fewer than half the individuals within each species having logCPM less than 1.5 (so want 2 humans AND 2 chimps with log2CPM >= 1.5)
lcpms <- cpm(dge$counts, log=TRUE) #Obtain log2CPM!
good.chimps <- which(rowSums(lcpms[,1:4]>=1.5)>=2) #Obtain good chimp indices
good.humans <- which(rowSums(lcpms[,5:8]>=1.5)>=2) #Obtain good human indices
filt <- good.humans[which(good.humans %in% good.chimps)] #Subsets us down to a solid 11,292 genes--will go for a similar percentage with RPKM cutoff vals! (25.6% of total)
#Repeat filtering step, this time on RPKMs. 0.4 was chosen as a cutoff as it obtains close to the same results as 1.5 lcpm (in terms of percentage of genes retained)
good.chimps <- which(rowSums(dge$E[,1:4]>=0.4)>=2) #Obtain good chimp indices.
good.humans <- which(rowSums(dge$E[,5:8]>=0.4)>=2) #Obtain good human indices.
RPKM_filt <- good.humans[which(good.humans %in% good.chimps)] #Still leaves us with 11,946 genes (27.1% of total)
#Do the actual filtering.
dge_filt <- dge[RPKM_filt,]
dge_filt$E <- dge$E[RPKM_filt,]
dge_filt$counts <- dge$counts[RPKM_filt,]
dge_final <- calcNormFactors(dge_filt, method="TMM") #Calculate normalization factors with trimmed mean of M-values (TMM).
dge_norm <- calcNormFactors(dge, method="TMM") #Calculate normalization factors with TMM on dge before filtering out lowly expressed genes, for normalization visualization.
#Quick visualization of the filtering I've just performed:
col <- brewer.pal(8, "Paired")
par(mfrow=c(1,2))
plot(density(dge$E[,1]), col=col[1], lwd=2, ylim=c(0,0.35), las=2,
main="", xlab="")
title(main="A. Raw data", xlab="Log-RPKM")
abline(v=1, lty=3)
for (i in 2:8){
den <- density(dge$E[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", colnames(dge$E[,1:8]), text.col=col, bty="n")
plot(density(dge_final$E[,1]), col=col[1], lwd=2, ylim=c(0,0.35), las=2,
main="", xlab="")
title(main="B. Filtered data", xlab="Log-RPKM")
abline(v=1, lty=3)
for (i in 2:8){
den <- density(dge_final$E[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", colnames(dge_final[,1:8]), text.col=col, bty="n")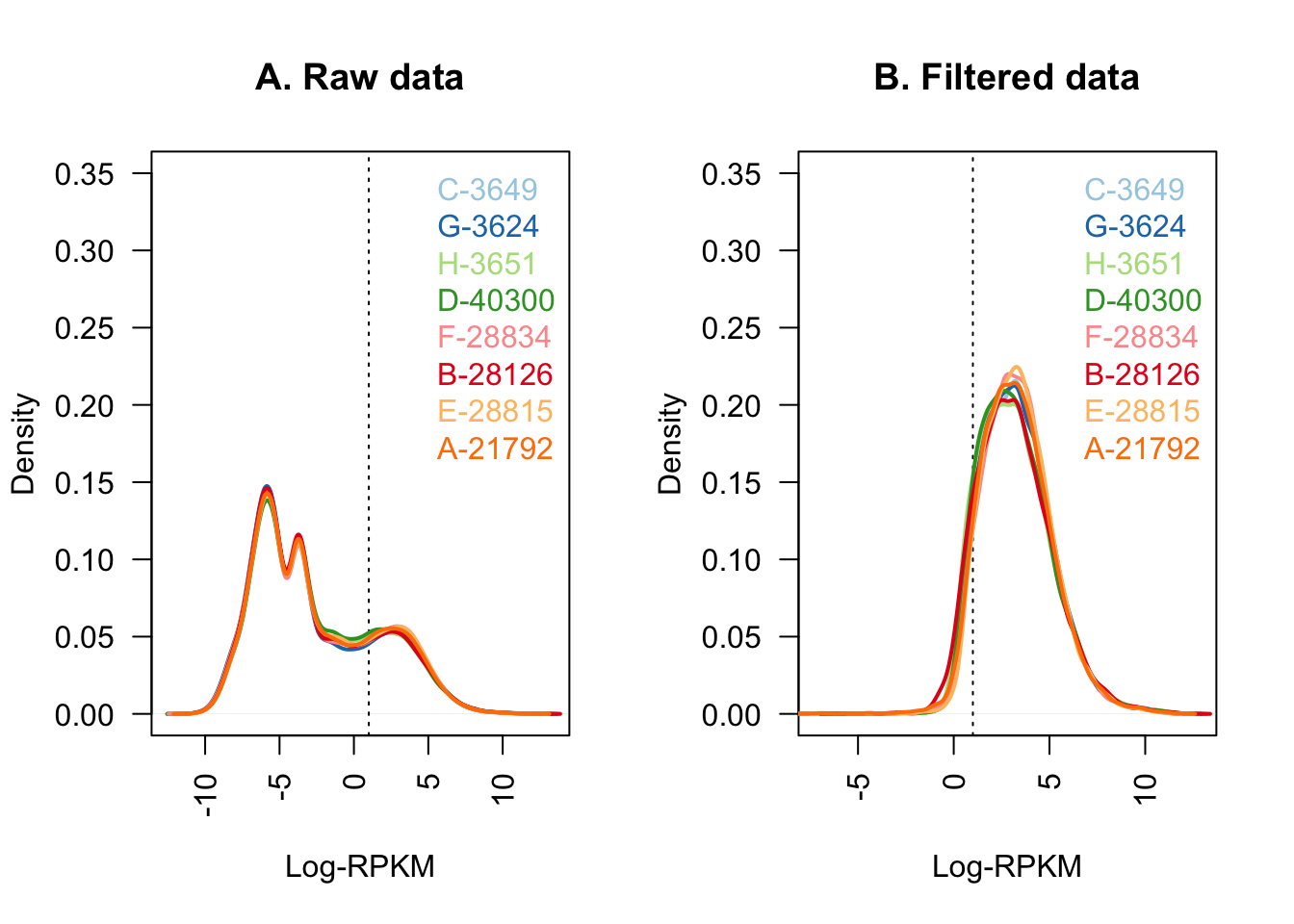
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
#Quick visualization of the normalization on the whole set of genes.
col <- brewer.pal(8, "Paired")
raw <- as.data.frame(dge$E[,1:8])
normed <- as.data.frame(dge_norm$E[,1:8])
par(mfrow=c(1,2))
boxplot(raw, las=2, col=col, main="")
title(main="Unnormalized data",ylab="Log-RPKM")
boxplot(normed, las=2, col=col, main="")
title(main="Normalized data",ylab="Log-RPKM")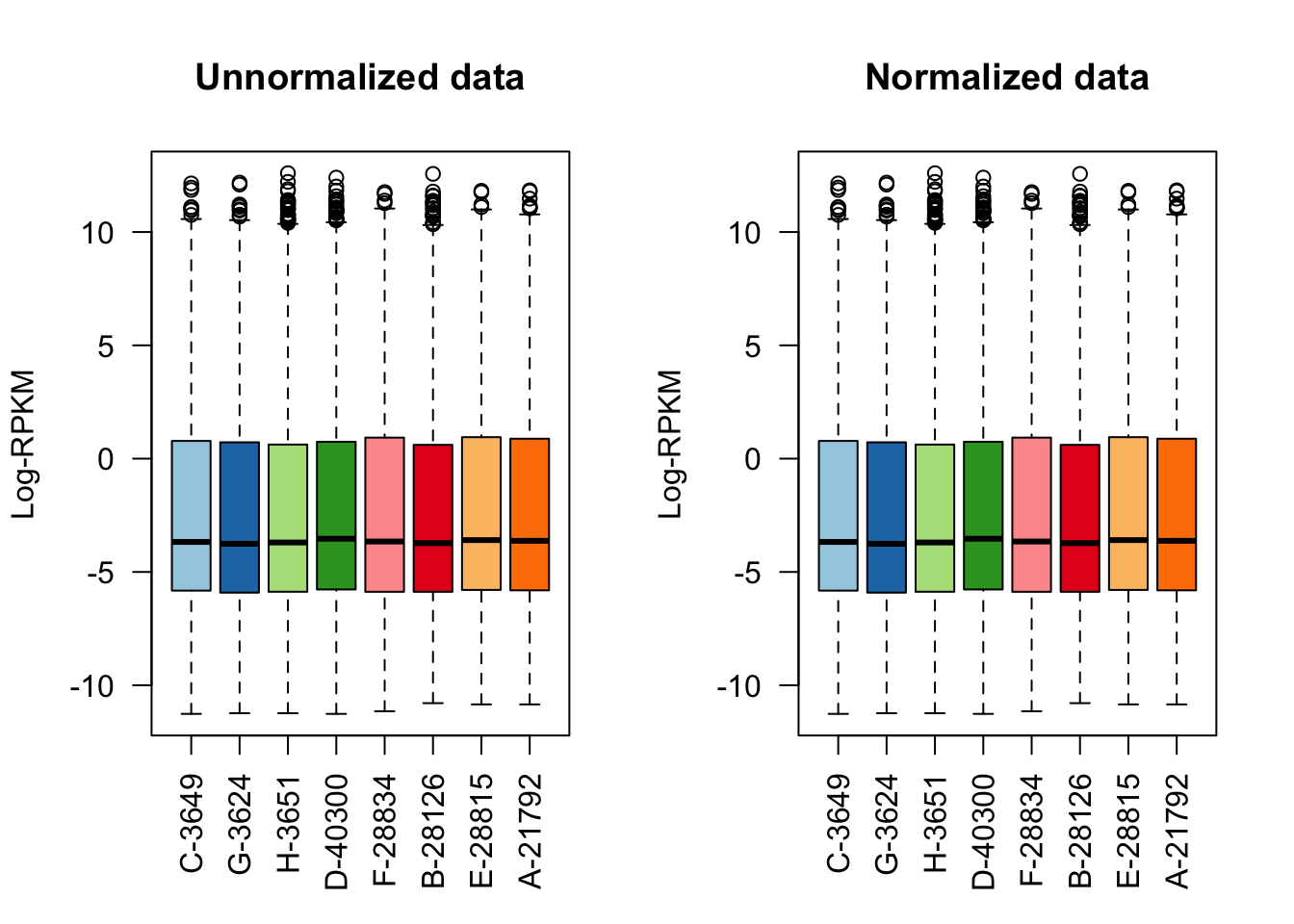
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
#Now, observe normalization on the filtered set of genes.
col <- brewer.pal(8, "Paired")
raw <- as.data.frame(dge_filt$E[,1:8])
normed <- as.data.frame(dge_final$E[,1:8])
par(mfrow=c(1,2))
boxplot(raw, las=2, col=col, main="")
title(main="Unnormalized data",ylab="Log-RPKM")
boxplot(normed, las=2, col=col, main="")
title(main="Normalized data",ylab="Log-RPKM")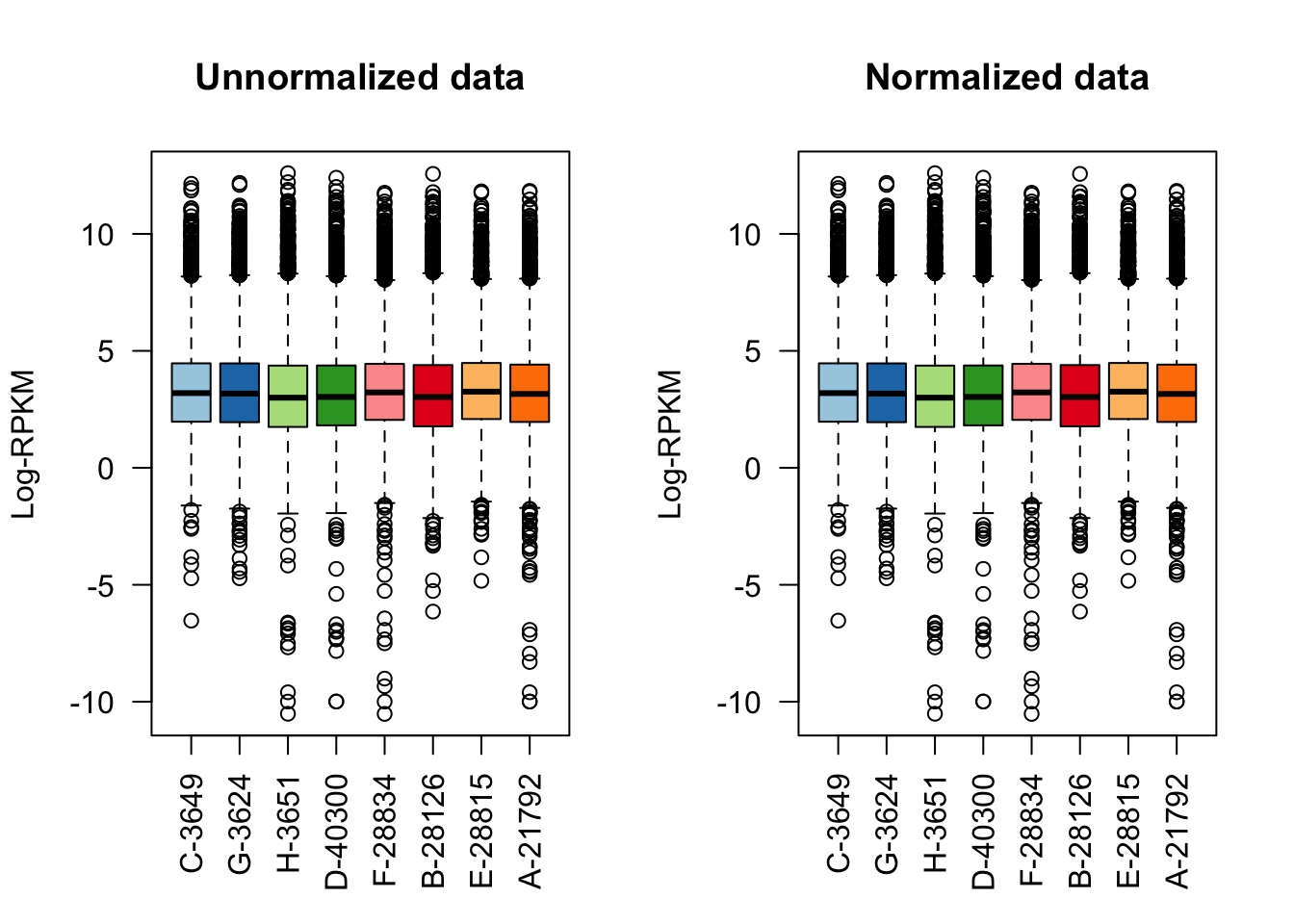
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
#Now, do some quick MDS plotting to make sure this expression data separates out species properly.
species <- c("C", "C", "C", "C", "H", "H", "H", "H")
color <- c(rep("red", 4), rep("blue", 4))
par(mfrow=c(1,1))
plotMDS(dge_final$E[,1:8], labels=species, col=color, main="MDS Plot") #Shows separation of the species along the logFC dimension representing the majority of the variance--orthogonal check to PCA, and looks great!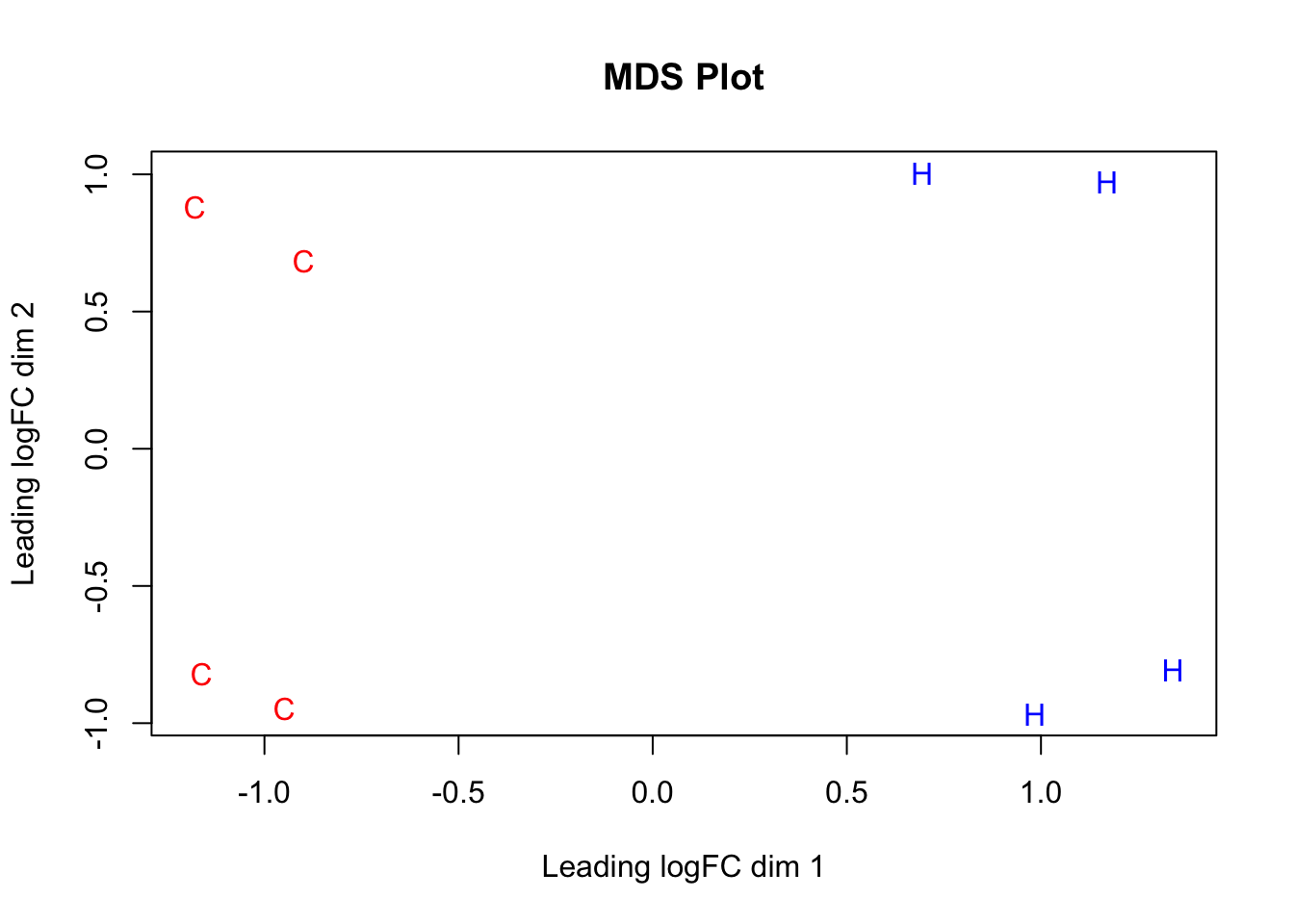
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
###Now, apply voom to get quality weights.
meta.exp.data <- data.frame("SP"=c("C", "C", "C", "C", "H", "H", "H", "H"), "SX"=c("M","M" ,"F","F","F", "M","M","F"))
SP <- factor(meta.exp.data$SP,levels = c("H","C"))
SX <- factor(meta.exp.data$SX, levels=c("M", "F"))
exp.design <- model.matrix(~0+SP+SX)#(~1+meta.exp.data$SP+meta.exp.data$SX)
colnames(exp.design) <- c("Human", "Chimp", "Sex")
weighted.data <- voom(dge_final, exp.design, plot=TRUE, normalize.method = "cyclicloess")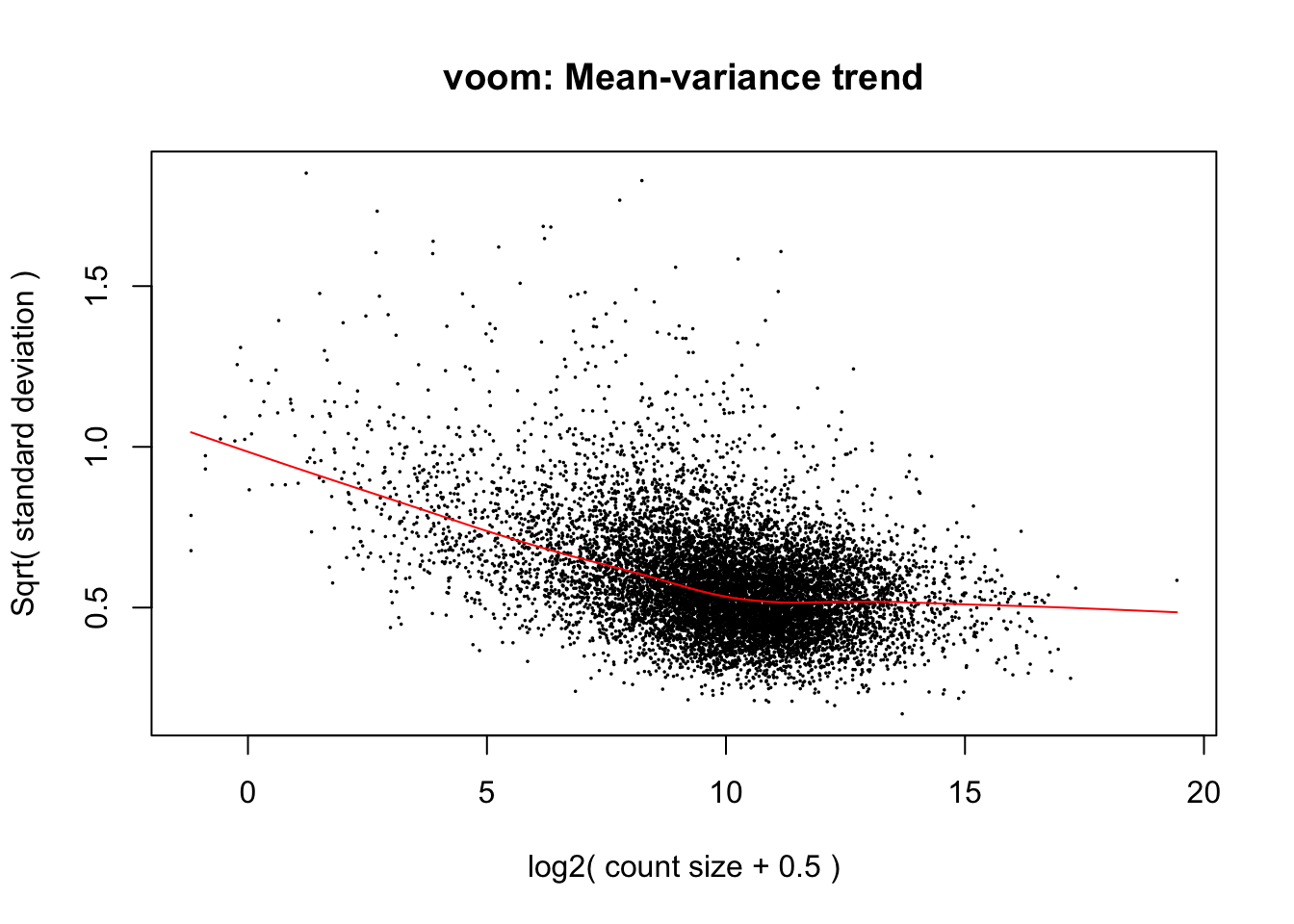
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
##Obtain rest of LM results, with particular eye to DE table!
vfit <- lmFit(weighted.data, exp.design)
efit <- eBayes(vfit)
mycon <- makeContrasts(HvC = Human-Chimp, levels = exp.design)
diff_species <- contrasts.fit(efit, mycon)
finalfit <- eBayes(diff_species)
detable <- topTable(finalfit, coef = 1, adjust.method = "BH", number = Inf, sort.by="none")
plotSA(efit, main="Final model: Mean−variance trend")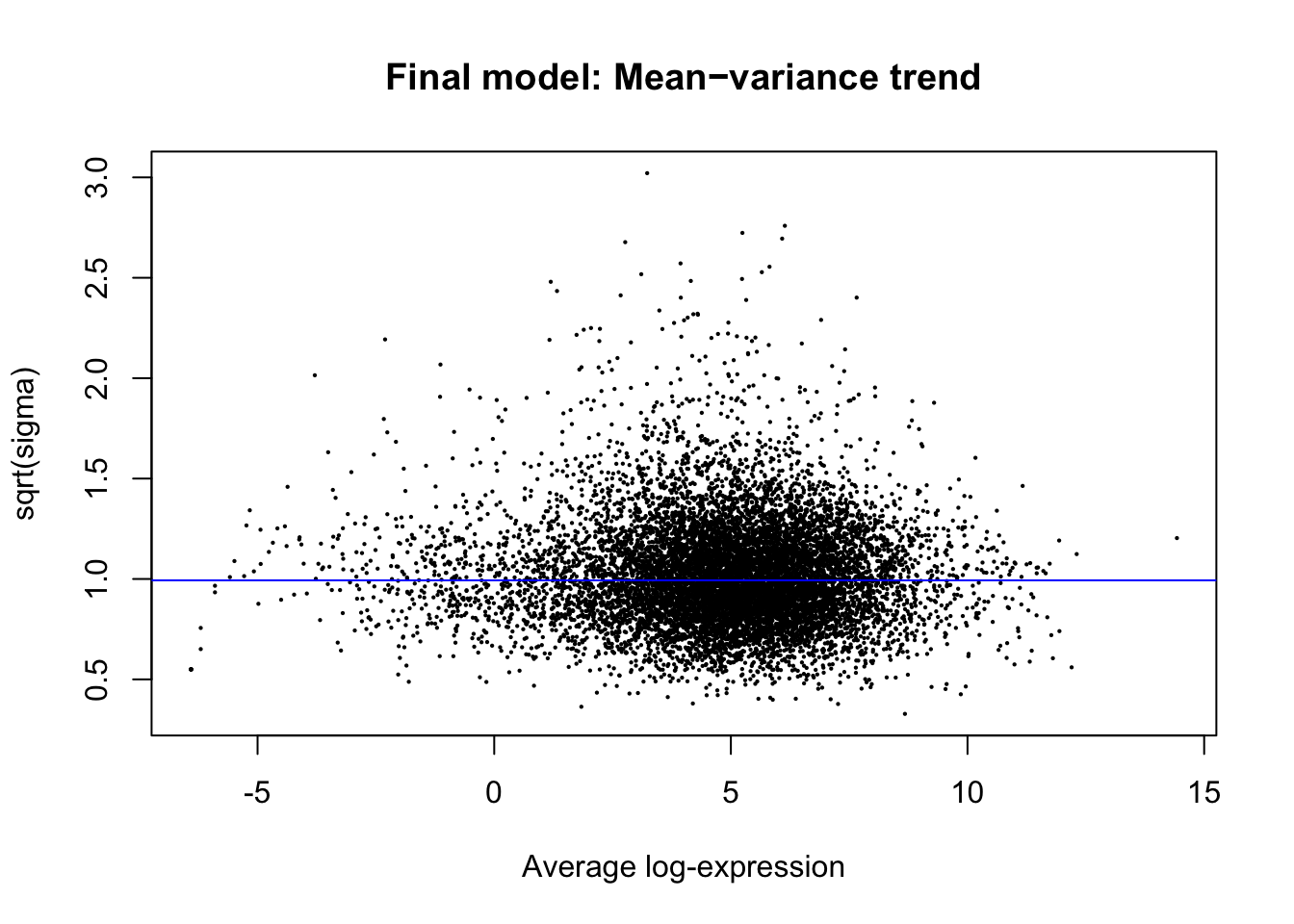
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
#Get lists of the DE and non-DE genes so I can run separate analyses on them at any point.
DEgenes <- detable$genes[which(detable$adj.P.Val<=0.05)]
nonDEgenes <- detable$genes[-which(detable$adj.P.Val<=0.05)]
#Rearrange RPKM and weight columns in voom object to be similar to the rest of my setup throughout in other dataframes.
weighted.data$E <- weighted.data$E[,c(8, 6, 1, 4, 7, 5, 2, 3)]
weighted.data$weights <- weighted.data$weights[,c(8, 6, 1, 4, 7, 5, 2, 3)]
RPKM <- weighted.data$E
rownames(RPKM) <- NULL #Just to match what I had before on midway2, about to write this out.
saveRDS(RPKM, file="output/juicer.IEE.RPKM.RDS") #Should be identical to non-juicer one, but just in case.
saveRDS(weighted.data, file="output/juicer.IEE_voom_object.RDS") #write this object out, can then be read in with readRDS.###Overlap Between Juicer Data and Orthogonal Gene Expression Data In this section I find the overlap between the 2 final filtered set of Hi-C Juicer significant hits and genes picked up on by an orthogonal RNA-seq experiment in the same set of cell lines. I utilize an in-house curated set of orthologous genes between humans and chimpanzees. Given that the resolution of the data is 10kb, I choose a simple and conservative approach and use a 1-nucleotide interval at the start of each gene as a proxy for the promoter. I then take a conservative pass and only use genes that had direct overlap with a bin from the Hi-C significant hits data, with more motivation explained below.
#Now, read in filtered data from linear_modeling_QC.Rmd.
data.KR <- fread("output/juicer.filt.KR.final", header=TRUE, data.table=FALSE, stringsAsFactors = FALSE, showProgress=FALSE)
data.VC <- fread("output/juicer.filt.VC.final", header=TRUE, data.table=FALSE, stringsAsFactors = FALSE, showProgress = FALSE)
#Quick VC df fix to make function work right (some col names have changed due to paper visualization):
data.VC$H1 <- gsub("Chr. ", "chr", data.VC$H1)
data.VC$H2 <- gsub("Chr. ", "chr", data.VC$H2)
data.VC$Hchr <- gsub("Chr. ", "chr", data.VC$Hchr)
meta.data <- data.frame("SP"=c("H", "H", "C", "C", "H", "H", "C", "C"), "SX"=c("F", "M", "M", "F", "M", "F", "M", "F"), "Batch"=c(1, 1, 1, 1, 2, 2, 2, 2))
#####GENE Hi-C Hit overlap: Already have hgenes and cgenes properly formatted and in the necessary folder from the HOMER version of this analysis. See HOMER version of gene_expression analysis for more details on how I obtained orthologous genes.
#Now, what I will need to do is prep bed files from the 2 sets of filtered data for each bin, in order to run bedtools-closest on them with the human and chimp gene data. This is for getting each bin's proximity to TSS by overlapping with the dfs I was just referring to (humgenes and chimpgenes). In the end this set of bedfiles is fairly useless, because really it would be preferable to get rid of duplicates so that I can merely group_by on a given bin afterwards and left_join as necessary. So somewhat deprecated, but I keep it here still:
hbin1KR <- data.frame(chr=data.KR$Hchr, start=as.numeric(gsub("chr.*-", "", data.KR$H1)), end=as.numeric(gsub("chr.*-", "", data.KR$H1))+10000)
hbin2KR <- data.frame(chr=data.KR$Hchr, start=as.numeric(gsub("chr.*-", "", data.KR$H2)), end=as.numeric(gsub("chr.*-", "", data.KR$H2))+10000)
cbin1KR <- data.frame(chr=data.KR$Cchr, start=as.numeric(gsub("chr.*-", "", data.KR$C1)), end=as.numeric(gsub("chr.*-", "", data.KR$C1))+10000)
cbin2KR <- data.frame(chr=data.KR$Cchr, start=as.numeric(gsub("chr.*-", "", data.KR$C2)), end=as.numeric(gsub("chr.*-", "", data.KR$C2))+10000)
hbin1VC <- data.frame(chr=data.VC$Hchr, start=as.numeric(gsub("chr.*-", "", data.VC$H1)), end=as.numeric(gsub("chr.*-", "", data.VC$H1))+10000)
hbin2VC <- data.frame(chr=data.VC$Hchr, start=as.numeric(gsub("chr.*-", "", data.VC$H2)), end=as.numeric(gsub("chr.*-", "", data.VC$H2))+10000)
cbin1VC <- data.frame(chr=data.VC$Cchr, start=as.numeric(gsub("chr.*-", "", data.VC$C1)), end=as.numeric(gsub("chr.*-", "", data.VC$C1))+10000)
cbin2VC <- data.frame(chr=data.VC$Cchr, start=as.numeric(gsub("chr.*-", "", data.VC$C2)), end=as.numeric(gsub("chr.*-", "", data.VC$C2))+10000)
#In most analyses, it will make more sense to have a single bed file for both sets of bins, and remove all duplicates. I create that here:
hbinsKR <- rbind(hbin1KR[!duplicated(hbin1KR),], hbin2KR[!duplicated(hbin2KR),])
hbinsKR <- hbinsKR[!duplicated(hbinsKR),]
cbinsKR <- rbind(cbin1KR[!duplicated(cbin1KR),], cbin2KR[!duplicated(cbin2KR),])
cbinsKR <- cbinsKR[!duplicated(cbinsKR),]
hbinsVC <- rbind(hbin1VC[!duplicated(hbin1VC),], hbin2VC[!duplicated(hbin2VC),])
hbinsVC <- hbinsVC[!duplicated(hbinsVC),]
cbinsVC <- rbind(cbin1VC[!duplicated(cbin1VC),], cbin2VC[!duplicated(cbin2VC),])
cbinsVC <- cbinsVC[!duplicated(cbinsVC),]
#Now, write all of these files out for analysis with bedtools.
options(scipen=999) #Don't want any scientific notation in these BED files, will mess up some bedtools analyses at times
write.table(hbin1KR, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/hbin1KR.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(hbin2KR, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/hbin2KR.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(cbin1KR, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/cbin1KR.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(cbin2KR, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/cbin2KR.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(hbinsKR, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/hbinsKR.bed", quote=FALSE, sep="\t", row.names=FALSE, col.names=FALSE)
write.table(cbinsKR, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/cbinsKR.bed", quote=FALSE, sep="\t", row.names=FALSE, col.names=FALSE)
write.table(hbin1VC, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/hbin1VC.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(hbin2VC, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/hbin2VC.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(cbin1VC, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/cbin1VC.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(cbin2VC, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/cbin2VC.bed", quote = FALSE, sep="\t", row.names = FALSE, col.names=FALSE)
write.table(hbinsVC, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/hbinsVC.bed", quote=FALSE, sep="\t", row.names=FALSE, col.names=FALSE)
write.table(cbinsVC, "data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/unsorted/cbinsVC.bed", quote=FALSE, sep="\t", row.names=FALSE, col.names=FALSE)
options(scipen=0)
#Read in new, simpler bedtools closest files for genes. This is after running two commands, after sorting the files w/ sort -k1,1 -k2,2n in.bed > out.bed:
#bedtools closest -D a -a cgenes.sorted.bed -b cbins.sorted.bed > cgene.hic.overlap
#bedtools closest -D a -a hgenes.sorted.bed -b hbins.sorted.bed > hgene.hic.overlap
hgene.hic.KR <- fread("data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/hgene.hic.KR.overlap", header=FALSE, stringsAsFactors = FALSE, data.table=FALSE)
cgene.hic.KR <- fread("data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/cgene.hic.KR.overlap", header=FALSE, stringsAsFactors = FALSE, data.table=FALSE)
hgene.hic.VC <- fread("data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/hgene.hic.VC.overlap", header=FALSE, stringsAsFactors = FALSE, data.table=FALSE)
cgene.hic.VC <- fread("data/hic_gene_overlap/juicer/juicer_10kb_filt_overlaps/cgene.hic.VC.overlap", header=FALSE, stringsAsFactors = FALSE, data.table=FALSE)
#Visualize the overlap of genes with bins and see how many genes we get back! Left out here b/c not super necessary, but code is collapsed here:
# hum.genelap <- data.frame(overlap=seq(0, 100000, 1000), perc.genes = NA, tot.genes=NA)
# for(row in 1:nrow(hum.genelap)){
# hum.genelap$perc.genes[row] <- sum(abs(hgene.hic$V10)<=hum.genelap$overlap[row])/length(hgene.hic$V10)
# hum.genelap$tot.genes[row] <- sum(abs(hgene.hic$V10)<=hum.genelap$overlap[row])
# }
#
# c.genelap <- data.frame(overlap=seq(0, 100000, 1000), perc.genes=NA, tot.genes=NA)
# for(row in 1:nrow(c.genelap)){
# c.genelap$perc.genes[row] <- sum(abs(cgene.hic$V10)<=c.genelap$overlap[row])/length(cgene.hic$V10)
# c.genelap$tot.genes[row] <- sum(abs(cgene.hic$V10)<=c.genelap$overlap[row])
# }
# c.genelap$type <- "chimp"
# hum.genelap$type <- "human"
#Examine what the potential gains are here if we are more lenient about the overlap/closeness to a TSS...
#ggoverlap <- rbind(hum.genelap, c.genelap)
#ggplot(data=ggoverlap) + geom_line(aes(x=overlap, y=perc.genes*100, color=type)) + ggtitle("Percent of Total Genes Picked Up | Min. Distance from TSS") + xlab("Distance from TSS") + ylab("Percentage of genes in ortho exon trios file (~44k)") + scale_color_discrete(guide=guide_legend(title="Species")) + coord_cartesian(xlim=c(0, 30000)) + scale_x_continuous(breaks=seq(0, 30000, 5000))
#ggplot(data=ggoverlap) + geom_line(aes(x=overlap, y=tot.genes, color=type)) + ggtitle("Total # Genes Picked Up | Min. Distance from TSS") + xlab("Distance from TSS") + ylab("Total # of Genes Picked up On (of ~44k)") + scale_color_discrete(guide=guide_legend(title="Species")) + coord_cartesian(xlim=c(0, 30000)) + scale_x_continuous(breaks=seq(0, 30000, 5000))
#Start with a conservative pass--only take those genes that had an actual overlap with a bin, not ones that were merely close to one. Allowing some leeway to include genes that are within 1kb, 2kb, 3kb etc. of a Hi-C bin adds an average of ~800 genes per 1kb. We can also examine the distribution manually to motivate this decision:
quantile(abs(hgene.hic.KR$V10), probs=seq(0, 1, 0.025)) 0% 2.5% 5% 7.5% 10% 12.5% 15%
0.0 0.0 0.0 0.0 1.0 1.0 111.2
17.5% 20% 22.5% 25% 27.5% 30% 32.5%
2801.8 5837.6 9170.0 12712.0 16555.3 20657.4 25122.3
35% 37.5% 40% 42.5% 45% 47.5% 50%
30415.8 35854.0 41976.0 48686.7 56315.6 64775.6 74086.0
52.5% 55% 57.5% 60% 62.5% 65% 67.5%
84493.0 96003.4 109333.4 124395.4 141705.5 161762.6 185550.4
70% 72.5% 75% 77.5% 80% 82.5% 85%
213137.8 245623.6 284011.0 326249.0 378968.0 446350.3 531628.2
87.5% 90% 92.5% 95% 97.5% 100%
644495.0 813443.4 1066258.7 1411418.6 2117706.0 8009993.0 quantile(abs(cgene.hic.KR$V10), probs=seq(0, 1, 0.025)) 0% 2.5% 5% 7.5% 10% 12.5% 15%
0.0 0.0 0.0 0.0 1.0 1.0 35.8
17.5% 20% 22.5% 25% 27.5% 30% 32.5%
2576.0 5543.2 9001.0 12613.0 16346.1 20659.2 25304.9
35% 37.5% 40% 42.5% 45% 47.5% 50%
30415.0 36090.5 42336.4 49350.3 57203.8 65644.9 74758.0
52.5% 55% 57.5% 60% 62.5% 65% 67.5%
85634.5 97537.4 110882.8 125779.8 143152.5 163577.6 187829.8
70% 72.5% 75% 77.5% 80% 82.5% 85%
216384.2 248610.8 288103.0 330921.3 384602.4 448229.0 536251.6
87.5% 90% 92.5% 95% 97.5% 100%
645908.0 806354.8 1046000.8 1398275.0 2071010.3 7529283.0 quantile(abs(hgene.hic.VC$V10), probs=seq(0, 1, 0.025)) 0% 2.5% 5% 7.5% 10% 12.5% 15%
0.0 0.0 0.0 0.0 0.0 748.5 2939.6
17.5% 20% 22.5% 25% 27.5% 30% 32.5%
5319.8 8011.8 10835.5 13955.0 16964.2 20339.0 24054.6
35% 37.5% 40% 42.5% 45% 47.5% 50%
28368.6 33051.5 38083.0 43953.0 50389.2 57444.3 64971.0
52.5% 55% 57.5% 60% 62.5% 65% 67.5%
73526.5 83118.4 94055.9 106232.2 119220.5 135994.6 153922.6
70% 72.5% 75% 77.5% 80% 82.5% 85%
175048.2 199192.6 228645.0 261707.1 302957.2 351916.0 413198.4
87.5% 90% 92.5% 95% 97.5% 100%
501881.5 641720.2 865347.1 1205121.8 1836870.5 6456300.0 quantile(abs(cgene.hic.VC$V10), probs=seq(0, 1, 0.025)) 0% 2.5% 5% 7.5% 10% 12.5% 15%
0.0 0.0 0.0 0.0 0.0 512.5 2649.0
17.5% 20% 22.5% 25% 27.5% 30% 32.5%
5010.2 7760.8 10585.5 13538.0 16670.4 20081.6 24187.6
35% 37.5% 40% 42.5% 45% 47.5% 50%
28398.8 33210.5 38408.6 44155.4 50593.8 57687.0 65471.0
52.5% 55% 57.5% 60% 62.5% 65% 67.5%
73931.1 83568.4 94835.9 107072.8 120344.5 136233.2 154759.7
70% 72.5% 75% 77.5% 80% 82.5% 85%
176847.0 202064.3 230537.0 264555.9 303717.0 353338.0 415156.6
87.5% 90% 92.5% 95% 97.5% 100%
501151.5 629124.0 840981.2 1183185.6 1767476.4 6500471.0 #So it looks as though we pick up on approximately 10% of 44k genes (~4.4k)
#Note I looked at proportion of overlap with DE and with non-DE genes just for curiosity, and roughly 66% of the DE genes have overlap with a Hi-C bin while roughly 70% of the non-DE genes do. Since this result isn't particularly interesting I have collapsed that analysis here.
#Also are interested in seeing how this differs for DE and non-DE genes.
sum(hgene.hic.KR$V10==0&(hgene.hic.KR$V4 %in% DEgenes)) #263 genes[1] 235sum(hgene.hic.KR$V10==0&(hgene.hic.KR$V4 %in% nonDEgenes)) #1011 genes[1] 1039sum(hgene.hic.KR$V10==0&(!hgene.hic.KR$V4 %in% DEgenes)&(!hgene.hic.KR$V4 %in% nonDEgenes)) #Checking which genes have direct overlap with bins, but were not picked up on in our RNAseq data. 2717[1] 2717sum(hgene.hic.VC$V10==0&(hgene.hic.VC$V4 %in% DEgenes)) #334 genes[1] 299sum(hgene.hic.VC$V10==0&(hgene.hic.VC$V4 %in% nonDEgenes)) #1296 genes[1] 1331sum(hgene.hic.VC$V10==0&(!hgene.hic.VC$V4 %in% DEgenes)&(!hgene.hic.VC$V4 %in% nonDEgenes)) #Checking which genes have direct overlap with bins, but were not picked up on in our RNAseq data. 3441[1] 3441###Linear Modeling Annotation In this next section I simply add information obtained from linear modeling on the Hi-C interaction frequencies to the appropriate genes having overlap with Hi-C bins. Because one Hi-C bin frequently shows up many times in the data, this means I must choose some kind of summary for Hi-C contact frequencies and linear modeling annotations for each gene. I toy with a variety of these summaries here, including choosing the minimum FDR contact, the maximum beta contact, the upstream contact, or summarizing all a bin’s contacts with the weighted Z-combine method or median FDR values.
hgene.hic.overlap.KR <- filter(hgene.hic.KR, V10==0) #Still leaves a solid ~4k genes.
cgene.hic.overlap.KR <- filter(cgene.hic.KR, V10==0) #Still leaves a solid ~4k genes.
hgene.hic.overlap.VC <- filter(hgene.hic.VC, V10==0) #Still leaves a solid ~5k genes.
cgene.hic.overlap.VC <- filter(cgene.hic.VC, V10==0) #Still leaves a solid ~5k genes.
#Add a column to both dfs indicating where along a bin the gene in question is found (from 0-10k):
hgene.hic.overlap.KR$bin_pos <- abs(hgene.hic.overlap.KR$V8-hgene.hic.overlap.KR$V2)
cgene.hic.overlap.KR$bin_pos <- abs(cgene.hic.overlap.KR$V8-cgene.hic.overlap.KR$V2)
hgene.hic.overlap.VC$bin_pos <- abs(hgene.hic.overlap.VC$V8-hgene.hic.overlap.VC$V2)
cgene.hic.overlap.VC$bin_pos <- abs(cgene.hic.overlap.VC$V8-cgene.hic.overlap.VC$V2)
#Rearrange columns and create another column of the bin ID.
hgene.hic.overlap.KR <- hgene.hic.overlap.KR[,c(4, 7:9, 6, 11, 1:2)]
hgene.hic.overlap.KR$HID <- paste(hgene.hic.overlap.KR$V7, hgene.hic.overlap.KR$V8, sep="-")
cgene.hic.overlap.KR <- cgene.hic.overlap.KR[,c(4, 7:9, 6, 11, 1:2)]
cgene.hic.overlap.KR$CID <- paste(cgene.hic.overlap.KR$V7, cgene.hic.overlap.KR$V8, sep="-")
colnames(hgene.hic.overlap.KR) <- c("genes", "HiC_chr", "H1start", "H1end", "Hstrand", "bin_pos", "genechr", "genepos", "HID")
colnames(cgene.hic.overlap.KR) <- c("genes", "HiC_chr", "C1start", "C1end", "Cstrand", "bin_pos", "genechr", "genepos", "CID")
hgene.hic.overlap.VC <- hgene.hic.overlap.VC[,c(4, 7:9, 6, 11, 1:2)]
hgene.hic.overlap.VC$HID <- paste(hgene.hic.overlap.VC$V7, hgene.hic.overlap.VC$V8, sep="-")
cgene.hic.overlap.VC <- cgene.hic.overlap.VC[,c(4, 7:9, 6, 11, 1:2)]
cgene.hic.overlap.VC$CID <- paste(cgene.hic.overlap.VC$V7, cgene.hic.overlap.VC$V8, sep="-")
colnames(hgene.hic.overlap.VC) <- c("genes", "HiC_chr", "H1start", "H1end", "Hstrand", "bin_pos", "genechr", "genepos", "HID")
colnames(cgene.hic.overlap.VC) <- c("genes", "HiC_chr", "C1start", "C1end", "Cstrand", "bin_pos", "genechr", "genepos", "CID")
#Before extracting some data from the data.KR and data.VC dfs, need to reformat their H1, H2, C1, and C2 columns to include chromosome (this was how homer data already was formatted so didn't have to think about it there)
data.KR$H1 <- paste(data.KR$Hchr, data.KR$H1, sep="-")
data.KR$H2 <- paste(data.KR$Hchr, data.KR$H2, sep="-")
data.VC$H1 <- paste(data.VC$Hchr, data.VC$H1, sep="-")
data.VC$H2 <- paste(data.VC$Hchr, data.VC$H2, sep="-")
data.KR$C1 <- paste(data.KR$Cchr, data.KR$C1, sep="-")
data.KR$C2 <- paste(data.KR$Cchr, data.KR$C2, sep="-")
data.VC$C1 <- paste(data.VC$Cchr, data.VC$C1, sep="-")
data.VC$C2 <- paste(data.VC$Cchr, data.VC$C2, sep="-")
hbindf.KR <- select(data.KR, "H1", "H2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "Hdist")
names(hbindf.KR) <- c("HID", "HID2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "distance") #I have confirmed that all the HID2s are higher numbered coordinates than the HID1s, the only instance in which this isn't the case is when the two bins are identical (this should have been filtered out long before now).
hbindf.VC <- select(data.VC, "H1", "H2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "Hdist")
names(hbindf.VC) <- c("HID", "HID2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "distance")
#This step is unnecessary in this paradigm as there are no hits like this, but keep it in here for the premise's sake:
hbindf.KR <- hbindf.KR[(which(hbindf.KR$distance!=0)),] #Removes pairs where the same bin represents both mates. These instances occur exclusively when liftOver of the genomic coordinates from one species to another, and the subsequent rounding to the nearest 10kb, results in a contact between adjacent bins in one species being mapped as a contact between the same bin in the other species. Because there are less than 50 instances of this total in the dataset I simply remove it here without further worry.
hbindf.VC <- hbindf.VC[(which(hbindf.VC$distance!=0)),]
#For explanations about the motivations and intents behind code in the rest of this chunk, please see the same file for the HOMER analysis.
group_by(hbindf.KR, HID) %>% summarise(DS_bin=HID2[which.min(distance)], DS_FDR=sp_BH_pval[which.min(distance)], DS_dist=distance[which.min(distance)]) -> hbin1.downstream.KR
group_by(hbindf.KR, HID2) %>% summarise(US_bin=HID[which.min(distance)], US_FDR=sp_BH_pval[which.min(distance)], US_dist=distance[which.min(distance)]) -> hbin2.upstream.KR
colnames(hbin2.upstream.KR) <- c("HID", "US_bin", "US_FDR", "US_dist")
Hstreams.KR <- full_join(hbin1.downstream.KR, hbin2.upstream.KR, by="HID")
hbindf.KR.flip <- hbindf.KR[,c(2, 1, 3:7)]
colnames(hbindf.KR.flip)[1:2] <- c("HID", "HID2")
hbindf.KR_x2 <- rbind(hbindf.KR[,1:7], hbindf.KR.flip)
as.data.frame(group_by(hbindf.KR_x2, HID) %>% summarise(min_FDR_bin=HID2[which.min(sp_BH_pval)], min_FDR=min(sp_BH_pval), min_FDR_pval=sp_pval[which.min(sp_BH_pval)], min_FDR_B=sp_beta[which.min(sp_BH_pval)], median_FDR=median(sp_BH_pval), weighted_Z.ALLvar=pnorm((sum((1/ALLvar)*((qnorm(1-sp_pval))))/sqrt(sum((1/ALLvar)^2))), lower.tail=FALSE), weighted_Z.s2post=pnorm(sum((1/(SE^2))*qnorm(1-sp_pval))/sqrt(sum(1/SE^2)), lower.tail=FALSE), fisher=-2*sum(log(sp_pval)), numcontacts=n(), max_B_bin=HID2[which.max(abs(sp_beta))], max_B_FDR=sp_BH_pval[which.max(abs(sp_beta))], max_B=sp_beta[which.max(abs(sp_beta))])) -> hbin.KR.info
full_join(hbin.KR.info, Hstreams.KR, by="HID") -> hbin.KR.full.info
left_join(hgene.hic.overlap.KR, hbin.KR.full.info, by="HID") -> humgenes.KR.full
colnames(humgenes.KR.full)[1:5] <- c("genes", "Hchr", "Hstart", "Hend", "Hstrand") #Fix column names for what was just created
group_by(hbindf.VC, HID) %>% summarise(DS_bin=HID2[which.min(distance)], DS_FDR=sp_BH_pval[which.min(distance)], DS_dist=distance[which.min(distance)]) -> hbin1.downstream.VC
group_by(hbindf.VC, HID2) %>% summarise(US_bin=HID[which.min(distance)], US_FDR=sp_BH_pval[which.min(distance)], US_dist=distance[which.min(distance)]) -> hbin2.upstream.VC
colnames(hbin2.upstream.VC) <- c("HID", "US_bin", "US_FDR", "US_dist")
Hstreams.VC <- full_join(hbin1.downstream.VC, hbin2.upstream.VC, by="HID")
hbindf.VC.flip <- hbindf.VC[,c(2, 1, 3:7)]
colnames(hbindf.VC.flip)[1:2] <- c("HID", "HID2")
hbindf.VC_x2 <- rbind(hbindf.VC[,1:7], hbindf.VC.flip)
as.data.frame(group_by(hbindf.VC_x2, HID) %>% summarise(min_FDR_bin=HID2[which.min(sp_BH_pval)], min_FDR=min(sp_BH_pval), min_FDR_pval=sp_pval[which.min(sp_BH_pval)], min_FDR_B=sp_beta[which.min(sp_BH_pval)], median_FDR=median(sp_BH_pval), weighted_Z.ALLvar=pnorm((sum((1/ALLvar)*((qnorm(1-sp_pval))))/sqrt(sum((1/ALLvar)^2))), lower.tail=FALSE), weighted_Z.s2post=pnorm(sum((1/(SE^2))*qnorm(1-sp_pval))/sqrt(sum(1/SE^2)), lower.tail=FALSE), fisher=-2*sum(log(sp_pval)), numcontacts=n(), max_B_bin=HID2[which.max(abs(sp_beta))], max_B_FDR=sp_BH_pval[which.max(abs(sp_beta))], max_B=sp_beta[which.max(abs(sp_beta))])) -> hbin.VC.info
full_join(hbin.VC.info, Hstreams.VC, by="HID") -> hbin.VC.full.info
left_join(hgene.hic.overlap.VC, hbin.VC.full.info, by="HID") -> humgenes.VC.full
colnames(humgenes.VC.full)[1:5] <- c("genes", "Hchr", "Hstart", "Hend", "Hstrand") #Fix column names for what was just created
###Do chimps in both as well, to maximize overlaps:
###KR
cbin.KR <- select(data.KR, "C1", "C2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "Cdist")
names(cbin.KR) <- c("CID", "CID2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "distance")
cbin.KR <- cbin.KR[(which(cbin.KR$dist!=0)),]
group_by(cbin.KR, CID) %>% summarise(DS_bin=CID2[which.min(distance)], DS_FDR=sp_BH_pval[which.min(distance)], DS_dist=distance[which.min(distance)]) -> cbin1.downstream.KR
group_by(cbin.KR, CID2) %>% summarise(US_bin=CID[which.min(distance)], US_FDR=sp_BH_pval[which.min(distance)], US_dist=distance[which.min(distance)]) -> cbin2.upstream.KR
colnames(cbin2.upstream.KR) <- c("CID", "US_bin", "US_FDR", "US_dist")
Cstreams.KR <- full_join(cbin1.downstream.KR, cbin2.upstream.KR, by="CID")
cbin.KR.flip <- cbin.KR[,c(2, 1, 3:7)]
colnames(cbin.KR.flip)[1:2] <- c("CID", "CID2")
cbin.KR_x2 <- rbind(cbin.KR[,1:7], cbin.KR.flip)
group_by(cbin.KR_x2, CID) %>% summarise(min_FDR_bin=CID2[which.min(sp_BH_pval)], min_FDR=min(sp_BH_pval), min_FDR_B=sp_beta[which.min(sp_BH_pval)], median_FDR=median(sp_BH_pval), weighted_Z.ALLvar=pnorm((sum((1/ALLvar)*((qnorm(1-sp_pval))))/sqrt(sum((1/ALLvar)^2))), lower.tail=FALSE), weighted_Z.s2post=pnorm(sum((1/(SE^2))*qnorm(1-sp_pval))/sqrt(sum(1/SE^2)), lower.tail=FALSE), fisher=-2*sum(log(sp_pval)), numcontacts=n(), max_B_bin=CID2[which.max(abs(sp_beta))], max_B_FDR=sp_BH_pval[which.max(abs(sp_beta))], max_B=sp_beta[which.max(abs(sp_beta))]) -> cbin.info.KR
full_join(cbin.info.KR, Cstreams.KR, by="CID") -> cbin.full.info.KR
left_join(cgene.hic.overlap.KR, cbin.full.info.KR, by="CID") -> chimpgenes.hic.KR
colnames(chimpgenes.hic.KR)[1:5] <- c("genes", "Hchr", "Hstart", "Hend", "Hstrand") #Fix column names for what was just created
###VC
cbin.VC <- select(data.VC, "C1", "C2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "Cdist")
names(cbin.VC) <- c("CID", "CID2", "ALLvar", "SE", "sp_beta", "sp_pval", "sp_BH_pval", "distance")
cbin.VC <- cbin.VC[(which(cbin.VC$dist!=0)),]
group_by(cbin.VC, CID) %>% summarise(DS_bin=CID2[which.min(distance)], DS_FDR=sp_BH_pval[which.min(distance)], DS_dist=distance[which.min(distance)]) -> cbin1.downstream.VC
group_by(cbin.VC, CID2) %>% summarise(US_bin=CID[which.min(distance)], US_FDR=sp_BH_pval[which.min(distance)], US_dist=distance[which.min(distance)]) -> cbin2.upstream.VC
colnames(cbin2.upstream.VC) <- c("CID", "US_bin", "US_FDR", "US_dist")
Cstreams.VC <- full_join(cbin1.downstream.VC, cbin2.upstream.VC, by="CID")
cbin.VC.flip <- cbin.VC[,c(2, 1, 3:7)]
colnames(cbin.VC.flip)[1:2] <- c("CID", "CID2")
cbin.VC_x2 <- rbind(cbin.VC[,1:7], cbin.VC.flip)
group_by(cbin.VC_x2, CID) %>% summarise(min_FDR_bin=CID2[which.min(sp_BH_pval)], min_FDR=min(sp_BH_pval), min_FDR_B=sp_beta[which.min(sp_BH_pval)], median_FDR=median(sp_BH_pval), weighted_Z.ALLvar=pnorm((sum((1/ALLvar)*((qnorm(1-sp_pval))))/sqrt(sum((1/ALLvar)^2))), lower.tail=FALSE), weighted_Z.s2post=pnorm(sum((1/(SE^2))*qnorm(1-sp_pval))/sqrt(sum(1/SE^2)), lower.tail=FALSE), fisher=-2*sum(log(sp_pval)), numcontacts=n(), max_B_bin=CID2[which.max(abs(sp_beta))], max_B_FDR=sp_BH_pval[which.max(abs(sp_beta))], max_B=sp_beta[which.max(abs(sp_beta))]) -> cbin.info.VC
full_join(cbin.info.VC, Cstreams.VC, by="CID") -> cbin.full.info.VC
left_join(cgene.hic.overlap.VC, cbin.full.info.VC, by="CID") -> chimpgenes.hic.VC
colnames(chimpgenes.hic.VC)[1:5] <- c("genes", "Hchr", "Hstart", "Hend", "Hstrand") #Fix column names for what was just created
#Now, combine chimpgenes.hic.full and humgenes.hic.full before a final left_join on detable:
full_join(humgenes.VC.full, chimpgenes.hic.VC, by="genes", suffix=c(".H", ".C")) -> genes.hic.VC
full_join(humgenes.KR.full, chimpgenes.hic.KR, by="genes", suffix=c(".H", ".C")) -> genes.hic.KR
###Final join of human and chimp values for both:
left_join(detable, genes.hic.VC, by="genes") -> gene.hic.VC
left_join(detable, genes.hic.KR, by="genes") -> gene.hic.KR
#Clean this dataframe up, removing rows where there is absolutely no Hi-C information for the gene.
filt.VC <- rowSums(is.na(gene.hic.VC)) #51 NA values are found when there is absolutely no Hi-C information.
filt.KR <- rowSums(is.na(gene.hic.KR)) #same.
filt.VC <- which(filt.VC==51)
filt.KR <- which(filt.KR==51)
gene.hic.VC <- gene.hic.VC[-filt.VC,]
gene.hic.KR <- gene.hic.KR[-filt.KR,]
saveRDS(gene.hic.KR, "output/gene.hic.filt.KR.RDS")
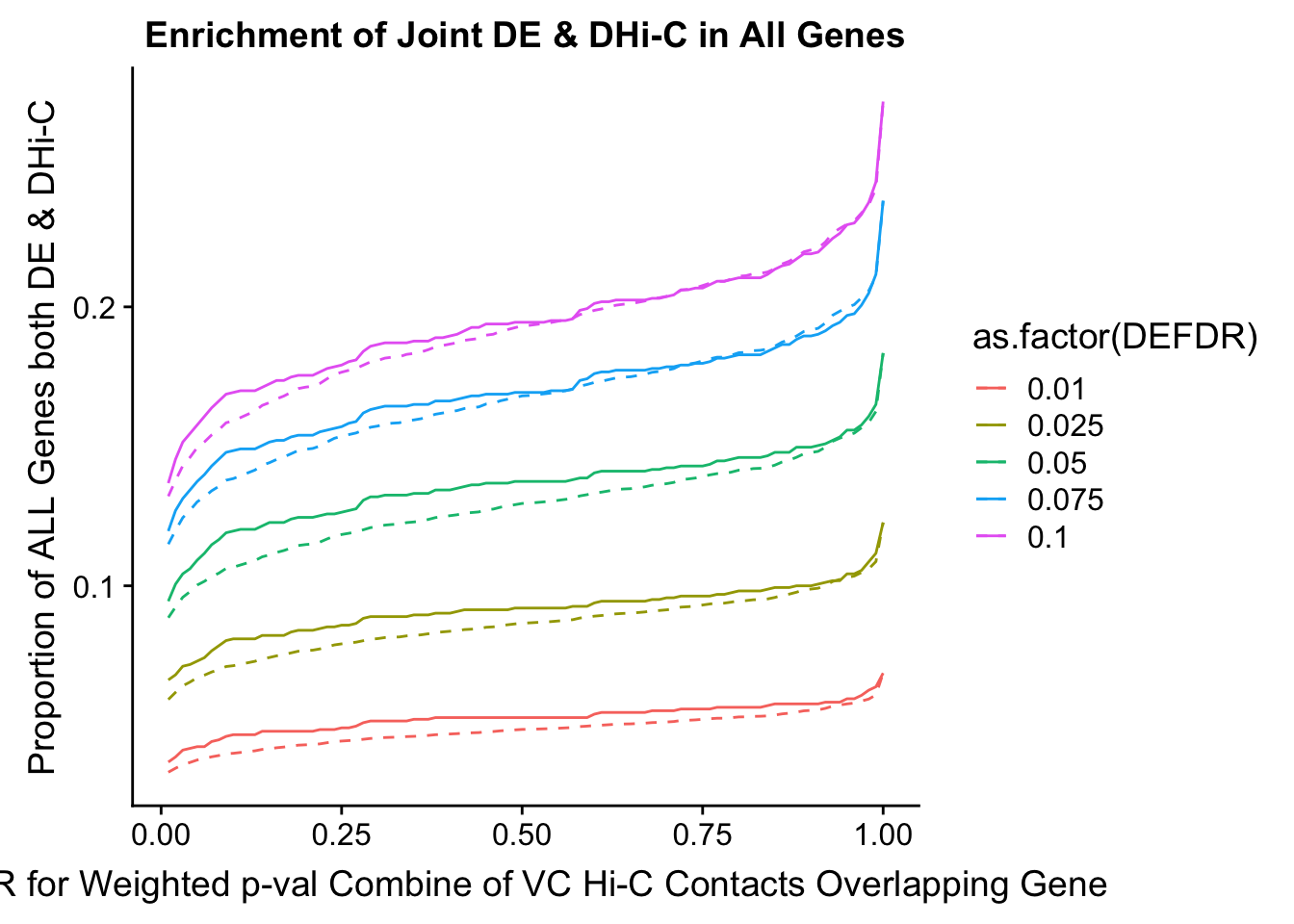
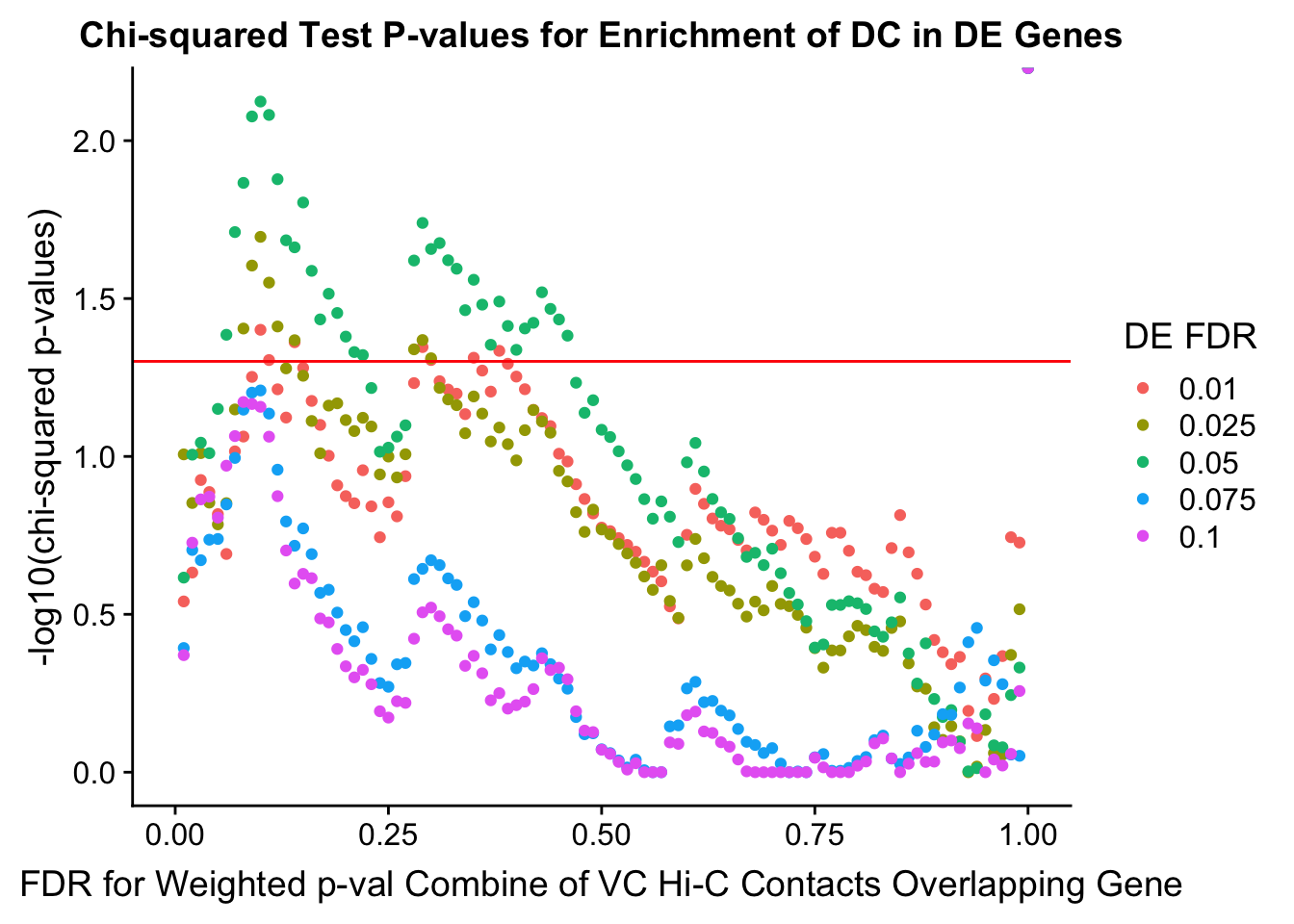
saveRDS(gene.hic.VC, "output/gene.hic.filt.VC.RDS")###Differential Expression-Differential Hi-C Enrichment Analyses Now I look for enrichment of DHi-C in DE genes using a variety of different metrics to call DHi-C. I now look to see if genes that are differentially expressed are also differential in Hi-C contacts (DHi-C). That is to say, are differentially expressed genes enriched in their overlapping bins for Hi-C contacts that are also differential between the species? To do this I utilize p-values from my prior linear modeling as well as previous RNA-seq analysis. I construct a function to calculate proportions of DE and DHi-C genes, as well as a function to plot this out in a variety of different ways.
####Enrichment analyses!
#A function for calculating proportion of DE genes that are DHi-C under a variety of different paradigms. Accounts for when no genes are DHi-C and when all genes are DHi-C. Returns the proportion of DE genes that are also DHi-C, as well as the expected proportion based on conditional probability alone.
prop.calculator <- function(de.vec, hic.vec, i, k){
my.result <- data.frame(prop=NA, exp.prop=NA, chisq.p=NA, Dneither=NA, DE=NA, DHiC=NA, Dboth=NA)
bad.indices <- which(is.na(hic.vec)) #First obtain indices where Hi-C info is missing, if there are any, then remove from both vectors.
if(length(bad.indices>0)){
de.vec <- de.vec[-bad.indices]
hic.vec <- hic.vec[-bad.indices]}
de.vec <- ifelse(de.vec<=i, 1, 0)
hic.vec <- ifelse(hic.vec<=k, 1, 0)
if(sum(hic.vec, na.rm=TRUE)==0){#The case where no genes show up as DHi-C.
my.result[1,] <- c(0, 0, 0, sum(de.vec==0, na.rm=TRUE), sum(de.vec==1, na.rm=TRUE), 0, 0) #Since no genes are DHi-C, the proportion is 0 and our expectation is 0, set p-val=0 since it's irrelevant.
}
else if(sum(hic.vec)==length(hic.vec)){ #The case where every gene shows up as DHi-C
my.result[1,] <- c(1, 1, 0, 0, 0, sum(hic.vec==1&de.vec==0, na.rm = TRUE), sum(de.vec==1&hic.vec==1, na.rm=TRUE)) #If every gene is DHi-C, the observed proportion of DE genes DHi-C is 1, and the expected proportion of DE genes also DHi-C would also be 1 (all DE genes are DHi-C, since all genes are). Again set p-val to 0 since irrelevant comparison.
}
else{#The typical case, where we get an actual table
mytable <- table(as.data.frame(cbind(de.vec, hic.vec)))
my.result[1,1] <- mytable[2,2]/sum(mytable[2,]) #The observed proportion of DE genes that are also DHi-C. # that are both/total # DE
my.result[1,2] <- (((sum(mytable[2,])/sum(mytable))*((sum(mytable[,2])/sum(mytable))))*sum(mytable))/sum(mytable[2,]) #The expected proportion: (p(DE) * p(DHiC)) * total # genes / # DE genes
my.result[1,3] <- chisq.test(mytable)$p.value
my.result[1,4] <- mytable[1,1]
my.result[1,5] <- mytable[2,1]
my.result[1,6] <- mytable[1,2]
my.result[1,7] <- mytable[2,2]
}
return(my.result)
}
#This is a function that computes observed and expected proportions of DE and DHiC enrichments, and spits out a variety of different visualizations for them. As input it takes a dataframe, the names of its DHiC and DE p-value columns, and a name to represent the type of Hi-C contact summary for the gene that ends up on the x-axis of all the plots.
enrichment.plotter <- function(df, HiC_col, DE_col, xlab, xmax=0.3, i=c(0.01, 0.025, 0.05, 0.075, 0.1), k=seq(0.01, 1, 0.01)){
enrich.table <- data.frame(DEFDR = c(rep(i[1], 100), rep(i[2], 100), rep(i[3], 100), rep(i[4], 100), rep(i[5], 100)), DHICFDR=rep(k, 5), prop.obs=NA, prop.exp=NA, chisq.p=NA, Dneither=NA, DE=NA, DHiC=NA, Dboth=NA)
for(de.FDR in i){
for(hic.FDR in k){
enrich.table[which(enrich.table$DEFDR==de.FDR&enrich.table$DHICFDR==hic.FDR), 3:9] <- prop.calculator(df[,DE_col], df[,HiC_col], de.FDR, hic.FDR)
}
}
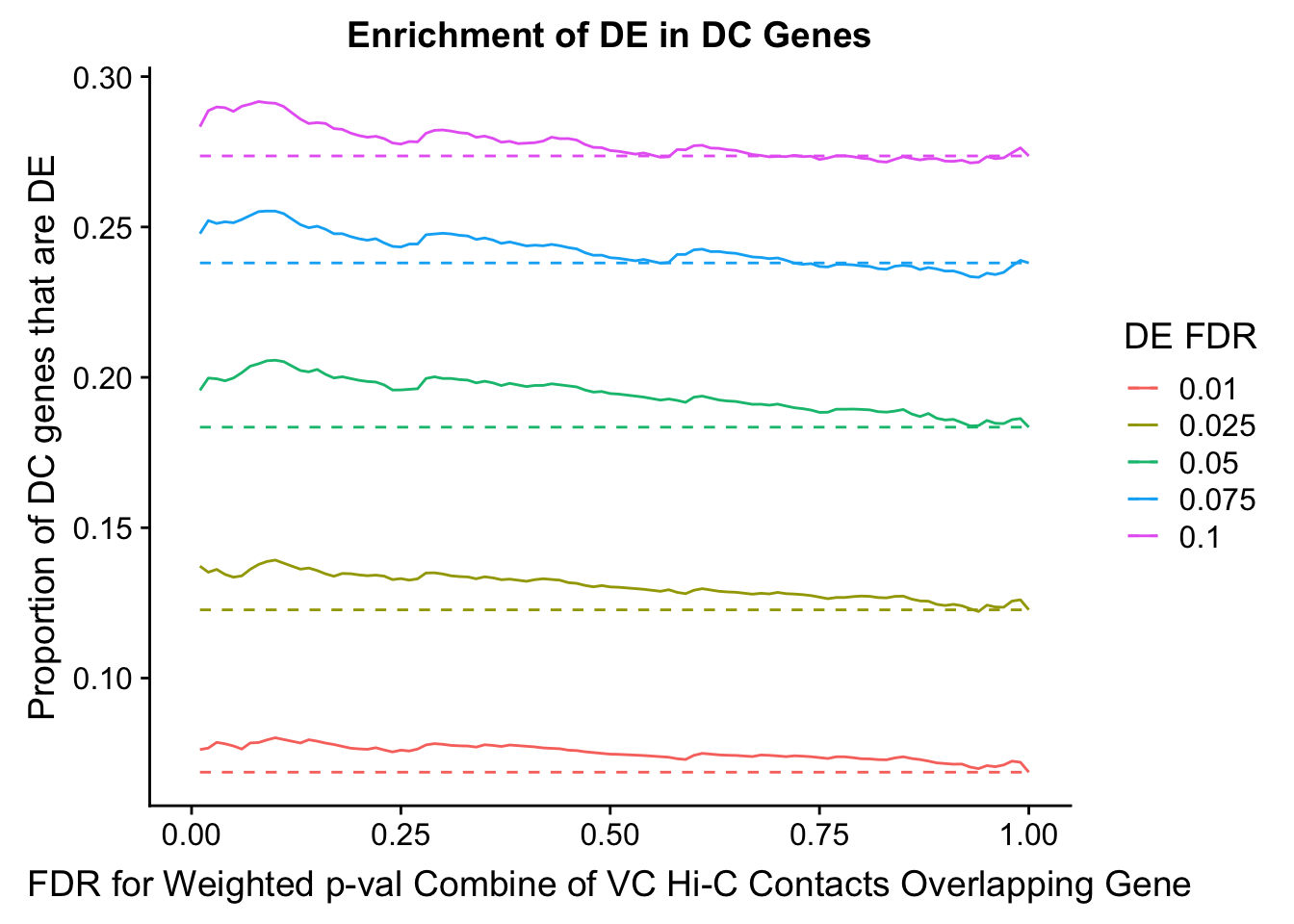
des.enriched <- ggplot(data=enrich.table, aes(x=DHICFDR, y=prop.obs, group=as.factor(DEFDR), color=as.factor(DEFDR))) +geom_line()+ geom_line(aes(y=prop.exp), linetype="dashed", size=0.5) + ggtitle("Enrichment of DC in DE Genes") + xlab(xlab) + ylab("Proportion of DE genes that are DC") + guides(color=guide_legend(title="FDR for DE Genes"))
dhics.enriched <- ggplot(data=enrich.table, aes(x=DHICFDR, y=Dboth/(Dboth+DHiC), group=as.factor(DEFDR), color=as.factor(DEFDR))) + geom_line() + geom_line(aes(y=(((((DE+Dboth)/(Dneither+DE+DHiC+Dboth))*((DHiC+Dboth)/(Dneither+DE+DHiC+Dboth)))*(Dneither+DE+DHiC+Dboth))/(DHiC+Dboth))), linetype="dashed") + ylab("Proportion of DC genes that are DE") +xlab(xlab) + ggtitle("Enrichment of DE in DC Genes") + coord_cartesian(xlim=c(0, xmax)) + guides(color=guide_legend(title="DE FDR"))
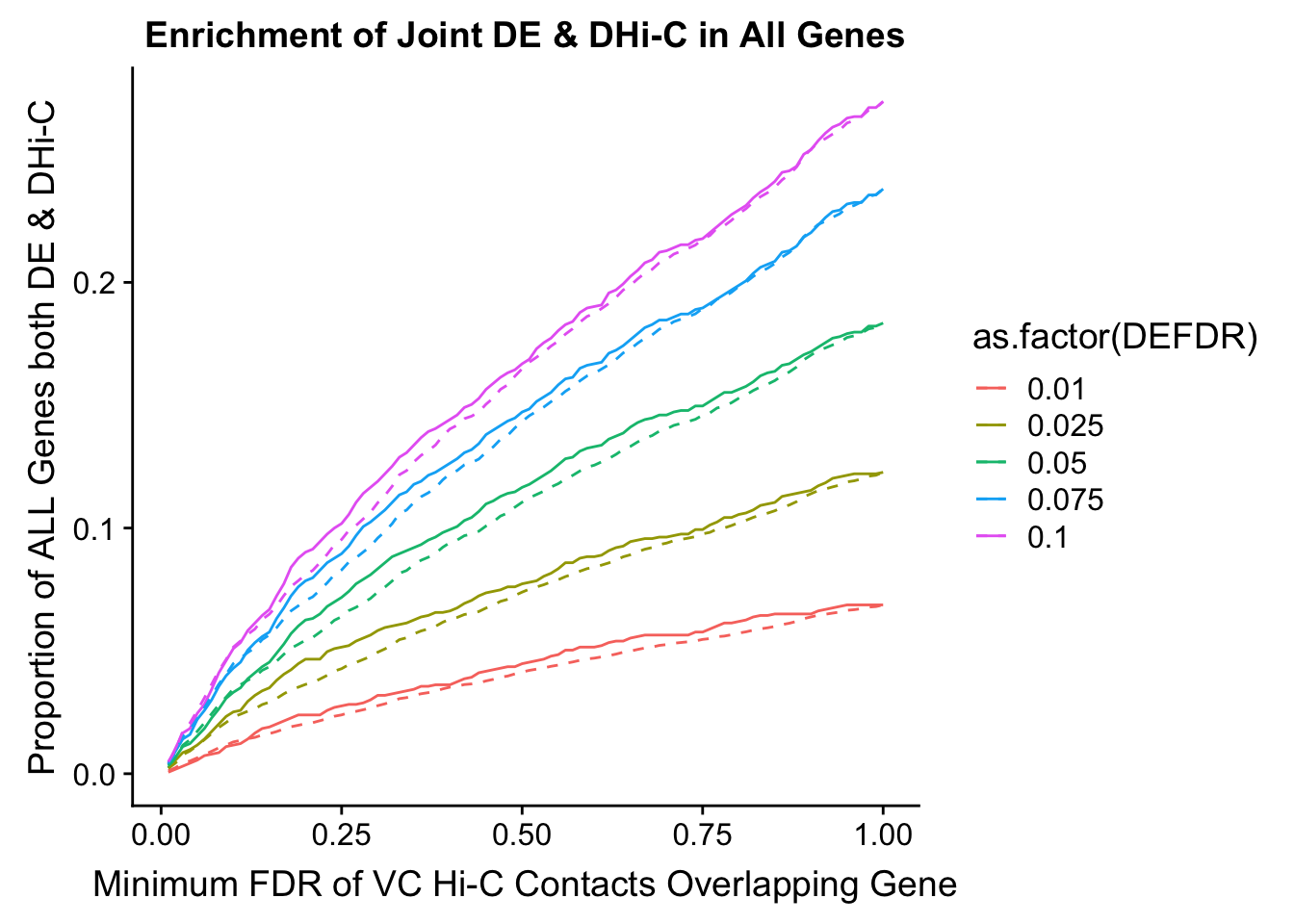
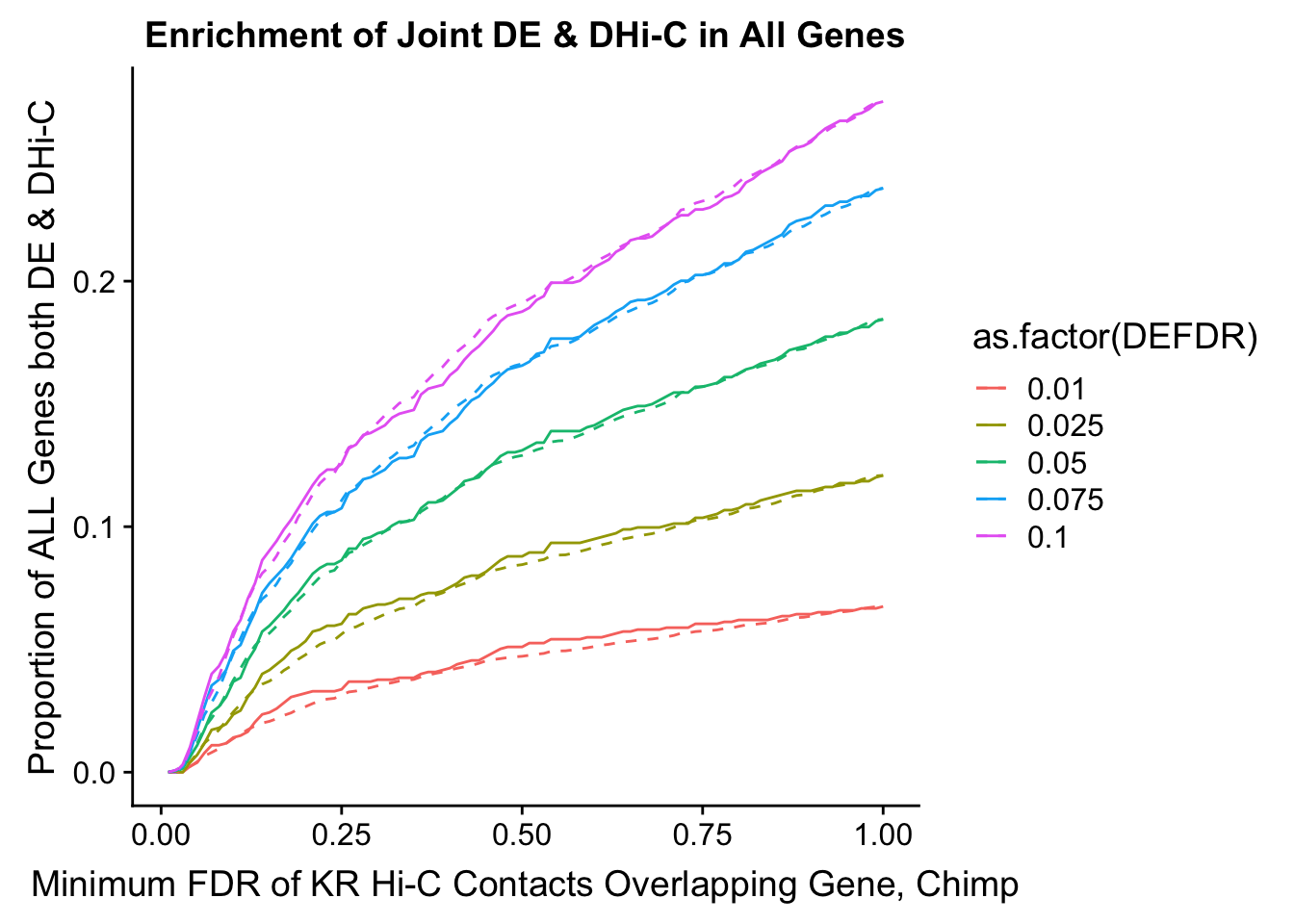
joint.enriched <- ggplot(data=enrich.table, aes(x=DHICFDR, y=Dboth/(Dneither+DE+DHiC+Dboth), group=as.factor(DEFDR), color=as.factor(DEFDR))) + geom_line() + ylab("Proportion of ALL Genes both DE & DHi-C") + xlab(xlab) + geom_line(aes(y=((DE+Dboth)/(Dneither+DE+DHiC+Dboth))*((DHiC+Dboth)/(Dneither+DE+DHiC+Dboth))), linetype="dashed") + ggtitle("Enrichment of Joint DE & DHi-C in All Genes")
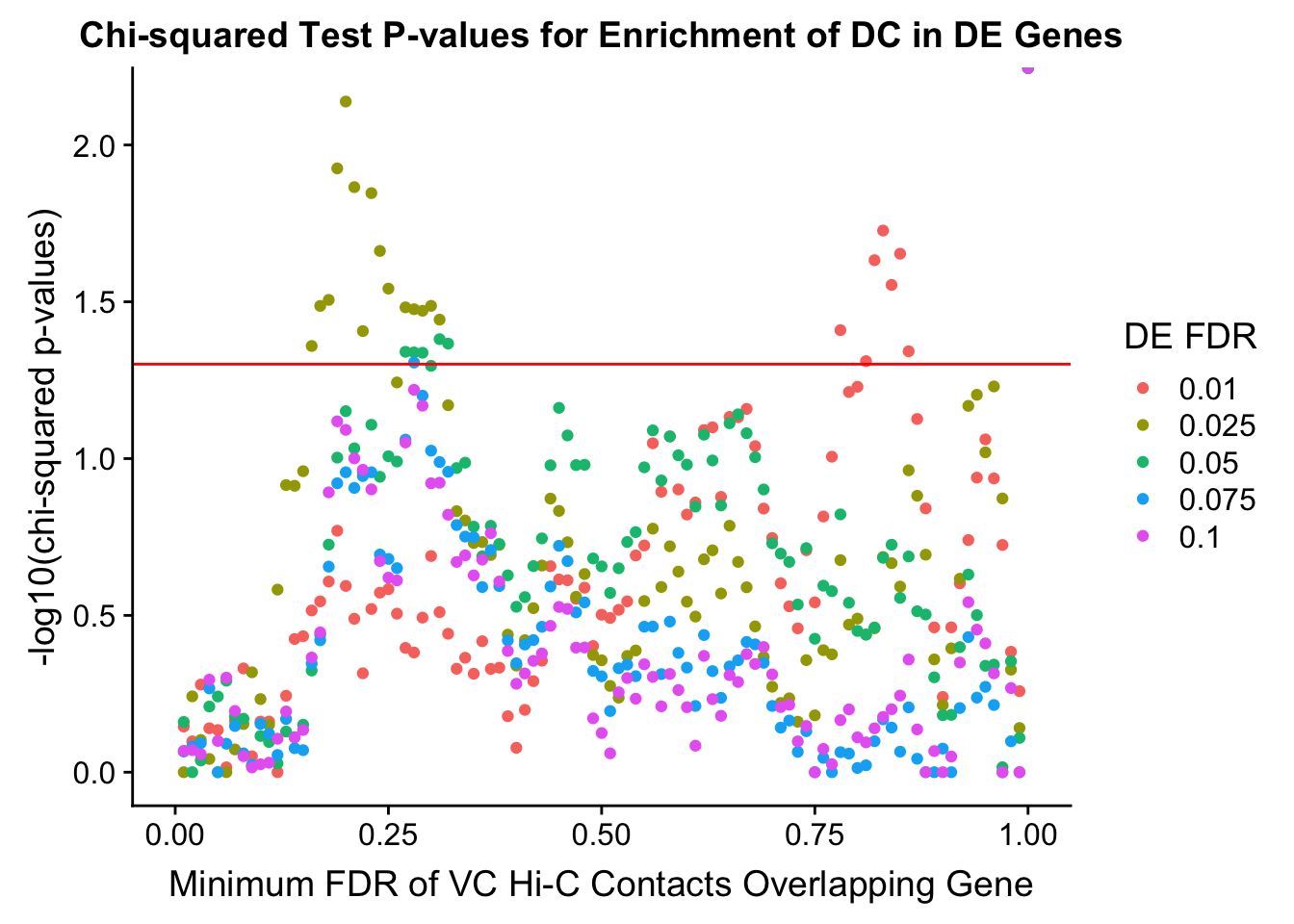
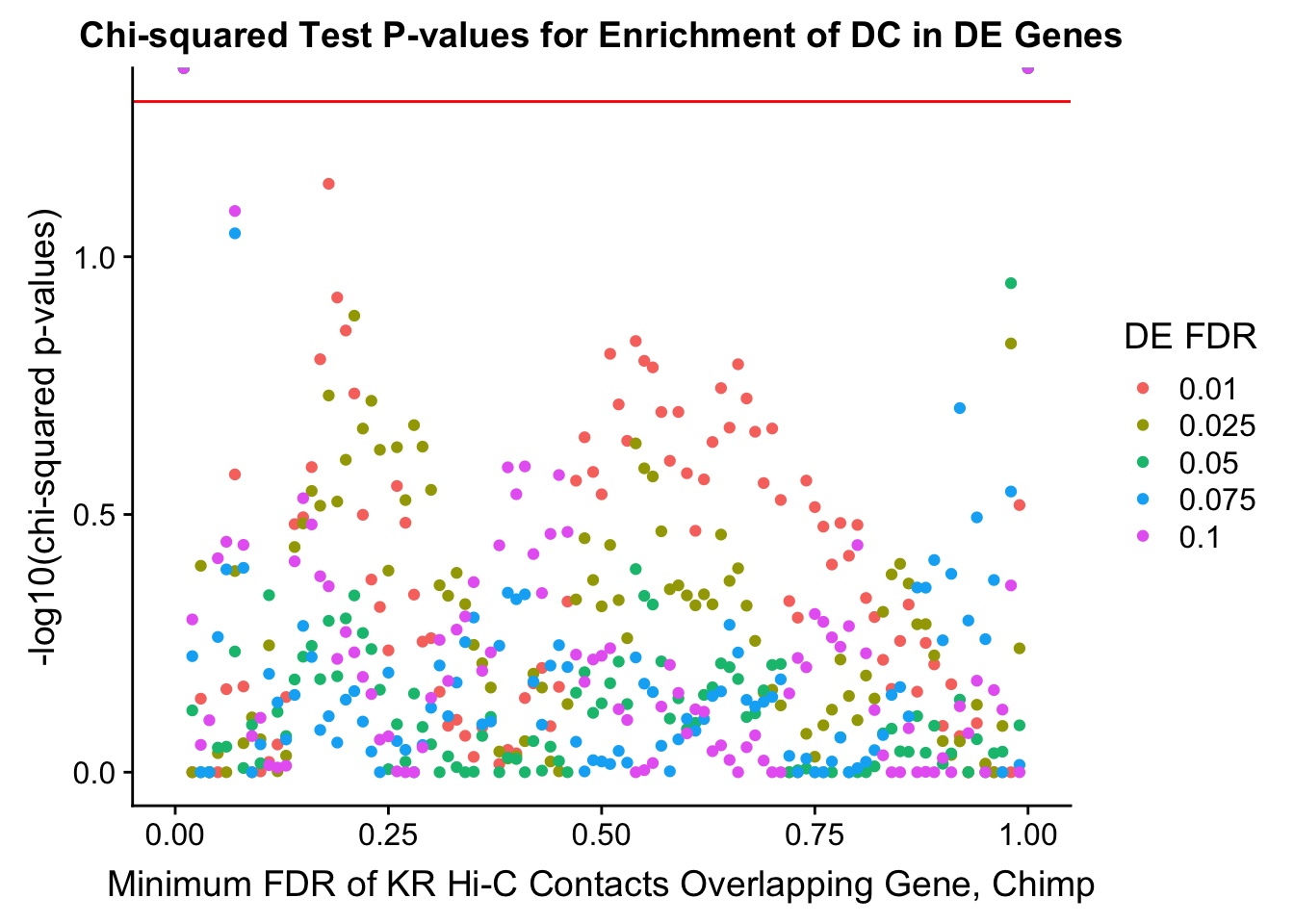
chisq.p <- ggplot(data=enrich.table, aes(x=DHICFDR, y=-log10(chisq.p), group=as.factor(DEFDR), color=as.factor(DEFDR))) + geom_point() + geom_hline(yintercept=-log10(0.05), color="red") + ggtitle("Chi-squared Test P-values for Enrichment of DC in DE Genes") + xlab(xlab) + ylab("-log10(chi-squared p-values)") + coord_cartesian(xlim=c(0, xmax)) + guides(color=guide_legend(title="DE FDR"))
print(des.enriched)
print(dhics.enriched)
print(joint.enriched)
print(chisq.p)
print(enrich.table[which(enrich.table$DEFDR==0.025),]) #Added to figure out comparison for the paper.
}
#Visualization of enrichment of DE/DC in one another. For most of these, using the gene.hic.filt df is sufficient as their Hi-C FDR numbers are the same. For the upstream genes it's a little more complicated because gene.hic.filt doesn't incorporate strand information on the genes, so use the specific US dfs for that, with the USFDR column.
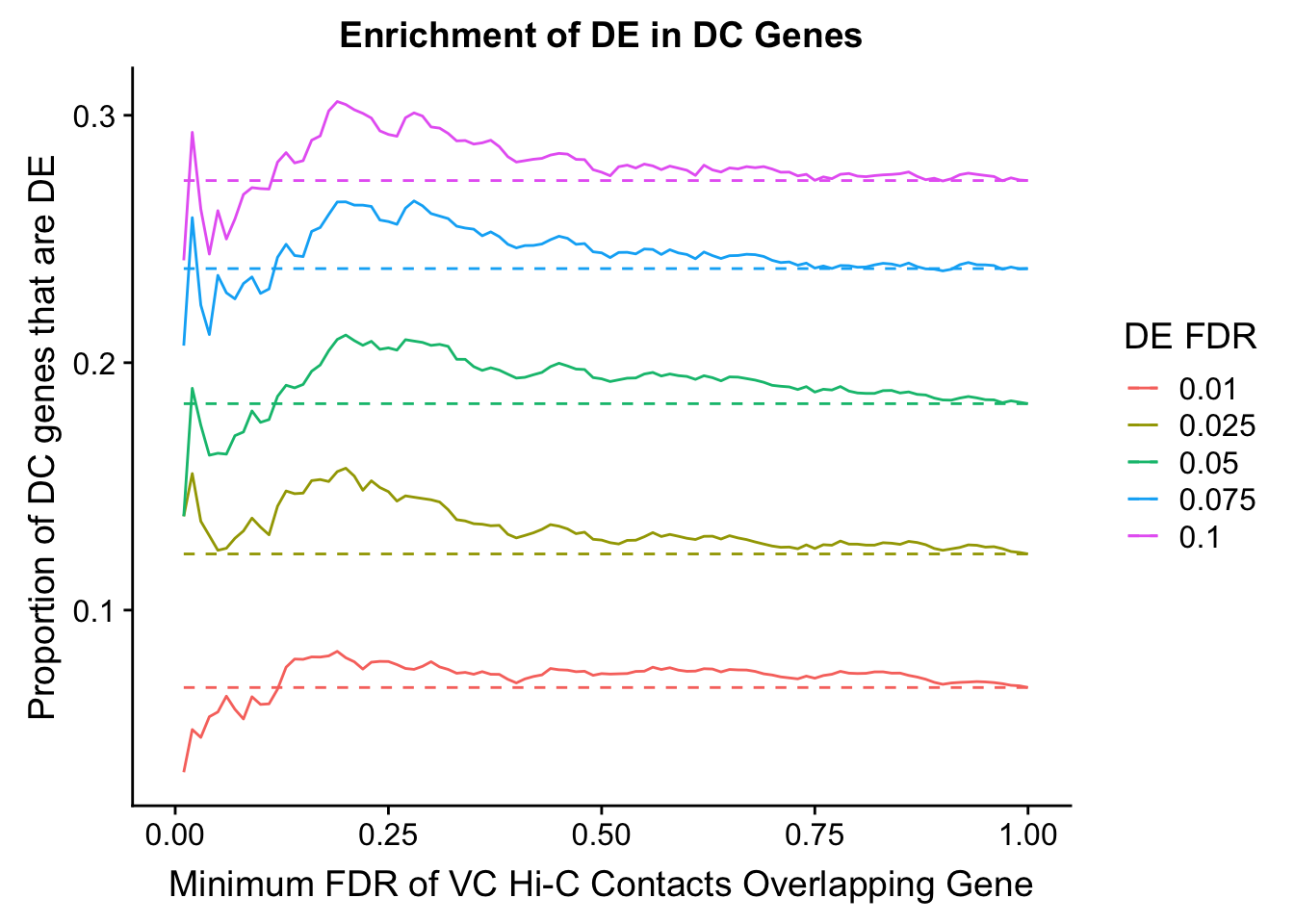
enrichment.plotter(gene.hic.VC, "min_FDR.H", "adj.P.Val", "Minimum FDR of VC Hi-C Contacts Overlapping Gene", xmax=1)Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect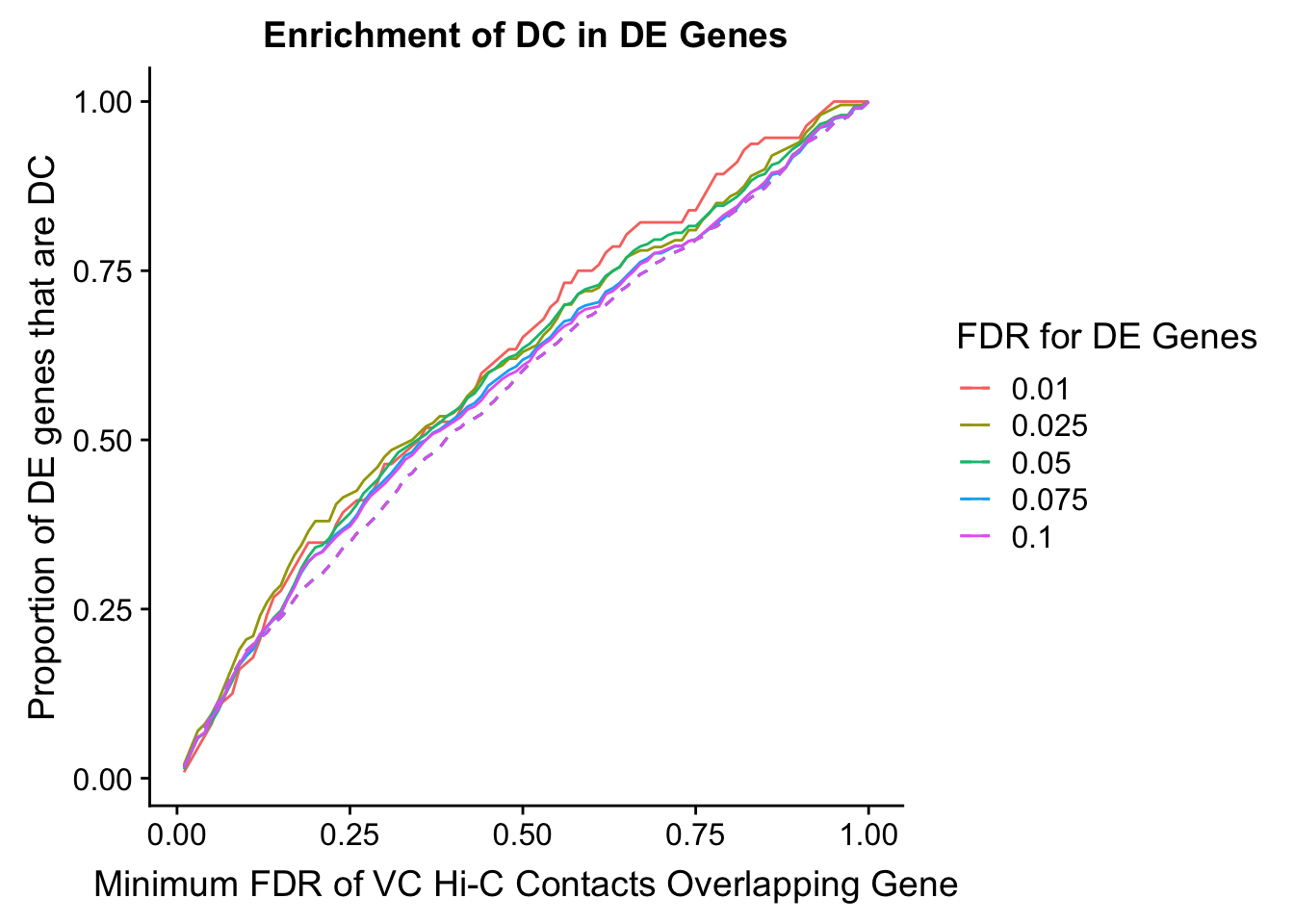
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
DEFDR DHICFDR prop.obs prop.exp chisq.p Dneither DE DHiC Dboth
101 0.025 0.01 0.020 0.01779141 1.000000000 1405 196 25 4
102 0.025 0.02 0.045 0.03558282 0.572897327 1381 191 49 9
103 0.025 0.03 0.070 0.06319018 0.789119683 1341 186 89 14
104 0.025 0.04 0.080 0.07546012 0.907171783 1323 184 107 16
105 0.025 0.05 0.095 0.09386503 1.000000000 1296 181 134 19
106 0.025 0.06 0.115 0.11288344 1.000000000 1269 177 161 23
107 0.025 0.07 0.140 0.13312883 0.845957833 1241 172 189 28
108 0.025 0.08 0.165 0.15337423 0.702184068 1213 167 217 33
109 0.025 0.09 0.190 0.16993865 0.480195424 1191 162 239 38
110 0.025 0.10 0.205 0.18834356 0.584598428 1164 159 266 41
111 0.025 0.11 0.210 0.19754601 0.705817097 1150 158 280 42
112 0.025 0.12 0.240 0.20736196 0.261685727 1140 152 290 48
113 0.025 0.13 0.260 0.21533742 0.121454368 1131 148 299 52
114 0.025 0.14 0.275 0.22944785 0.122119104 1111 145 319 55
115 0.025 0.15 0.285 0.23742331 0.109705391 1100 143 330 57
116 0.025 0.16 0.310 0.24969325 0.043748558 1085 138 345 62
117 0.025 0.17 0.330 0.26503067 0.032589830 1064 134 366 66
118 0.025 0.18 0.345 0.27852761 0.031184315 1045 131 385 69
119 0.025 0.19 0.365 0.28711656 0.011875824 1035 127 395 73
120 0.025 0.20 0.380 0.29631902 0.007268675 1023 124 407 76
121 0.025 0.21 0.380 0.30245399 0.013628675 1013 124 417 76
122 0.025 0.22 0.380 0.31411043 0.039206989 994 124 436 76
123 0.025 0.23 0.405 0.32638037 0.014240464 979 119 451 81
124 0.025 0.24 0.415 0.34049080 0.021768586 958 117 472 83
125 0.025 0.25 0.420 0.34846626 0.028704091 946 116 484 84
126 0.025 0.26 0.425 0.36196319 0.057174173 925 115 505 85
127 0.025 0.27 0.440 0.36932515 0.032938065 916 112 514 88
128 0.025 0.28 0.450 0.37914110 0.033391020 902 110 528 90
129 0.025 0.29 0.460 0.38895706 0.033767432 888 108 542 92
130 0.025 0.30 0.475 0.40306748 0.032578922 868 105 562 95
131 0.025 0.31 0.485 0.41411043 0.036050770 852 103 578 97
132 0.025 0.32 0.490 0.42760736 0.067569499 831 102 599 98
133 0.025 0.33 0.495 0.44478528 0.147133471 804 101 626 99
134 0.025 0.34 0.500 0.45092025 0.157533070 795 100 635 100
135 0.025 0.35 0.510 0.46380368 0.185838639 776 98 654 102
136 0.025 0.36 0.520 0.47361963 0.184533517 762 96 668 104
137 0.025 0.37 0.525 0.48036810 0.202926038 752 95 678 105
138 0.025 0.38 0.535 0.48895706 0.188439927 740 93 690 107
139 0.025 0.39 0.535 0.50245399 0.364234990 718 93 712 107
140 0.025 0.40 0.540 0.51288344 0.457115337 702 92 728 108
141 0.025 0.41 0.550 0.51840491 0.379295853 695 90 735 110
142 0.025 0.42 0.565 0.52822086 0.299830652 682 87 748 113
143 0.025 0.43 0.575 0.53190184 0.219272327 678 85 752 115
144 0.025 0.44 0.590 0.53803681 0.134131986 671 82 759 118
145 0.025 0.45 0.600 0.54969325 0.146828106 654 80 776 120
146 0.025 0.46 0.605 0.55889571 0.184849313 640 79 790 121
147 0.025 0.47 0.610 0.57177914 0.275726286 620 78 810 122
148 0.025 0.48 0.620 0.57852761 0.233395796 611 76 819 124
149 0.025 0.49 0.620 0.59141104 0.422942689 590 76 840 124
150 0.025 0.50 0.630 0.60245399 0.439686449 574 74 856 126
151 0.025 0.51 0.635 0.61226994 0.530721470 559 73 871 127
152 0.025 0.52 0.640 0.61963190 0.578407394 548 72 882 128
153 0.025 0.53 0.655 0.62699387 0.425837350 539 69 891 131
154 0.025 0.54 0.665 0.63619632 0.409075796 526 67 904 133
155 0.025 0.55 0.680 0.64355828 0.284616408 517 64 913 136
156 0.025 0.56 0.700 0.65398773 0.167251299 504 60 926 140
157 0.025 0.57 0.700 0.66196319 0.256677086 491 60 939 140
158 0.025 0.58 0.715 0.67177914 0.190410091 478 57 952 143
159 0.025 0.59 0.720 0.68036810 0.229270801 465 56 965 144
160 0.025 0.60 0.720 0.68466258 0.285950334 458 56 972 144
161 0.025 0.61 0.725 0.69202454 0.318901469 447 55 983 145
162 0.025 0.62 0.740 0.69938650 0.209465132 438 52 992 148
163 0.025 0.63 0.750 0.70858896 0.196047167 425 50 1005 150
164 0.025 0.64 0.755 0.71963190 0.269232821 408 49 1022 151
165 0.025 0.65 0.770 0.72638037 0.163733087 400 46 1030 154
166 0.025 0.66 0.775 0.73619632 0.213569338 385 45 1045 155
167 0.025 0.67 0.780 0.74478528 0.257229890 372 44 1058 156
168 0.025 0.68 0.780 0.75030675 0.342828567 363 44 1067 156
169 0.025 0.69 0.785 0.76012270 0.428799898 348 43 1082 157
170 0.025 0.70 0.785 0.76503067 0.533865096 340 43 1090 157
171 0.025 0.71 0.790 0.77300613 0.601372349 328 42 1102 158
172 0.025 0.72 0.795 0.77730061 0.581232387 322 41 1108 159
173 0.025 0.73 0.795 0.78159509 0.690253305 315 41 1115 159
174 0.025 0.74 0.810 0.78650307 0.439131395 310 38 1120 162
175 0.025 0.75 0.810 0.79570552 0.658715418 295 38 1135 162
176 0.025 0.76 0.825 0.80061350 0.408180594 290 35 1140 165
177 0.025 0.77 0.835 0.81165644 0.420867538 274 33 1156 167
178 0.025 0.78 0.850 0.81533742 0.210749352 271 30 1159 170
179 0.025 0.79 0.850 0.82331288 0.338313613 258 30 1172 170
180 0.025 0.80 0.860 0.83312883 0.323694782 244 28 1186 172
181 0.025 0.81 0.865 0.84049080 0.364099263 233 27 1197 173
182 0.025 0.82 0.875 0.85030675 0.347611740 219 25 1211 175
183 0.025 0.83 0.890 0.85828221 0.205902238 209 22 1221 178
184 0.025 0.84 0.895 0.86441718 0.215508013 200 21 1230 179
185 0.025 0.85 0.900 0.87239264 0.255880294 188 20 1242 180
186 0.025 0.86 0.920 0.88343558 0.108985363 174 16 1256 184
187 0.025 0.87 0.925 0.89141104 0.131364623 162 15 1268 185
188 0.025 0.88 0.930 0.90245399 0.202463161 145 14 1285 186
189 0.025 0.89 0.935 0.91840491 0.436906929 120 13 1310 187
190 0.025 0.90 0.940 0.92883436 0.610818332 104 12 1326 188
191 0.025 0.91 0.955 0.93926380 0.402743212 90 9 1340 191
192 0.025 0.92 0.965 0.94478528 0.241572480 83 7 1347 193
193 0.025 0.93 0.980 0.95153374 0.067902504 75 4 1355 196
194 0.025 0.94 0.985 0.95766871 0.062589817 66 3 1364 197
195 0.025 0.95 0.990 0.96809816 0.095529255 50 2 1380 198
196 0.025 0.96 0.995 0.97177914 0.058864046 45 1 1385 199
197 0.025 0.97 0.995 0.97791411 0.133995196 35 1 1395 199
198 0.025 0.98 0.995 0.98711656 0.471047426 20 1 1410 199
199 0.025 0.99 0.995 0.99018405 0.722823657 15 1 1415 199
200 0.025 1.00 1.000 1.00000000 0.000000000 0 0 1430 200enrichment.plotter(gene.hic.KR, "min_FDR.H", "adj.P.Val", "Minimum FDR of KR Hi-C Contacts Overlapping Gene, Chimp", xmax=1)Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect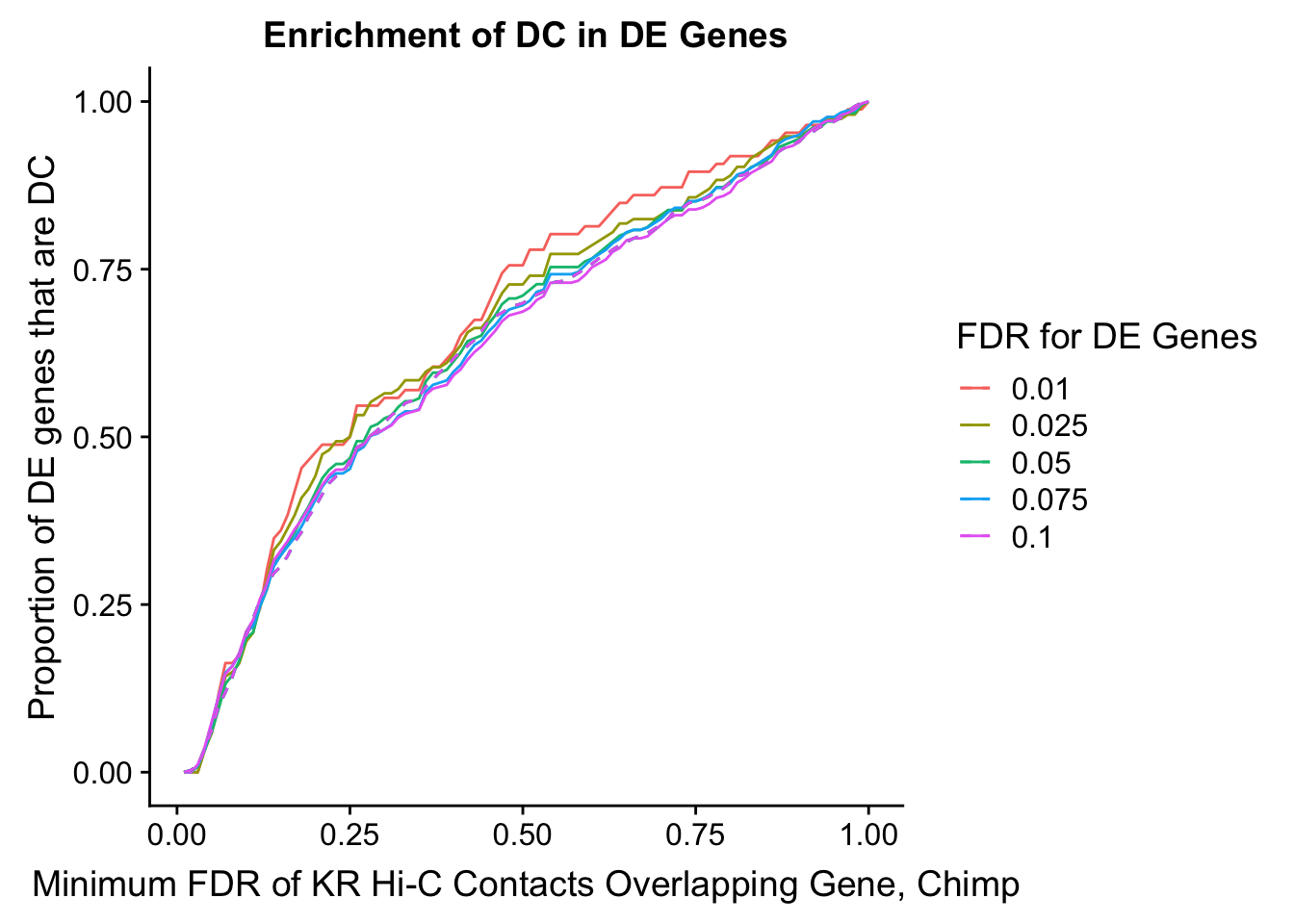
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
Warning: Removed 5 rows containing missing values (geom_path).Warning: Removed 5 rows containing missing values (geom_path).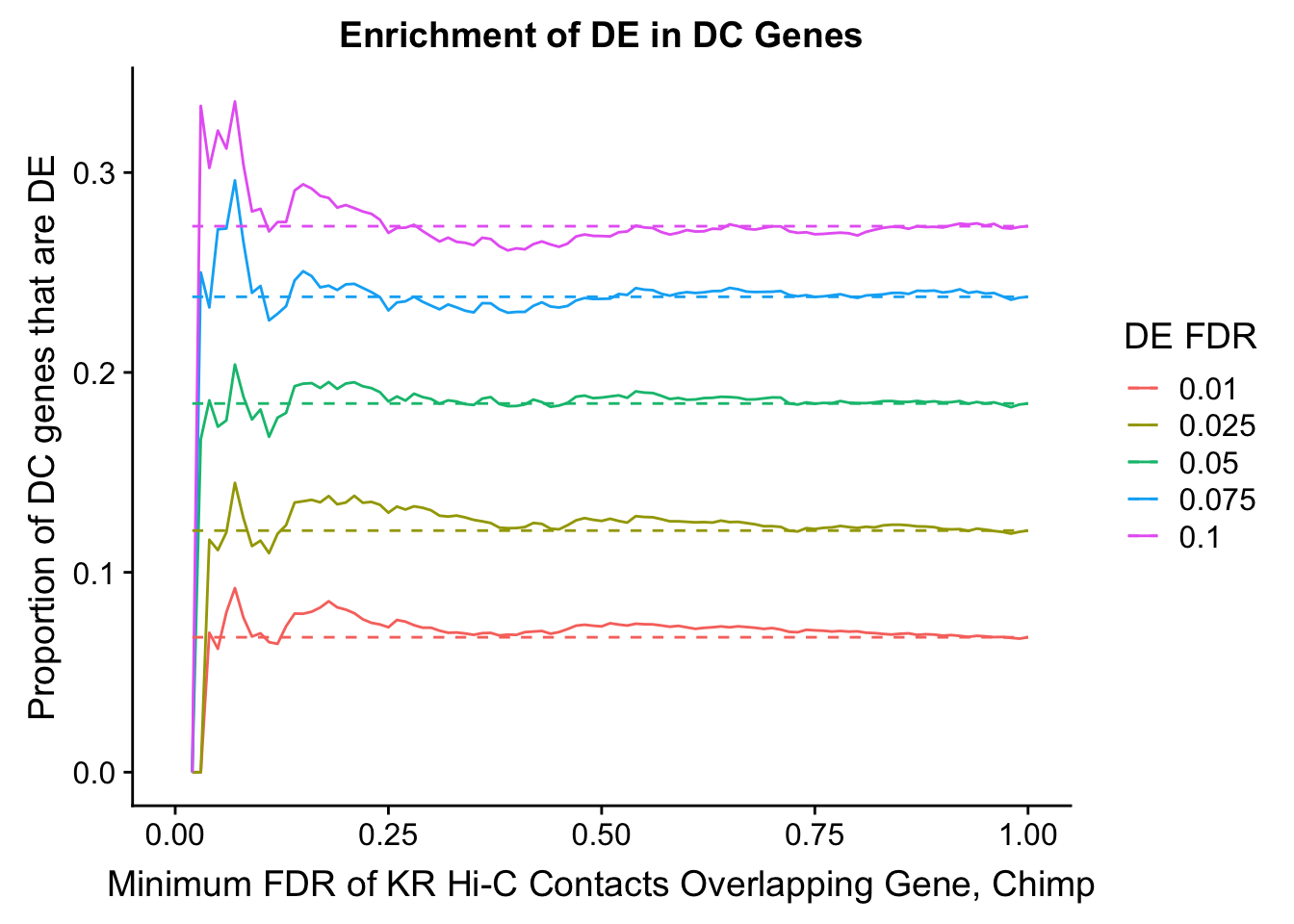
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
DEFDR DHICFDR prop.obs prop.exp chisq.p Dneither DE DHiC Dboth
101 0.025 0.01 0.00000000 0.000000000 0.0000000 1120 154 0 0
102 0.025 0.02 0.00000000 0.003139717 1.0000000 1116 154 4 0
103 0.025 0.03 0.00000000 0.009419152 0.3976953 1108 154 12 0
104 0.025 0.04 0.03246753 0.033751962 1.0000000 1082 149 38 5
105 0.025 0.05 0.05844156 0.063579278 0.9183031 1048 145 72 9
106 0.025 0.06 0.09740260 0.098116170 1.0000000 1010 139 110 15
107 0.025 0.07 0.14285714 0.119309262 0.4071553 990 132 130 22
108 0.025 0.08 0.14935065 0.142072214 0.8785229 962 131 158 23
109 0.025 0.09 0.16233766 0.173469388 0.7828475 924 129 196 25
110 0.025 0.10 0.19480519 0.203296703 0.8630574 891 124 229 30
111 0.025 0.11 0.20779221 0.229199372 0.5674219 860 122 260 32
112 0.025 0.12 0.25324675 0.256671900 0.9956871 832 115 288 39
113 0.025 0.13 0.28571429 0.279434851 0.9287234 808 110 312 44
114 0.025 0.14 0.33116883 0.296703297 0.3657160 793 103 327 51
115 0.025 0.15 0.34415584 0.306907378 0.3291895 782 101 338 53
116 0.025 0.16 0.36363636 0.322605965 0.2847310 765 98 355 56
117 0.025 0.17 0.38311688 0.343014129 0.3041534 742 95 378 59
118 0.025 0.18 0.40909091 0.357927786 0.1858653 727 91 393 63
119 0.025 0.19 0.42207792 0.380690738 0.2985086 700 89 420 65
120 0.025 0.20 0.44155844 0.395604396 0.2476931 684 86 436 68
121 0.025 0.21 0.47402597 0.414442700 0.1301291 665 81 455 73
122 0.025 0.22 0.48051948 0.430926217 0.2154553 645 80 475 74
123 0.025 0.23 0.49350649 0.441130298 0.1903307 634 78 486 76
124 0.025 0.24 0.49350649 0.445839874 0.2368939 628 78 492 76
125 0.025 0.25 0.50000000 0.465463108 0.4063940 604 77 516 77
126 0.025 0.26 0.53246753 0.484301413 0.2341898 585 72 535 82
127 0.025 0.27 0.53246753 0.489795918 0.2965675 578 72 542 82
128 0.025 0.28 0.55194805 0.501569859 0.2121739 566 69 554 85
129 0.025 0.29 0.55844156 0.510204082 0.2335807 556 68 564 86
130 0.025 0.30 0.56493506 0.521193093 0.2833148 543 67 577 87
131 0.025 0.31 0.56493506 0.532182104 0.4338186 529 67 591 87
132 0.025 0.32 0.57142857 0.540031397 0.4547242 520 66 600 88
133 0.025 0.33 0.58441558 0.550235479 0.4105120 509 64 611 90
134 0.025 0.34 0.58441558 0.554160126 0.4720350 504 64 616 90
135 0.025 0.35 0.58441558 0.559654631 0.5662427 497 64 623 90
136 0.025 0.36 0.59740260 0.575353218 0.6146340 479 62 641 92
137 0.025 0.37 0.60389610 0.585557300 0.6851261 467 61 653 93
138 0.025 0.38 0.60389610 0.596546311 0.9118592 453 61 667 93
139 0.025 0.39 0.61038961 0.604395604 0.9407234 444 60 676 94
140 0.025 0.40 0.62337662 0.616954474 0.9311060 430 58 690 96
141 0.025 0.41 0.63636364 0.627158556 0.8704528 419 56 701 98
142 0.025 0.42 0.65584416 0.635792779 0.6439357 411 53 709 101
143 0.025 0.43 0.66233766 0.644427002 0.6851487 401 52 719 102
144 0.025 0.44 0.66233766 0.656985871 0.9531993 385 52 735 102
145 0.025 0.45 0.67532468 0.671899529 0.9959877 368 50 752 104
146 0.025 0.46 0.69480519 0.679748823 0.7376207 361 47 759 107
147 0.025 0.47 0.71428571 0.685243328 0.4622518 357 44 763 110
148 0.025 0.48 0.72727273 0.691522763 0.3516342 351 42 769 112
149 0.025 0.49 0.72727273 0.696232339 0.4237710 345 42 775 112
150 0.025 0.50 0.72727273 0.699372057 0.4766950 341 42 779 112
151 0.025 0.51 0.74025974 0.705651491 0.3624187 335 40 785 114
152 0.025 0.52 0.74025974 0.711930926 0.4635295 327 40 793 114
153 0.025 0.53 0.74025974 0.716640502 0.5496016 321 40 799 114
154 0.025 0.54 0.77272727 0.729199372 0.2302373 310 35 810 119
155 0.025 0.55 0.77272727 0.731554160 0.2573285 307 35 813 119
156 0.025 0.56 0.77272727 0.732339089 0.2668634 306 35 814 119
157 0.025 0.57 0.77272727 0.737833595 0.3409159 299 35 821 119
158 0.025 0.58 0.77272727 0.744113030 0.4416371 291 35 829 119
159 0.025 0.59 0.77922078 0.750392465 0.4340206 284 34 836 120
160 0.025 0.60 0.78571429 0.758241758 0.4539223 275 33 845 121
161 0.025 0.61 0.79220779 0.766091052 0.4745762 266 32 854 122
162 0.025 0.62 0.79870130 0.771585557 0.4517403 260 31 860 123
163 0.025 0.63 0.80519481 0.779434851 0.4723591 251 30 869 124
164 0.025 0.64 0.81818182 0.785714286 0.3459172 245 28 875 126
165 0.025 0.65 0.81818182 0.790423862 0.4254069 239 28 881 126
166 0.025 0.66 0.82467532 0.795918367 0.4021718 233 27 887 127
167 0.025 0.67 0.82467532 0.799843014 0.4752136 228 27 892 127
168 0.025 0.68 0.82467532 0.803767661 0.5561493 223 27 897 127
169 0.025 0.69 0.82467532 0.810047096 0.7009619 215 27 905 127
170 0.025 0.70 0.83116883 0.816326531 0.6918514 208 26 912 128
171 0.025 0.71 0.83766234 0.824960754 0.7419211 198 25 922 129
172 0.025 0.72 0.83766234 0.838304553 1.0000000 181 25 939 129
173 0.025 0.73 0.83766234 0.840659341 1.0000000 178 25 942 129
174 0.025 0.74 0.85714286 0.848508634 0.8423539 171 22 949 132
175 0.025 0.75 0.85714286 0.851648352 0.9332976 167 22 953 132
176 0.025 0.76 0.86363636 0.854003140 0.8108077 165 21 955 133
177 0.025 0.77 0.87012987 0.858712716 0.7562121 160 20 960 134
178 0.025 0.78 0.88311688 0.866562009 0.6044720 152 18 968 136
179 0.025 0.79 0.88311688 0.870486656 0.7114716 147 18 973 136
180 0.025 0.80 0.88961039 0.879905808 0.7926030 136 17 984 137
181 0.025 0.81 0.90259740 0.888540031 0.6493524 127 15 993 139
182 0.025 0.82 0.90259740 0.890894819 0.7196186 124 15 996 139
183 0.025 0.83 0.91558442 0.896389325 0.4885422 119 13 1001 141
184 0.025 0.84 0.92207792 0.900313972 0.4133100 115 12 1005 142
185 0.025 0.85 0.92857143 0.906593407 0.3942483 108 11 1012 143
186 0.025 0.86 0.93506494 0.915227630 0.4305068 98 10 1022 144
187 0.025 0.87 0.94155844 0.925431711 0.5163797 86 9 1034 145
188 0.025 0.88 0.94805195 0.932496075 0.5161159 78 8 1042 146
189 0.025 0.89 0.94805195 0.934850863 0.5934424 75 8 1045 146
190 0.025 0.90 0.94805195 0.941915228 0.8701028 66 8 1054 146
191 0.025 0.91 0.95454545 0.949764521 0.9259348 57 7 1063 147
192 0.025 0.92 0.96103896 0.955259027 0.8711665 51 6 1069 148
193 0.025 0.93 0.96103896 0.962323391 1.0000000 42 6 1078 148
194 0.025 0.94 0.97402597 0.966248038 0.7398224 39 4 1081 150
195 0.025 0.95 0.97402597 0.970172684 0.9623608 34 4 1086 150
196 0.025 0.96 0.97402597 0.975667190 1.0000000 27 4 1093 150
197 0.025 0.97 0.98051948 0.985871272 0.8133811 15 3 1105 151
198 0.025 0.98 0.98051948 0.992935636 0.1473268 6 3 1114 151
199 0.025 0.99 0.99350649 0.998430141 0.5750706 1 1 1119 153
200 0.025 1.00 1.00000000 1.000000000 0.0000000 0 0 1120 154#enrichment.plotter(h_US, "USFDR", "adj.P.Val", "FDR for Closest Upstream Hi-C Contact Overlapping Gene, Human") #These two are ugly, and can't be run anyway until next chunk is complete to create their DFs. It's OK without them.
#enrichment.plotter(c_US, "USFDR", "adj.P.Val", "FDR for Closest Upstream Hi-C Contact Overlapping Gene, Chimp")
#enrichment.plotter(gene.hic.filt, "max_B_FDR.H", "adj.P.Val", "FDR for Hi-C Contact Overlapping Gene w/ Strongest Effect Size, Human")
#enrichment.plotter(gene.hic.filt, "max_B_FDR.C", "adj.P.Val", "FDR for Hi-C Contact Overlapping Gene w/ Strongest Effect Size, Chimp")
#enrichment.plotter(gene.hic.filt, "median_FDR.H", "adj.P.Val", "Median FDR of Hi-C Contacts Overlapping Gene, Human")
#enrichment.plotter(gene.hic.filt, "median_FDR.C", "adj.P.Val", "Median FDR of Hi-C Contacts Overlapping Gene, Chimp")
enrichment.plotter(gene.hic.VC, "weighted_Z.s2post.H", "adj.P.Val", "FDR for Weighted p-val Combine of VC Hi-C Contacts Overlapping Gene", xmax=1)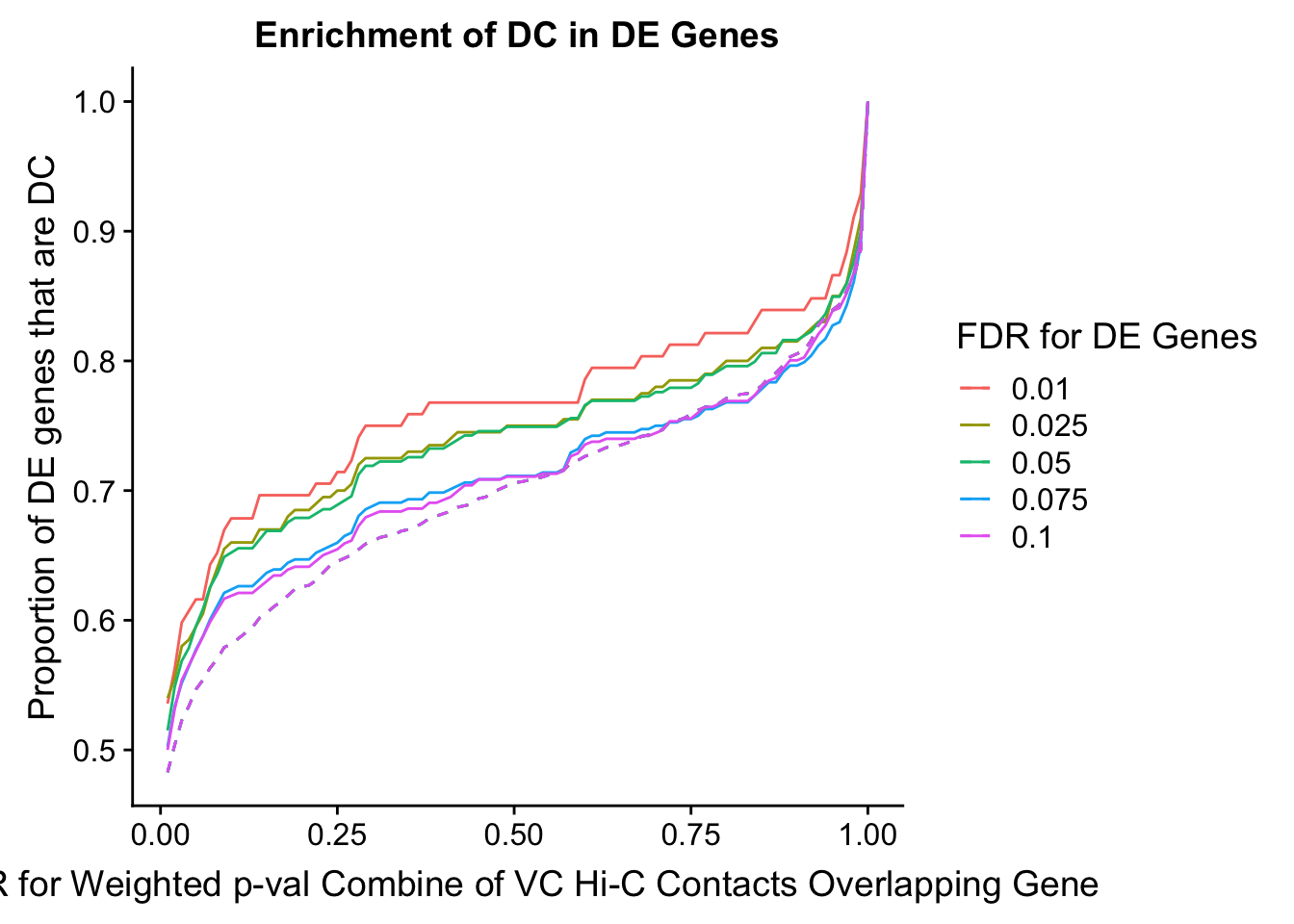
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
DEFDR DHICFDR prop.obs prop.exp chisq.p Dneither DE DHiC Dboth
101 0.025 0.01 0.540 0.4828221 0.09851197 751 92 679 108
102 0.025 0.02 0.555 0.5036810 0.14041388 720 89 710 111
103 0.025 0.03 0.580 0.5226994 0.09761075 694 84 736 116
104 0.025 0.04 0.585 0.5337423 0.14001822 677 83 753 117
105 0.025 0.05 0.595 0.5466258 0.16411957 658 81 772 119
106 0.025 0.06 0.605 0.5539877 0.14059911 648 79 782 121
107 0.025 0.07 0.625 0.5631902 0.07099899 637 75 793 125
108 0.025 0.08 0.640 0.5699387 0.03935656 629 72 801 128
109 0.025 0.09 0.655 0.5791411 0.02486225 617 69 813 131
110 0.025 0.10 0.660 0.5815951 0.02016402 614 68 816 132
111 0.025 0.11 0.660 0.5858896 0.02815759 607 68 823 132
112 0.025 0.12 0.660 0.5901840 0.03876550 600 68 830 132
113 0.025 0.13 0.660 0.5944785 0.05262293 593 68 837 132
114 0.025 0.14 0.670 0.6018405 0.04284623 583 66 847 134
115 0.025 0.15 0.670 0.6055215 0.05552805 577 66 853 134
116 0.025 0.16 0.670 0.6104294 0.07722448 569 66 861 134
117 0.025 0.17 0.670 0.6141104 0.09773563 563 66 867 134
118 0.025 0.18 0.680 0.6190184 0.06902426 557 64 873 136
119 0.025 0.19 0.685 0.6239264 0.06788893 550 63 880 137
120 0.025 0.20 0.685 0.6257669 0.07670891 547 63 883 137
121 0.025 0.21 0.685 0.6269939 0.08310023 545 63 885 137
122 0.025 0.22 0.690 0.6306748 0.07544256 540 62 890 138
123 0.025 0.23 0.695 0.6368098 0.08039011 531 61 899 139
124 0.025 0.24 0.695 0.6423313 0.11402637 522 61 908 139
125 0.025 0.25 0.700 0.6453988 0.10009589 518 60 912 140
126 0.025 0.26 0.700 0.6478528 0.11655314 514 60 916 140
127 0.025 0.27 0.705 0.6503067 0.09842421 511 59 919 141
128 0.025 0.28 0.720 0.6546012 0.04579717 507 56 923 144
129 0.025 0.29 0.725 0.6588957 0.04279546 501 55 929 145
130 0.025 0.30 0.725 0.6607362 0.04887697 498 55 932 145
131 0.025 0.31 0.725 0.6638037 0.06065379 493 55 937 145
132 0.025 0.32 0.725 0.6650307 0.06599507 491 55 939 145
133 0.025 0.33 0.725 0.6656442 0.06881079 490 55 940 145
134 0.025 0.34 0.725 0.6687117 0.08444351 485 55 945 145
135 0.025 0.35 0.730 0.6699387 0.06456853 484 54 946 146
136 0.025 0.36 0.730 0.6717791 0.07318306 481 54 949 146
137 0.025 0.37 0.730 0.6748466 0.08966824 476 54 954 146
138 0.025 0.38 0.735 0.6785276 0.08101186 471 53 959 147
139 0.025 0.39 0.735 0.6803681 0.09142997 468 53 962 147
140 0.025 0.40 0.735 0.6822086 0.10292917 465 53 965 147
141 0.025 0.41 0.740 0.6840491 0.08256892 463 52 967 148
142 0.025 0.42 0.745 0.6871166 0.07131101 459 51 971 149
143 0.025 0.43 0.745 0.6883436 0.07749172 457 51 973 149
144 0.025 0.44 0.745 0.6895706 0.08411411 455 51 975 149
145 0.025 0.45 0.745 0.6938650 0.11109463 448 51 982 149
146 0.025 0.46 0.745 0.6950920 0.11998338 446 51 984 149
147 0.025 0.47 0.745 0.6987730 0.15013681 440 51 990 149
148 0.025 0.48 0.745 0.7012270 0.17336638 436 51 994 149
149 0.025 0.49 0.750 0.7036810 0.14736799 433 50 997 150
150 0.025 0.50 0.750 0.7061350 0.17035639 429 50 1001 150
151 0.025 0.51 0.750 0.7067485 0.17651928 428 50 1002 150
152 0.025 0.52 0.750 0.7079755 0.18936217 426 50 1004 150
153 0.025 0.53 0.750 0.7092025 0.20291108 424 50 1006 150
154 0.025 0.54 0.750 0.7104294 0.21718490 422 50 1008 150
155 0.025 0.55 0.750 0.7122699 0.23999321 419 50 1011 150
156 0.025 0.56 0.750 0.7141104 0.26452600 416 50 1014 150
157 0.025 0.57 0.755 0.7159509 0.22106122 414 49 1016 151
158 0.025 0.58 0.755 0.7208589 0.28686952 406 49 1024 151
159 0.025 0.59 0.755 0.7233129 0.32458154 402 49 1028 151
160 0.025 0.60 0.765 0.7263804 0.22122125 399 47 1031 153
161 0.025 0.61 0.770 0.7282209 0.18249857 397 46 1033 154
162 0.025 0.62 0.770 0.7306748 0.21006530 393 46 1037 154
163 0.025 0.63 0.770 0.7331288 0.24069177 389 46 1041 154
164 0.025 0.64 0.770 0.7343558 0.25719823 387 46 1043 154
165 0.025 0.65 0.770 0.7349693 0.26575685 386 46 1044 154
166 0.025 0.66 0.770 0.7368098 0.29267509 383 46 1047 154
167 0.025 0.67 0.770 0.7386503 0.32148737 380 46 1050 154
168 0.025 0.68 0.775 0.7417178 0.28829181 376 45 1054 155
169 0.025 0.69 0.775 0.7429448 0.30719019 374 45 1056 155
170 0.025 0.70 0.780 0.7447853 0.25722989 372 44 1058 156
171 0.025 0.71 0.780 0.7472393 0.29311087 368 44 1062 156
172 0.025 0.72 0.785 0.7527607 0.29795001 360 43 1070 157
173 0.025 0.73 0.785 0.7539877 0.31751994 358 43 1072 157
174 0.025 0.74 0.785 0.7558282 0.34854337 355 43 1075 157
175 0.025 0.75 0.785 0.7588957 0.40474382 350 43 1080 157
176 0.025 0.76 0.785 0.7619632 0.46655886 345 43 1085 157
177 0.025 0.77 0.790 0.7644172 0.41148528 342 42 1088 158
178 0.025 0.78 0.790 0.7644172 0.41148528 342 42 1088 158
179 0.025 0.79 0.795 0.7674847 0.37126795 338 41 1092 159
180 0.025 0.80 0.800 0.7711656 0.34388428 333 40 1097 160
181 0.025 0.81 0.800 0.7717791 0.35478827 332 40 1098 160
182 0.025 0.82 0.800 0.7742331 0.40076361 328 40 1102 160
183 0.025 0.83 0.800 0.7748466 0.41284809 327 40 1103 160
184 0.025 0.84 0.805 0.7766871 0.34936028 325 39 1105 161
185 0.025 0.85 0.810 0.7809816 0.33298636 319 38 1111 162
186 0.025 0.86 0.810 0.7871166 0.45214491 309 38 1121 162
187 0.025 0.87 0.810 0.7907975 0.53524526 303 38 1127 162
188 0.025 0.88 0.815 0.7963190 0.54409373 295 37 1135 163
189 0.025 0.89 0.815 0.8030675 0.72025043 284 37 1146 163
190 0.025 0.90 0.815 0.8055215 0.79007473 280 37 1150 163
191 0.025 0.91 0.820 0.8079755 0.71503958 277 36 1153 164
192 0.025 0.92 0.825 0.8159509 0.79859516 265 35 1165 165
193 0.025 0.93 0.830 0.8276074 1.00000000 247 34 1183 166
194 0.025 0.94 0.830 0.8337423 0.95981835 237 34 1193 166
195 0.025 0.95 0.850 0.8392638 0.73492610 232 30 1198 170
196 0.025 0.96 0.850 0.8435583 0.86986526 225 30 1205 170
197 0.025 0.97 0.860 0.8539877 0.88062368 210 28 1220 172
198 0.025 0.98 0.885 0.8644172 0.42514762 198 23 1232 177
199 0.025 0.99 0.910 0.8858896 0.30477538 168 18 1262 182
200 0.025 1.00 1.000 1.0000000 0.00000000 0 0 1430 200enrichment.plotter(gene.hic.KR, "weighted_Z.s2post.H", "adj.P.Val", "FDR for Weighted p-val Combine of KR Hi-C Contacts Overlapping Gene", xmax=1)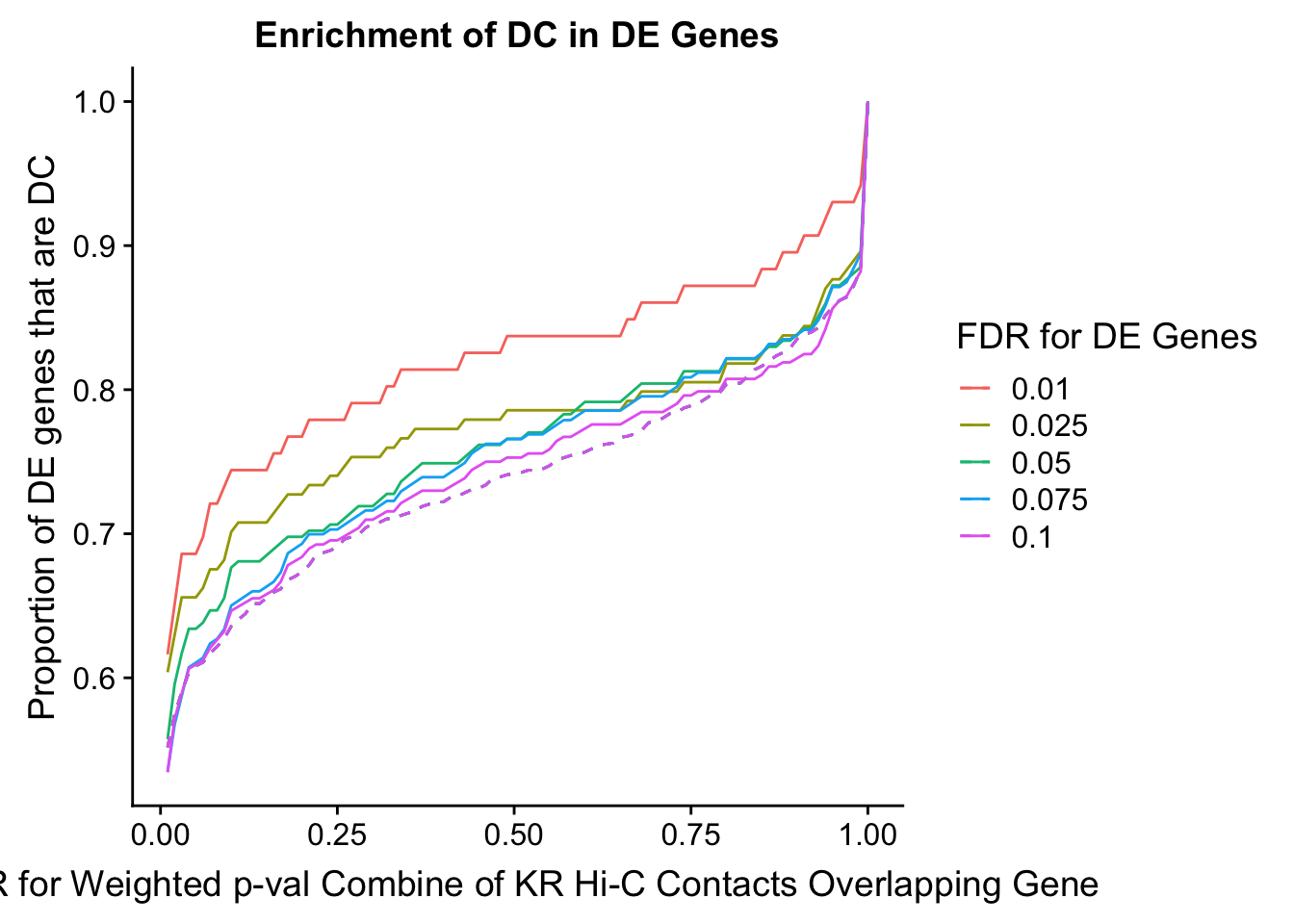
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
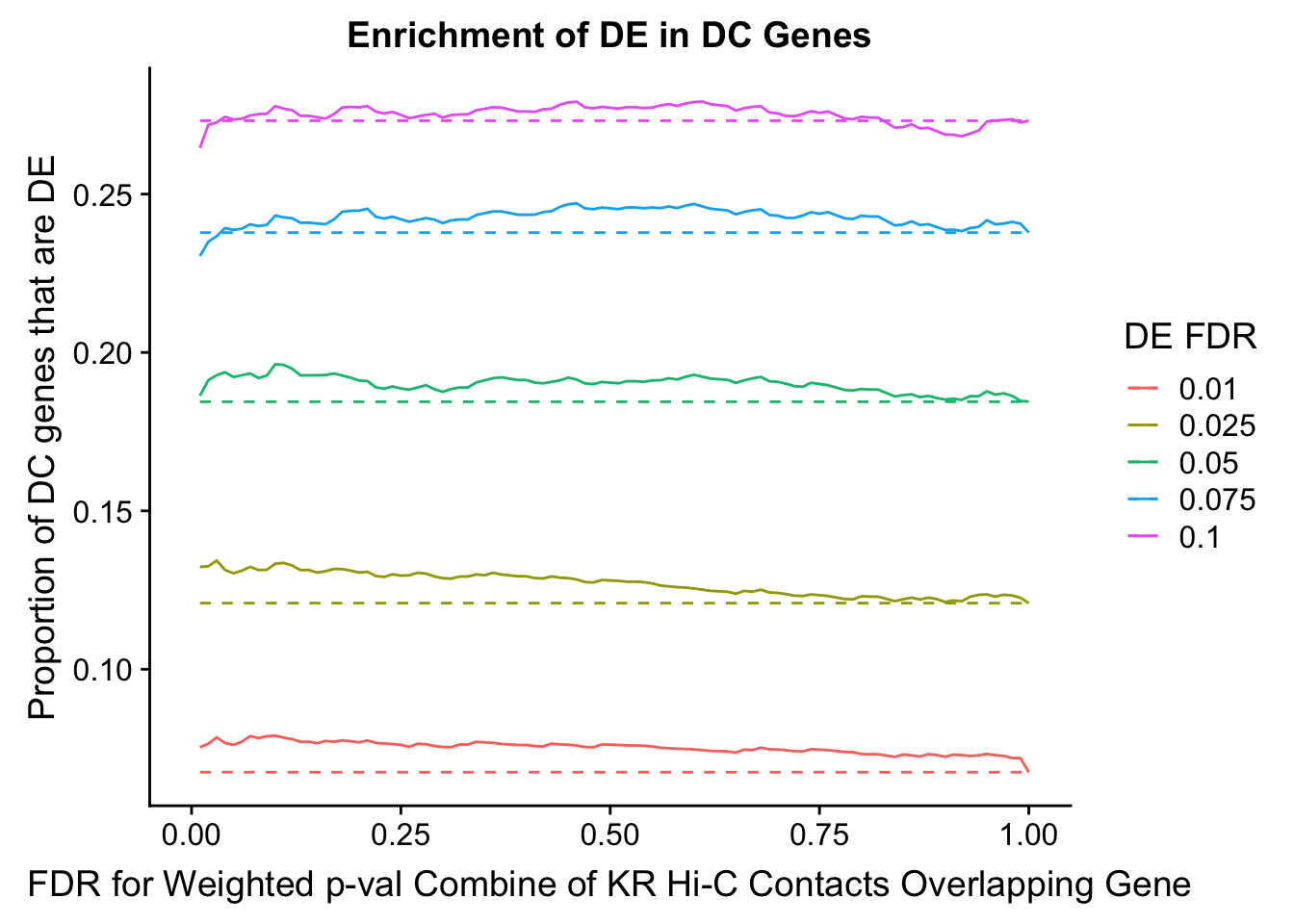
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
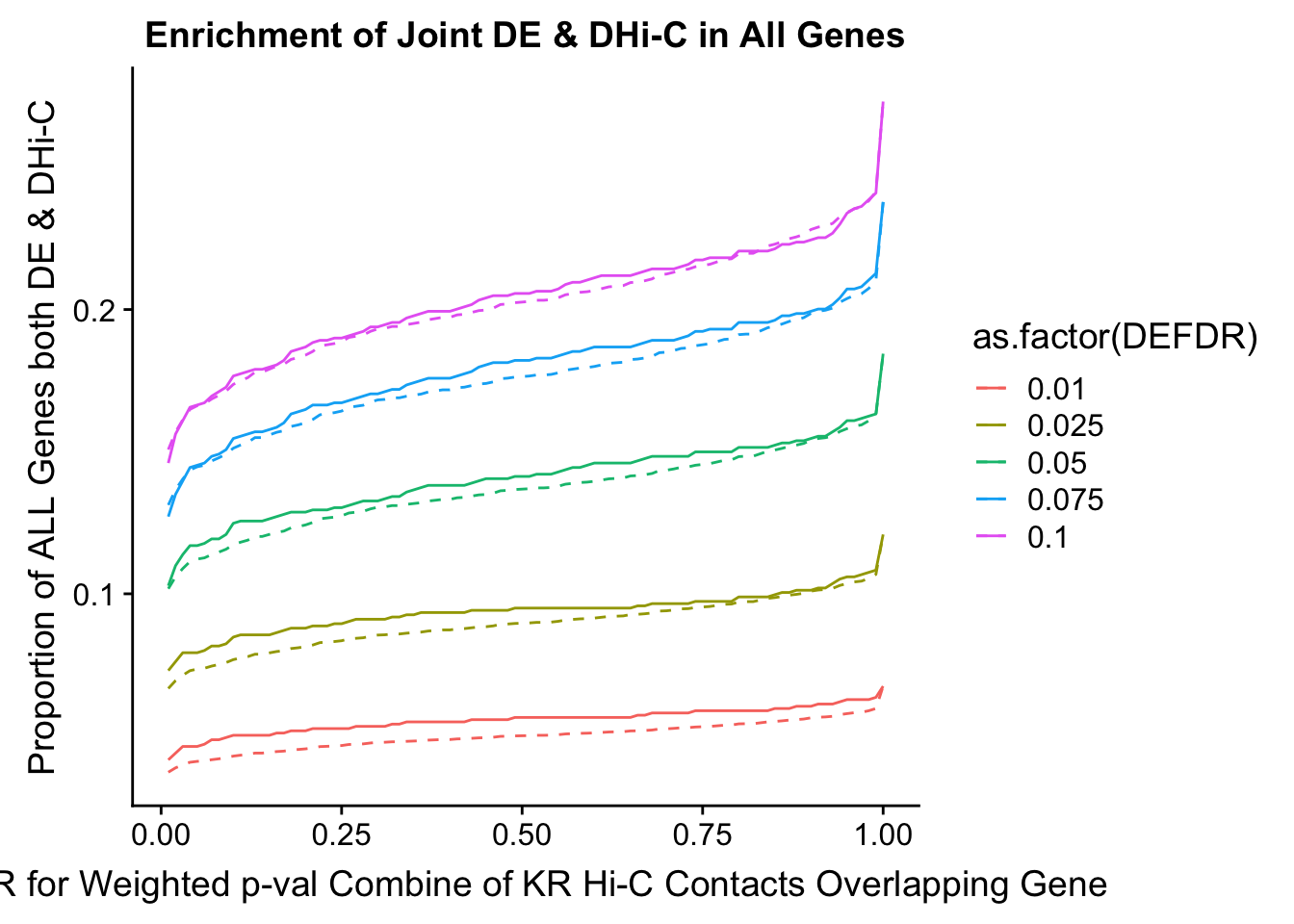
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
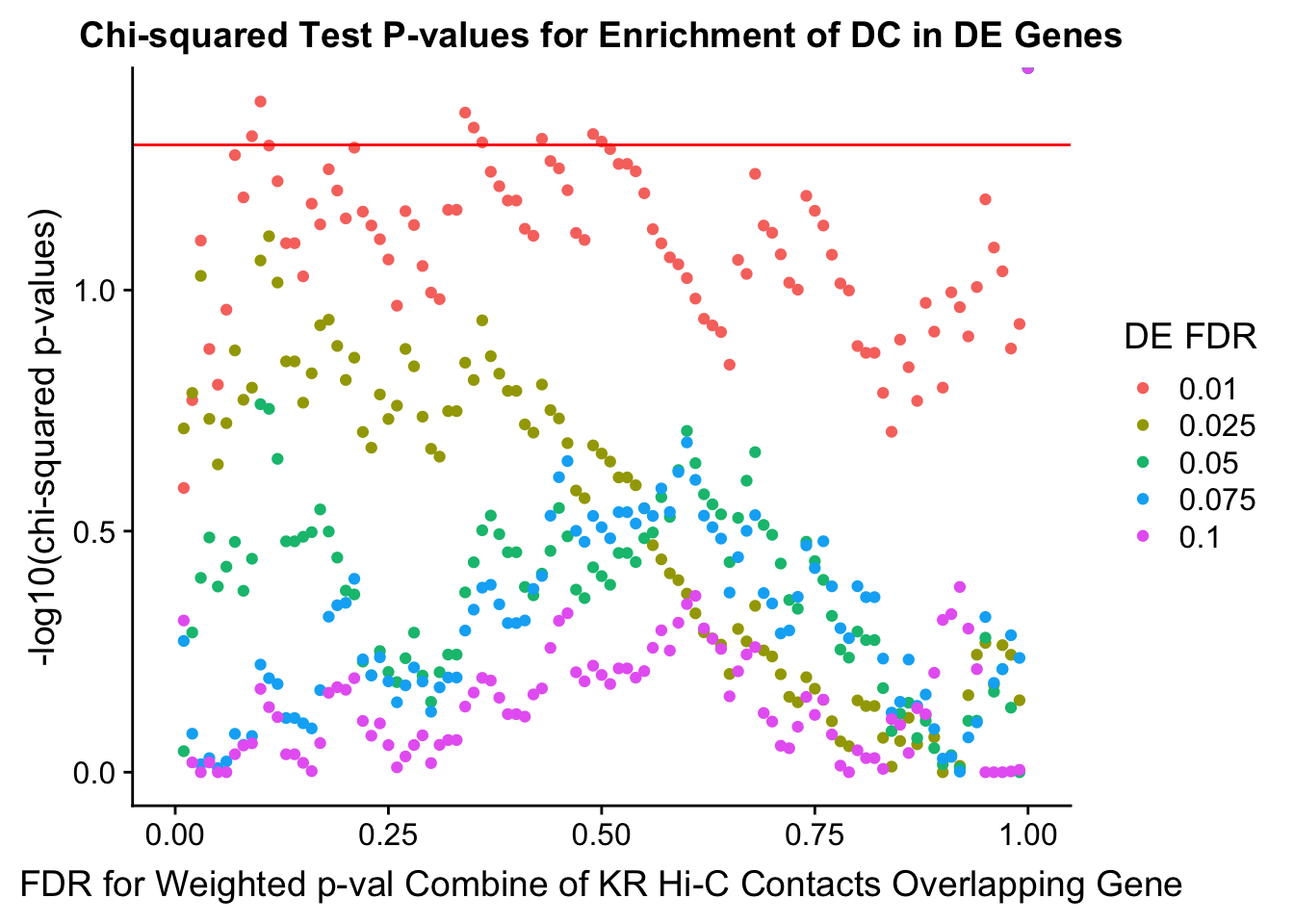
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
DEFDR DHICFDR prop.obs prop.exp chisq.p Dneither DE DHiC Dboth
101 0.025 0.01 0.6038961 0.5518053 0.19362378 510 61 610 93
102 0.025 0.02 0.6298701 0.5745683 0.16346233 485 57 635 97
103 0.025 0.03 0.6558442 0.5902669 0.09344506 469 53 651 101
104 0.025 0.04 0.6558442 0.6036107 0.18501010 452 53 668 101
105 0.025 0.05 0.6558442 0.6083203 0.22992174 446 53 674 101
106 0.025 0.06 0.6623377 0.6106750 0.18877776 444 52 676 102
107 0.025 0.07 0.6753247 0.6169545 0.13340921 438 50 682 104
108 0.025 0.08 0.6753247 0.6216641 0.16886970 432 50 688 104
109 0.025 0.09 0.6818182 0.6271586 0.15936680 426 49 694 105
110 0.025 0.10 0.7012987 0.6357928 0.08682146 418 46 702 108
111 0.025 0.11 0.7077922 0.6405024 0.07732129 413 45 707 109
112 0.025 0.12 0.7077922 0.6444270 0.09646552 408 45 712 109
113 0.025 0.13 0.7077922 0.6514914 0.14057634 399 45 721 109
114 0.025 0.14 0.7077922 0.6514914 0.14057634 399 45 721 109
115 0.025 0.15 0.7077922 0.6554160 0.17122756 394 45 726 109
116 0.025 0.16 0.7142857 0.6593407 0.14880439 390 44 730 110
117 0.025 0.17 0.7207792 0.6616954 0.11829284 388 43 732 111
118 0.025 0.18 0.7272727 0.6679749 0.11519447 381 42 739 112
119 0.025 0.19 0.7272727 0.6703297 0.13058301 378 42 742 112
120 0.025 0.20 0.7272727 0.6734694 0.15360930 374 42 746 112
121 0.025 0.21 0.7337662 0.6781790 0.13811656 369 41 751 113
122 0.025 0.22 0.7337662 0.6852433 0.19694136 360 41 760 113
123 0.025 0.23 0.7337662 0.6868132 0.21230007 358 41 762 113
124 0.025 0.24 0.7402597 0.6883830 0.16462667 357 40 763 114
125 0.025 0.25 0.7402597 0.6907378 0.18512255 354 40 766 114
126 0.025 0.26 0.7467532 0.6962323 0.17365846 348 39 772 115
127 0.025 0.27 0.7532468 0.6978022 0.13246669 347 38 773 116
128 0.025 0.28 0.7532468 0.6993721 0.14391637 345 38 775 116
129 0.025 0.29 0.7532468 0.7040816 0.18304081 339 38 781 116
130 0.025 0.30 0.7532468 0.7072214 0.21339902 335 38 785 116
131 0.025 0.31 0.7532468 0.7080063 0.22155452 334 38 786 116
132 0.025 0.32 0.7597403 0.7103611 0.17827316 332 37 788 117
133 0.025 0.33 0.7597403 0.7103611 0.17827316 332 37 788 117
134 0.025 0.34 0.7662338 0.7127159 0.14144849 330 36 790 118
135 0.025 0.35 0.7662338 0.7142857 0.15362662 328 36 792 118
136 0.025 0.36 0.7727273 0.7158556 0.11555790 327 35 793 119
137 0.025 0.37 0.7727273 0.7189953 0.13713275 323 35 797 119
138 0.025 0.38 0.7727273 0.7205651 0.14907835 321 35 799 119
139 0.025 0.39 0.7727273 0.7221350 0.16184095 319 35 801 119
140 0.025 0.40 0.7727273 0.7221350 0.16184095 319 35 801 119
141 0.025 0.41 0.7727273 0.7252747 0.18994753 315 35 805 119
142 0.025 0.42 0.7727273 0.7260597 0.19753486 314 35 806 119
143 0.025 0.43 0.7792208 0.7284144 0.15699680 312 34 808 120
144 0.025 0.44 0.7792208 0.7307692 0.17738072 309 34 811 120
145 0.025 0.45 0.7792208 0.7315542 0.18461907 308 34 812 120
146 0.025 0.46 0.7792208 0.7339089 0.20772066 305 34 815 120
147 0.025 0.47 0.7792208 0.7386185 0.26048957 299 34 821 120
148 0.025 0.48 0.7792208 0.7394035 0.27017359 298 34 822 120
149 0.025 0.49 0.7857143 0.7409733 0.20999672 297 33 823 121
150 0.025 0.50 0.7857143 0.7417582 0.21829411 296 33 824 121
151 0.025 0.51 0.7857143 0.7425432 0.22683967 295 33 825 121
152 0.025 0.52 0.7857143 0.7441130 0.24468917 293 33 827 121
153 0.025 0.53 0.7857143 0.7441130 0.24468917 293 33 827 121
154 0.025 0.54 0.7857143 0.7448980 0.25399968 292 33 828 121
155 0.025 0.55 0.7857143 0.7472527 0.28351012 289 33 831 121
156 0.025 0.56 0.7857143 0.7511774 0.33810518 284 33 836 121
157 0.025 0.57 0.7857143 0.7527473 0.36188040 282 33 838 121
158 0.025 0.58 0.7857143 0.7543171 0.38677595 280 33 840 121
159 0.025 0.59 0.7857143 0.7551020 0.39964489 279 33 841 121
160 0.025 0.60 0.7857143 0.7566719 0.42622458 277 33 843 121
161 0.025 0.61 0.7857143 0.7590267 0.46818744 274 33 846 121
162 0.025 0.62 0.7857143 0.7613815 0.51262611 271 33 849 121
163 0.025 0.63 0.7857143 0.7621664 0.52797843 270 33 850 121
164 0.025 0.64 0.7857143 0.7629513 0.54359540 269 33 851 121
165 0.025 0.65 0.7857143 0.7668760 0.62551045 264 33 856 121
166 0.025 0.66 0.7922078 0.7676609 0.50443204 264 32 856 122
167 0.025 0.67 0.7922078 0.7692308 0.53538924 262 32 858 122
168 0.025 0.68 0.7987013 0.7715856 0.45174028 260 31 860 123
169 0.025 0.69 0.7987013 0.7770801 0.55901129 253 31 867 123
170 0.025 0.70 0.7987013 0.7778650 0.57544229 252 31 868 123
171 0.025 0.71 0.7987013 0.7802198 0.62630594 249 31 871 123
172 0.025 0.72 0.7987013 0.7833595 0.69757837 245 31 875 123
173 0.025 0.73 0.7987013 0.7841444 0.71596929 244 31 876 123
174 0.025 0.74 0.8051948 0.7872841 0.63531171 241 30 879 124
175 0.025 0.75 0.8051948 0.7888540 0.67109957 239 30 881 124
176 0.025 0.76 0.8051948 0.7904239 0.70784390 237 30 883 124
177 0.025 0.77 0.8051948 0.7935636 0.78395017 233 30 887 124
178 0.025 0.78 0.8051948 0.7967033 0.86305735 229 30 891 124
179 0.025 0.79 0.8051948 0.7974882 0.88322566 228 30 892 124
180 0.025 0.80 0.8181818 0.8037677 0.70976859 222 28 898 126
181 0.025 0.81 0.8181818 0.8045526 0.72894209 221 28 899 126
182 0.025 0.82 0.8181818 0.8045526 0.72894209 221 28 899 126
183 0.025 0.83 0.8181818 0.8092622 0.84844127 215 28 905 126
184 0.025 0.84 0.8181818 0.8139717 0.97386178 209 28 911 126
185 0.025 0.85 0.8246753 0.8163265 0.86155795 207 27 913 127
186 0.025 0.86 0.8311688 0.8194662 0.77107460 204 26 916 128
187 0.025 0.87 0.8311688 0.8233909 0.87503428 199 26 921 128
188 0.025 0.88 0.8376623 0.8257457 0.76226567 197 25 923 129
189 0.025 0.89 0.8376623 0.8288854 0.84590172 193 25 927 129
190 0.025 0.90 0.8376623 0.8351648 1.00000000 185 25 935 129
191 0.025 0.91 0.8441558 0.8383046 0.92540274 182 24 938 130
192 0.025 0.92 0.8441558 0.8398744 0.97021185 180 24 940 130
193 0.025 0.93 0.8571429 0.8430141 0.69217168 178 22 942 132
194 0.025 0.94 0.8701299 0.8516484 0.57052573 169 20 951 134
195 0.025 0.95 0.8766234 0.8571429 0.53920655 163 19 957 135
196 0.025 0.96 0.8766234 0.8618524 0.65846260 157 19 963 135
197 0.025 0.97 0.8831169 0.8642072 0.54508104 155 18 965 136
198 0.025 0.98 0.8896104 0.8720565 0.57078171 146 17 974 137
199 0.025 0.99 0.8961039 0.8838305 0.70926028 132 16 988 138
200 0.025 1.00 1.0000000 1.0000000 0.00000000 0 0 1120 154enrichment.plotter(gene.hic.VC, "weighted_Z.ALLvar.H", "adj.P.Val", "FDR for Weighted p-val Combine of VC Hi-C Contacts Overlapping Gene", xmax=1)Warning in chisq.test(mytable): Chi-squared approximation may be incorrectWarning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect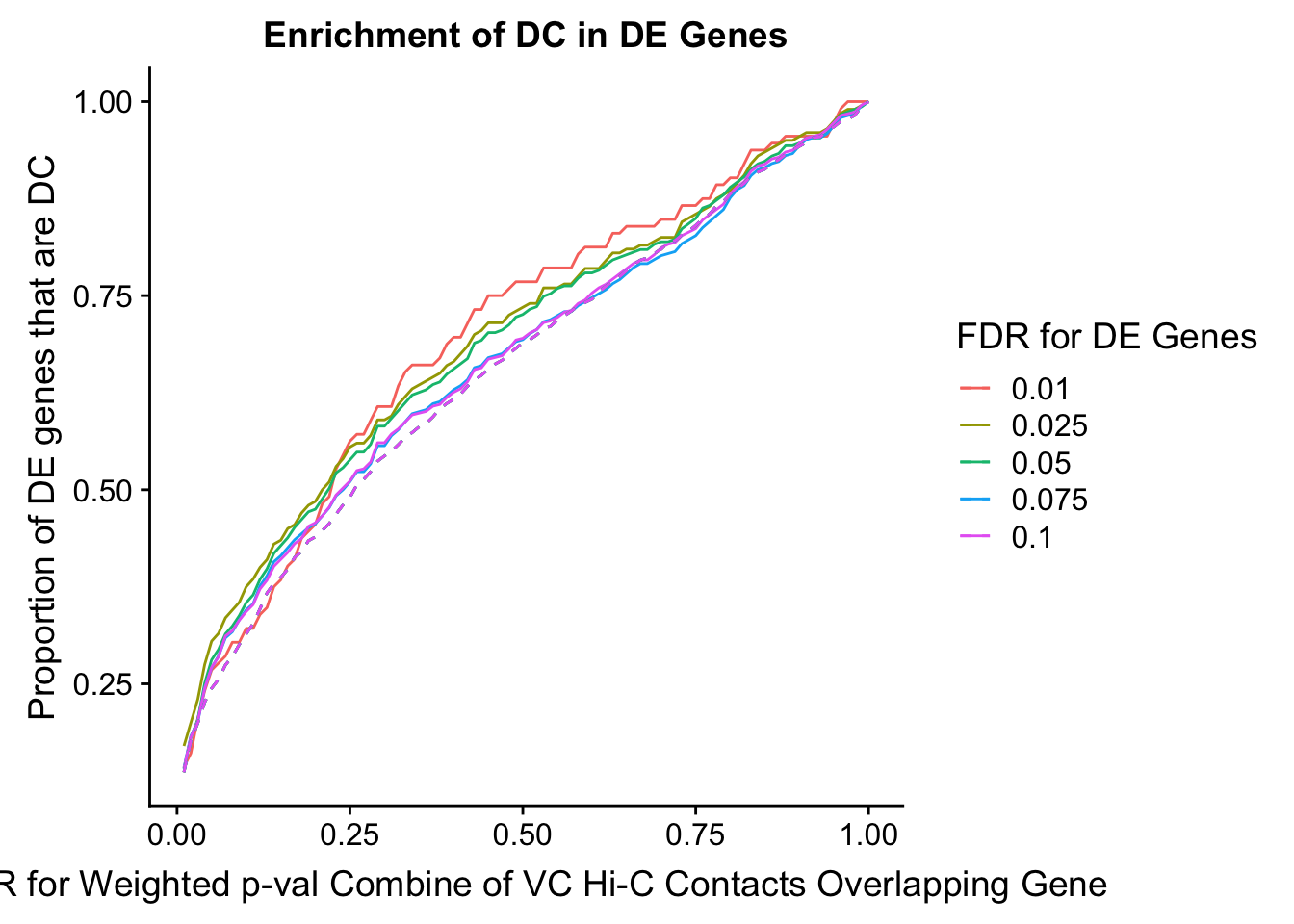
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
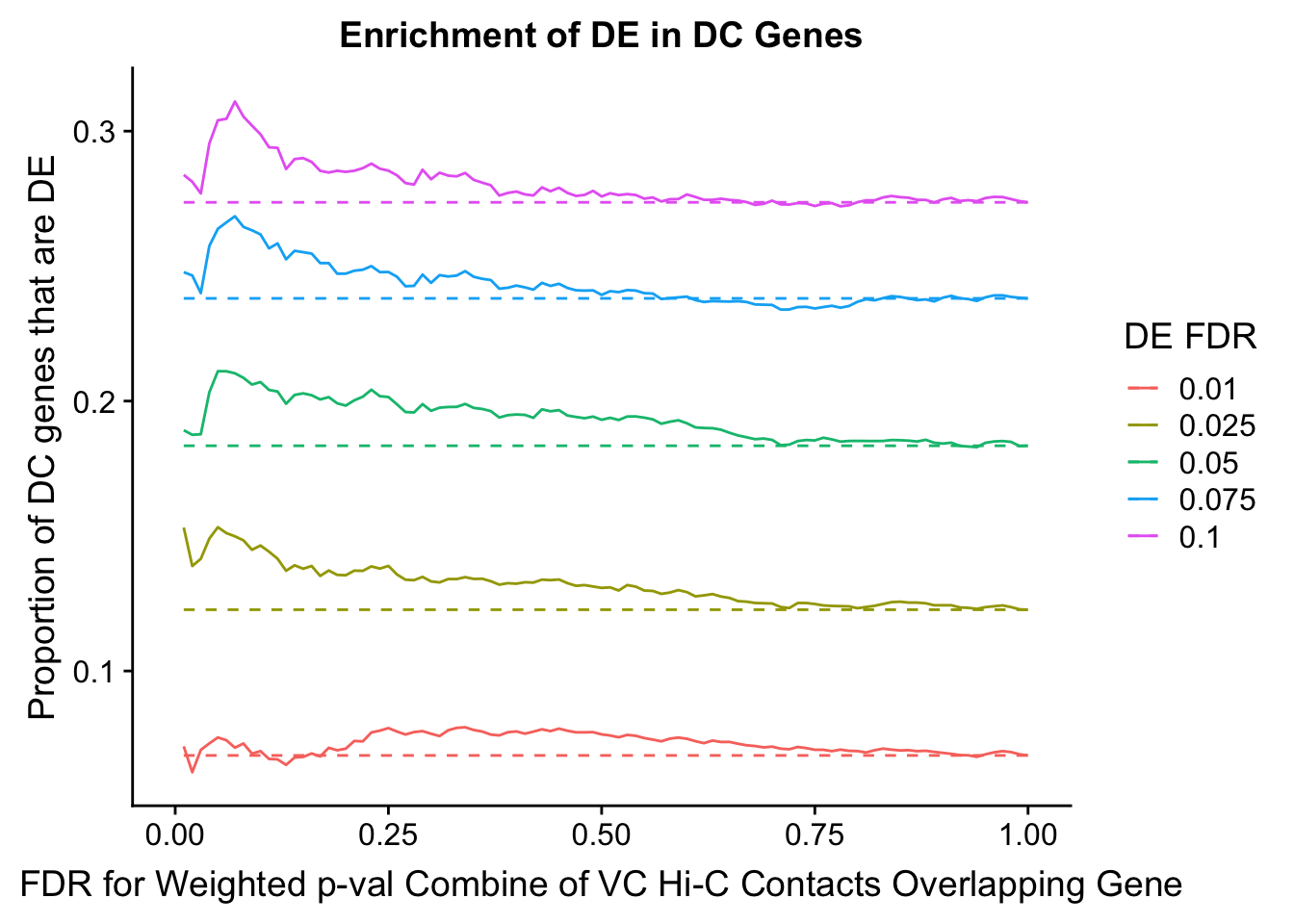
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
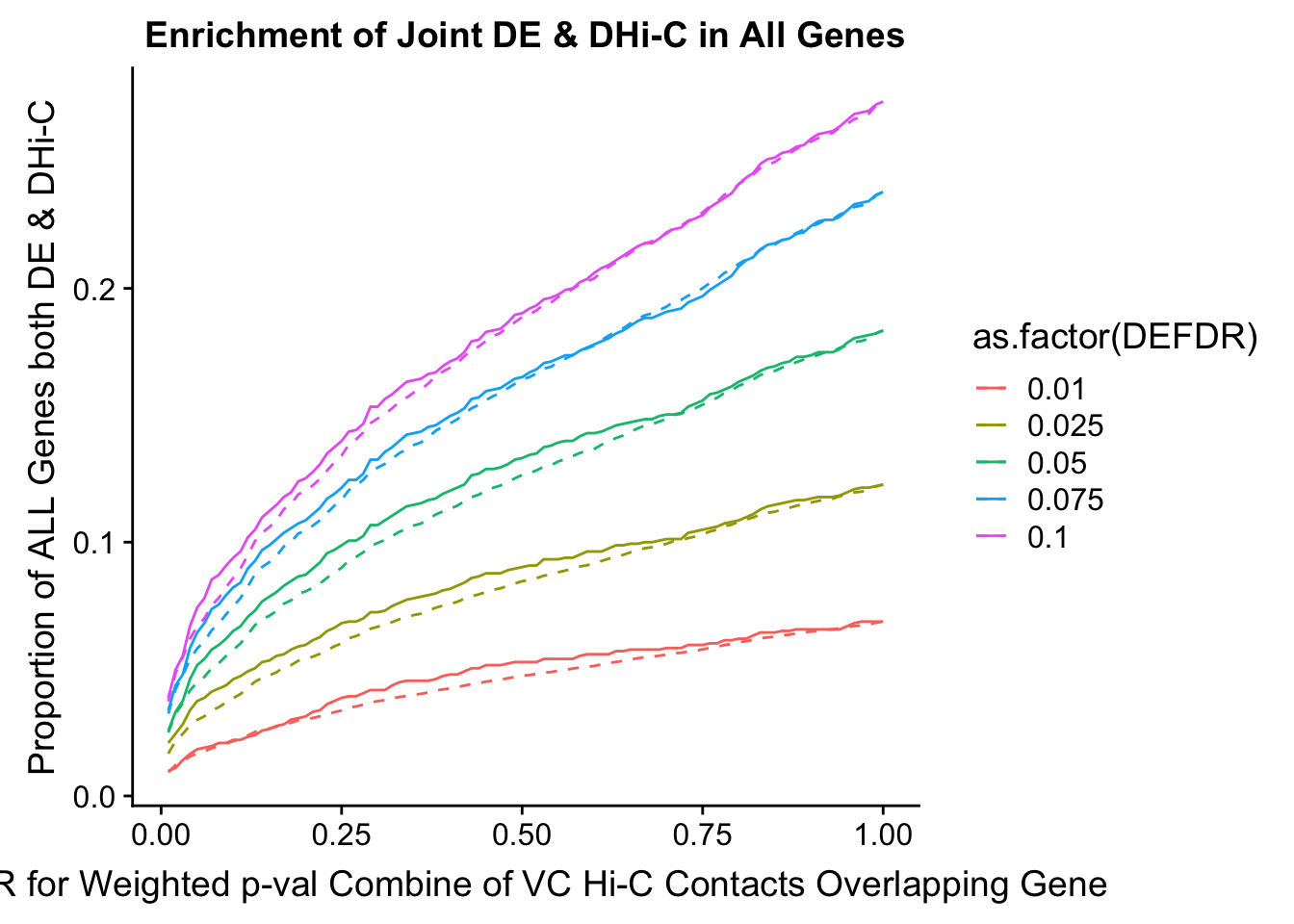
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
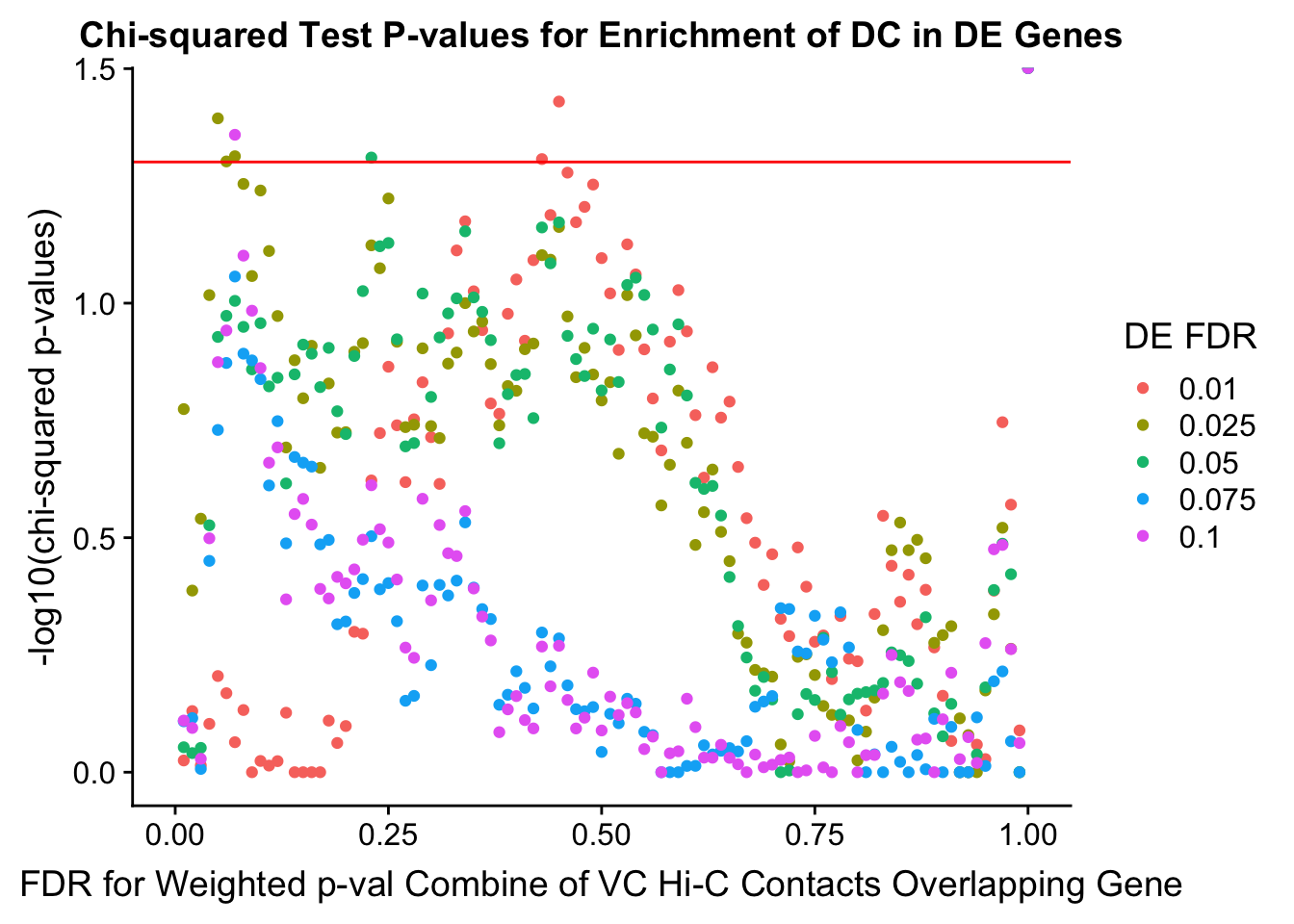
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
DEFDR DHICFDR prop.obs prop.exp chisq.p Dneither DE DHiC Dboth
101 0.025 0.01 0.170 0.1361963 0.16820646 1242 166 188 34
102 0.025 0.02 0.200 0.1766871 0.40998168 1182 160 248 40
103 0.025 0.03 0.230 0.1993865 0.28804497 1151 154 279 46
104 0.025 0.04 0.275 0.2263804 0.09611996 1116 145 314 55
105 0.025 0.05 0.305 0.2441718 0.04036150 1093 139 337 61
106 0.025 0.06 0.315 0.2558282 0.04986890 1076 137 354 63
107 0.025 0.07 0.335 0.2742331 0.04861114 1050 133 380 67
108 0.025 0.08 0.345 0.2852761 0.05569076 1034 131 396 69
109 0.025 0.09 0.355 0.3006135 0.08753096 1011 129 419 71
110 0.025 0.10 0.375 0.3141104 0.05751687 993 125 437 75
111 0.025 0.11 0.385 0.3276074 0.07741230 973 123 457 77
112 0.025 0.12 0.400 0.3466258 0.10650832 945 120 485 80
113 0.025 0.13 0.410 0.3668712 0.20307639 914 118 516 82
114 0.025 0.14 0.430 0.3791411 0.13233854 898 114 532 86
115 0.025 0.15 0.435 0.3871166 0.15949057 886 113 544 87
116 0.025 0.16 0.450 0.3975460 0.12327131 872 110 558 90
117 0.025 0.17 0.455 0.4128834 0.22440299 848 109 582 91
118 0.025 0.18 0.470 0.4202454 0.14832384 839 106 591 94
119 0.025 0.19 0.480 0.4343558 0.18877181 818 104 612 96
120 0.025 0.20 0.485 0.4392638 0.18838729 811 103 619 97
121 0.025 0.21 0.500 0.4472393 0.12694290 801 100 629 100
122 0.025 0.22 0.510 0.4564417 0.12169070 788 98 642 102
123 0.025 0.23 0.530 0.4687117 0.07528138 772 94 658 106
124 0.025 0.24 0.540 0.4803681 0.08424471 755 92 675 108
125 0.025 0.25 0.555 0.4901840 0.05981596 742 89 688 111
126 0.025 0.26 0.560 0.5061350 0.12085100 717 88 713 112
127 0.025 0.27 0.560 0.5134969 0.18376011 705 88 725 112
128 0.025 0.28 0.570 0.5233129 0.18161708 691 86 739 114
129 0.025 0.29 0.590 0.5368098 0.12481303 673 82 757 118
130 0.025 0.30 0.590 0.5435583 0.18286234 662 82 768 118
131 0.025 0.31 0.595 0.5496933 0.19391371 653 81 777 119
132 0.025 0.32 0.610 0.5582822 0.13453521 642 78 788 122
133 0.025 0.33 0.620 0.5674847 0.12743754 629 76 801 124
134 0.025 0.34 0.630 0.5736196 0.09997385 621 74 809 126
135 0.025 0.35 0.635 0.5809816 0.11490102 610 73 820 127
136 0.025 0.36 0.640 0.5852761 0.10949204 604 72 826 128
137 0.025 0.37 0.645 0.5938650 0.13485231 591 71 839 129
138 0.025 0.38 0.650 0.6042945 0.18218993 575 70 855 130
139 0.025 0.39 0.660 0.6110429 0.15020219 566 68 864 132
140 0.025 0.40 0.665 0.6165644 0.15374036 558 67 872 133
141 0.025 0.41 0.675 0.6233129 0.12535714 549 65 881 135
142 0.025 0.42 0.685 0.6331288 0.12193110 535 63 895 137
143 0.025 0.43 0.700 0.6417178 0.07900054 524 60 906 140
144 0.025 0.44 0.705 0.6472393 0.08078255 516 59 914 141
145 0.025 0.45 0.715 0.6552147 0.06879333 505 57 925 143
146 0.025 0.46 0.715 0.6619632 0.10673215 494 57 936 143
147 0.025 0.47 0.715 0.6668712 0.14382821 486 57 944 143
148 0.025 0.48 0.725 0.6748466 0.12454137 475 55 955 145
149 0.025 0.49 0.730 0.6822086 0.14191980 464 54 966 146
150 0.025 0.50 0.735 0.6895706 0.16122648 453 53 977 147
151 0.025 0.51 0.740 0.6932515 0.14739909 448 52 982 148
152 0.025 0.52 0.740 0.6993865 0.20946513 438 52 992 148
153 0.025 0.53 0.760 0.7073620 0.09613652 429 48 1001 152
154 0.025 0.54 0.760 0.7104294 0.11712869 424 48 1006 152
155 0.025 0.55 0.760 0.7184049 0.18938560 411 48 1019 152
156 0.025 0.56 0.765 0.7239264 0.19264943 403 47 1027 153
157 0.025 0.57 0.765 0.7300613 0.26989942 393 47 1037 153
158 0.025 0.58 0.775 0.7368098 0.22106323 384 45 1046 155
159 0.025 0.59 0.785 0.7411043 0.15360830 379 43 1051 157
160 0.025 0.60 0.785 0.7453988 0.19848078 372 43 1058 157
161 0.025 0.61 0.785 0.7546012 0.32763762 357 43 1073 157
162 0.025 0.62 0.795 0.7619632 0.27897785 347 41 1083 159
163 0.025 0.63 0.805 0.7687117 0.22631770 338 39 1092 161
164 0.025 0.64 0.805 0.7742331 0.30733598 329 39 1101 161
165 0.025 0.65 0.810 0.7822086 0.35486570 317 38 1113 162
166 0.025 0.66 0.810 0.7895706 0.50660137 305 38 1125 162
167 0.025 0.67 0.815 0.7957055 0.52939442 296 37 1134 163
168 0.025 0.68 0.815 0.7987730 0.60518037 291 37 1139 163
169 0.025 0.69 0.820 0.8042945 0.61527420 283 36 1147 164
170 0.025 0.70 0.825 0.8098160 0.62555090 275 35 1155 165
171 0.025 0.71 0.825 0.8184049 0.87257649 261 35 1169 165
172 0.025 0.72 0.825 0.8208589 0.94847915 257 35 1173 165
173 0.025 0.73 0.845 0.8282209 0.56759982 249 31 1181 169
174 0.025 0.74 0.850 0.8331288 0.56060003 242 30 1188 170
175 0.025 0.75 0.855 0.8404908 0.62044691 231 29 1199 171
176 0.025 0.76 0.860 0.8490798 0.72247372 218 28 1212 172
177 0.025 0.77 0.865 0.8552147 0.75458565 209 27 1221 173
178 0.025 0.78 0.875 0.8656442 0.76148539 194 25 1236 175
179 0.025 0.79 0.880 0.8711656 0.77527575 186 24 1244 176
180 0.025 0.80 0.885 0.8809816 0.94355633 171 23 1259 177
181 0.025 0.81 0.895 0.8877301 0.81954560 162 21 1268 179
182 0.025 0.82 0.905 0.8944785 0.69341806 153 19 1277 181
183 0.025 0.83 0.920 0.9042945 0.49792688 140 16 1290 184
184 0.025 0.84 0.930 0.9092025 0.33628193 134 14 1296 186
185 0.025 0.85 0.935 0.9128834 0.29358873 129 13 1301 187
186 0.025 0.86 0.940 0.9202454 0.33622659 118 12 1312 188
187 0.025 0.87 0.945 0.9251534 0.31958057 111 11 1319 189
188 0.025 0.88 0.950 0.9319018 0.34984405 101 10 1329 190
189 0.025 0.89 0.950 0.9374233 0.52988575 92 10 1338 190
190 0.025 0.90 0.955 0.9423313 0.51013994 85 9 1345 191
191 0.025 0.91 0.960 0.9472393 0.48830875 78 8 1352 192
192 0.025 0.92 0.960 0.9533742 0.76764143 68 8 1362 192
193 0.025 0.93 0.960 0.9546012 0.83347787 66 8 1364 192
194 0.025 0.94 0.965 0.9625767 1.00000000 54 7 1376 193
195 0.025 0.95 0.975 0.9674847 0.66941736 48 5 1382 195
196 0.025 0.96 0.985 0.9748466 0.46054429 38 3 1392 197
197 0.025 0.97 0.990 0.9773006 0.30116605 35 2 1395 198
198 0.025 0.98 0.990 0.9822086 0.54559553 27 2 1403 198
199 0.025 0.99 0.995 0.9938650 1.00000000 9 1 1421 199
200 0.025 1.00 1.000 1.0000000 0.00000000 0 0 1430 200enrichment.plotter(gene.hic.KR, "weighted_Z.ALLvar.H", "adj.P.Val", "FDR for Weighted p-val Combine of KR Hi-C Contacts Overlapping Gene", xmax=1)Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect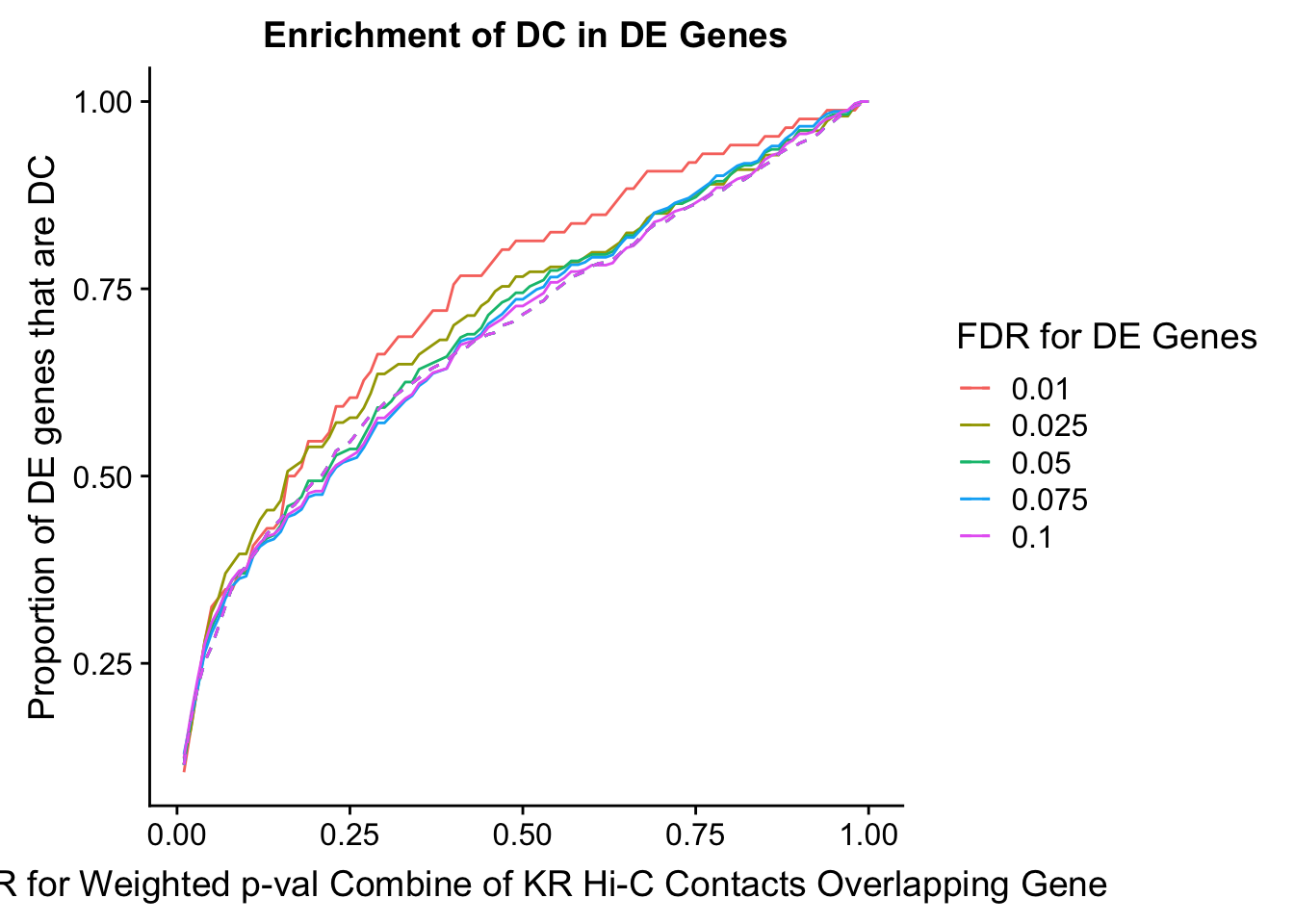
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
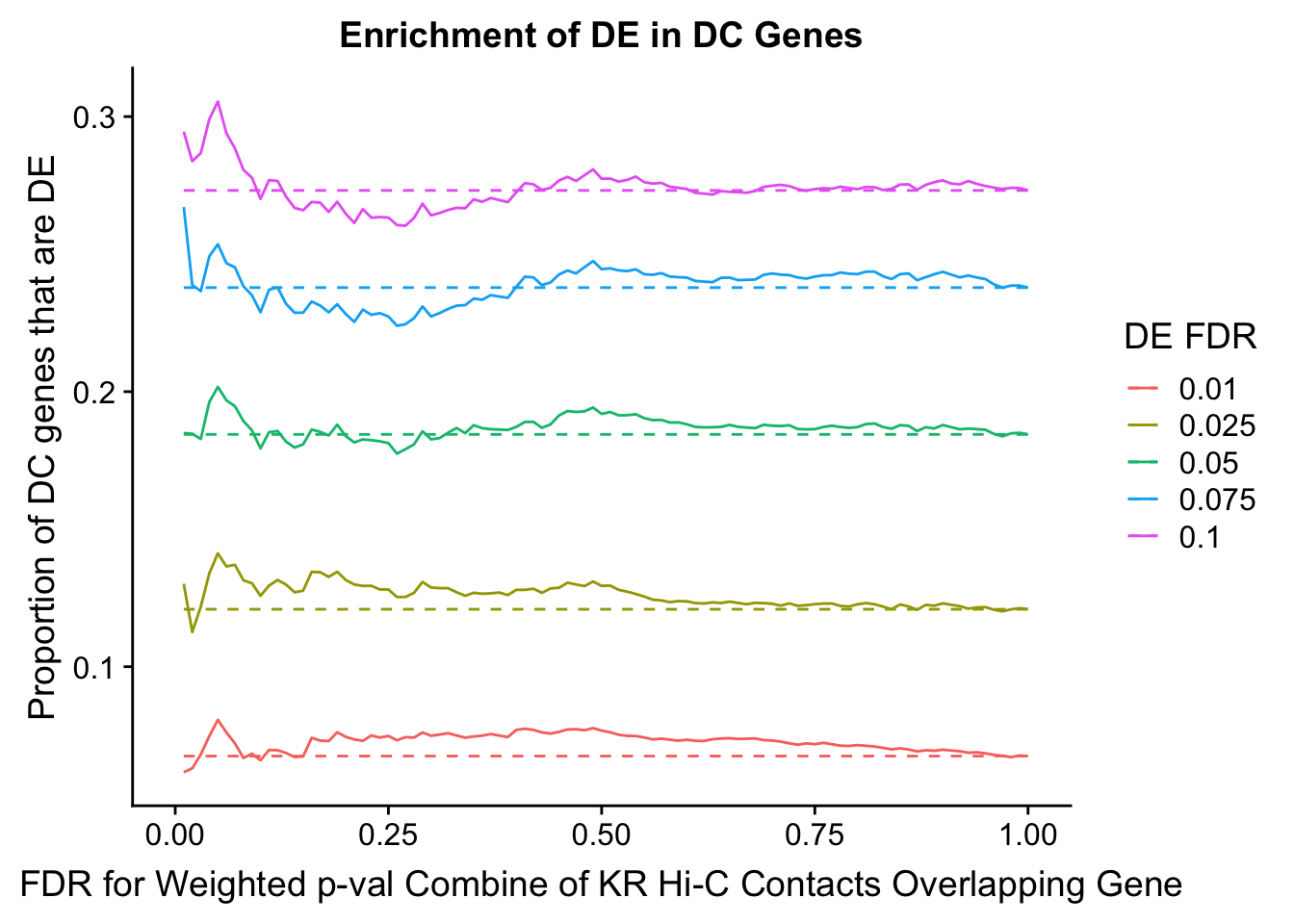
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
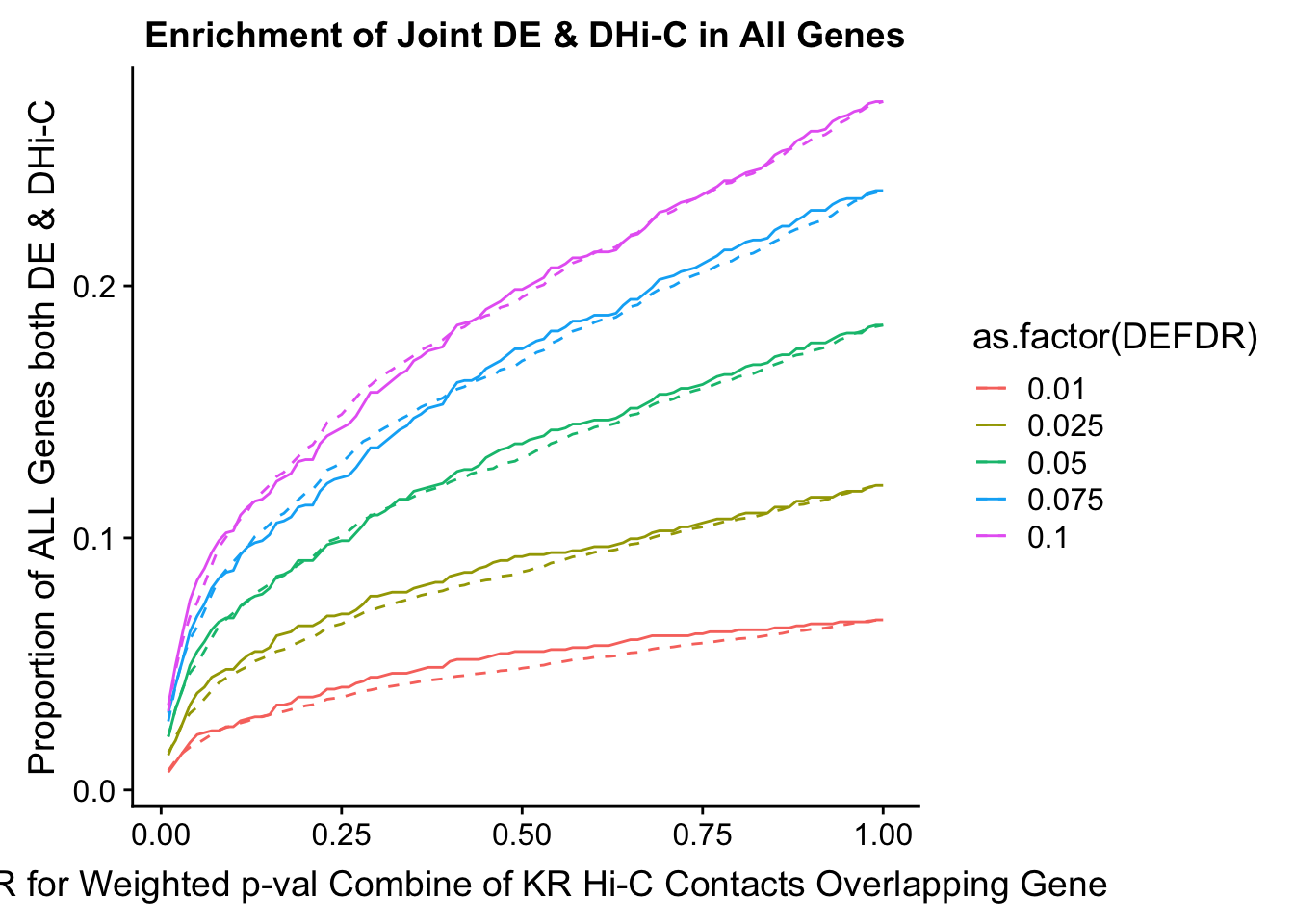
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
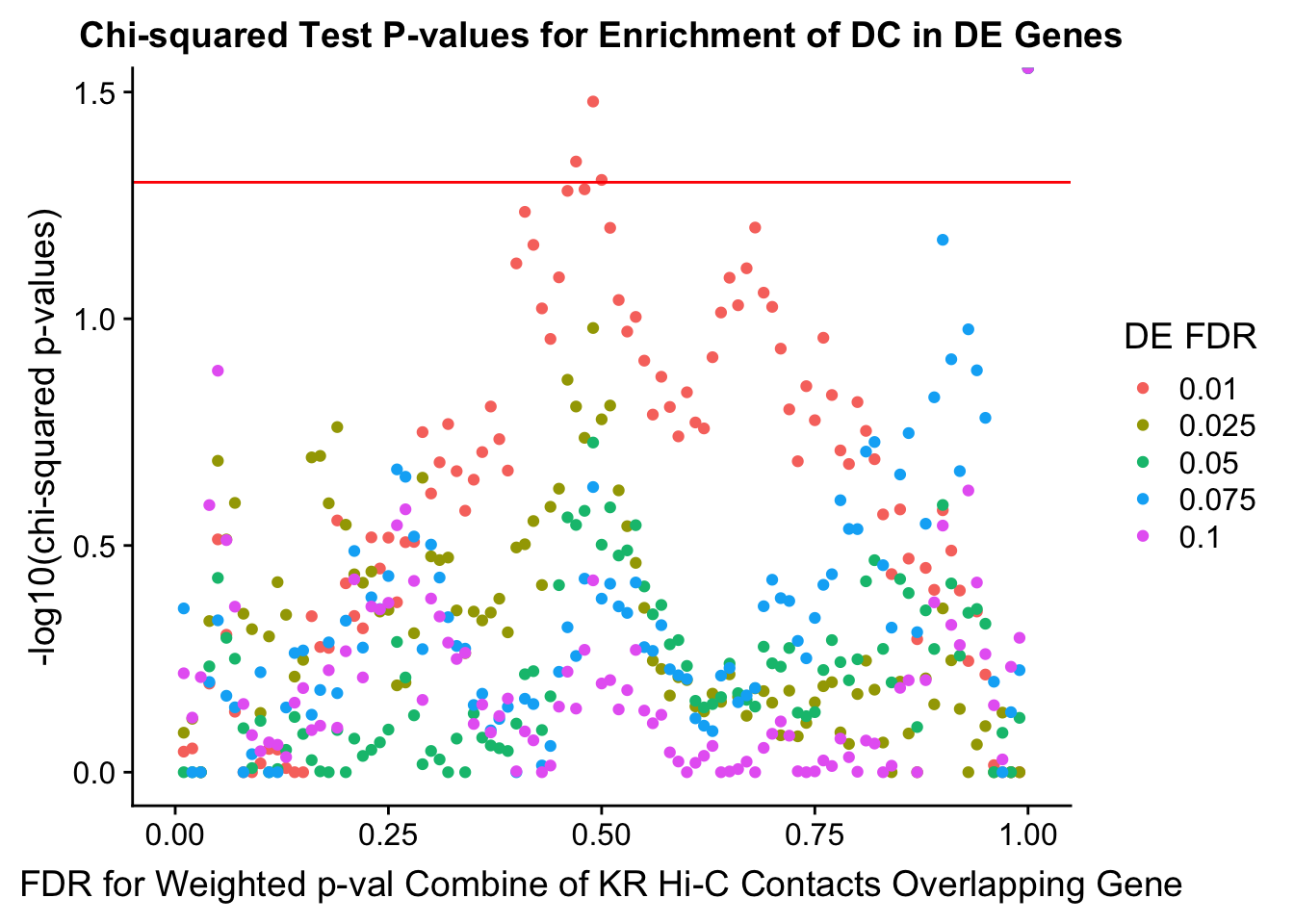
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
DEFDR DHICFDR prop.obs prop.exp chisq.p Dneither DE DHiC Dboth
101 0.025 0.01 0.1233766 0.1145997 0.8182620 993 135 127 19
102 0.025 0.02 0.1623377 0.1742543 0.7622657 923 129 197 25
103 0.025 0.03 0.2207792 0.2189953 1.0000000 875 120 245 34
104 0.025 0.04 0.2792208 0.2519623 0.4641496 842 111 278 43
105 0.025 0.05 0.3181818 0.2723705 0.2057056 822 105 298 49
106 0.025 0.06 0.3376623 0.2990581 0.3067275 791 102 329 52
107 0.025 0.07 0.3701299 0.3265306 0.2547448 761 97 359 57
108 0.025 0.08 0.3831169 0.3524333 0.4471751 730 95 390 59
109 0.025 0.09 0.3961039 0.3673469 0.4836948 713 93 407 61
110 0.025 0.10 0.3961039 0.3806907 0.7401660 696 93 424 61
111 0.025 0.11 0.4220779 0.3940345 0.5018118 683 89 437 65
112 0.025 0.12 0.4415584 0.4058085 0.3809923 671 86 449 68
113 0.025 0.13 0.4545455 0.4230769 0.4496179 651 84 469 70
114 0.025 0.14 0.4545455 0.4324961 0.6154436 639 84 481 70
115 0.025 0.15 0.4675325 0.4427002 0.5651729 628 82 492 72
116 0.025 0.16 0.5064935 0.4552590 0.2021724 618 76 502 78
117 0.025 0.17 0.5129870 0.4615385 0.2006405 611 75 509 79
118 0.025 0.18 0.5194805 0.4733124 0.2552115 597 74 523 80
119 0.025 0.19 0.5389610 0.4843014 0.1733216 586 71 534 83
120 0.025 0.20 0.5389610 0.4952904 0.2845757 572 71 548 83
121 0.025 0.21 0.5389610 0.5015699 0.3660841 564 71 556 83
122 0.025 0.22 0.5519481 0.5156986 0.3820981 548 69 572 85
123 0.025 0.23 0.5714286 0.5337520 0.3609968 528 66 592 88
124 0.025 0.24 0.5714286 0.5392465 0.4423027 521 66 599 88
125 0.025 0.25 0.5779221 0.5455259 0.4384429 514 65 606 89
126 0.025 0.26 0.5779221 0.5572998 0.6433700 499 65 621 89
127 0.025 0.27 0.5909091 0.5698587 0.6341147 485 63 635 91
128 0.025 0.28 0.6103896 0.5816327 0.4936861 473 60 647 94
129 0.025 0.29 0.6363636 0.5879121 0.2241600 469 56 651 98
130 0.025 0.30 0.6363636 0.5973312 0.3341709 457 56 663 98
131 0.025 0.31 0.6428571 0.6043956 0.3405045 449 55 671 99
132 0.025 0.32 0.6493506 0.6106750 0.3362088 442 54 678 100
133 0.025 0.33 0.6493506 0.6177394 0.4397866 433 54 687 100
134 0.025 0.34 0.6493506 0.6240188 0.5461988 425 54 695 100
135 0.025 0.35 0.6623377 0.6310832 0.4423350 418 52 702 102
136 0.025 0.36 0.6688312 0.6389325 0.4626945 409 51 711 103
137 0.025 0.37 0.6753247 0.6444270 0.4445516 403 50 717 104
138 0.025 0.38 0.6818182 0.6491366 0.4143167 398 49 722 105
139 0.025 0.39 0.6818182 0.6538462 0.4915362 392 49 728 105
140 0.025 0.40 0.7012987 0.6624804 0.3194243 384 46 736 108
141 0.025 0.41 0.7077922 0.6687598 0.3142592 377 45 743 109
142 0.025 0.42 0.7142857 0.6726845 0.2793232 373 44 747 110
143 0.025 0.43 0.7142857 0.6805338 0.3865395 363 44 757 110
144 0.025 0.44 0.7272727 0.6844584 0.2597976 360 42 760 112
145 0.025 0.45 0.7337662 0.6891680 0.2370065 355 41 765 113
146 0.025 0.46 0.7467532 0.6915228 0.1363123 354 39 766 115
147 0.025 0.47 0.7532468 0.7009419 0.1561415 343 38 777 116
148 0.025 0.48 0.7532468 0.7040816 0.1830408 339 38 781 116
149 0.025 0.49 0.7662338 0.7072214 0.1047996 337 36 783 118
150 0.025 0.50 0.7662338 0.7158556 0.1666235 326 36 794 118
151 0.025 0.51 0.7727273 0.7213501 0.1553555 320 35 800 119
152 0.025 0.52 0.7727273 0.7299843 0.2390183 309 35 811 119
153 0.025 0.53 0.7727273 0.7339089 0.2867058 304 35 816 119
154 0.025 0.54 0.7792208 0.7448980 0.3454087 291 34 829 120
155 0.025 0.55 0.7792208 0.7503925 0.4340206 284 34 836 120
156 0.025 0.56 0.7792208 0.7574568 0.5674631 275 34 845 120
157 0.025 0.57 0.7857143 0.7653061 0.5919963 266 33 854 121
158 0.025 0.58 0.7857143 0.7692308 0.6775451 261 33 859 121
159 0.025 0.59 0.7922078 0.7731554 0.6174154 257 32 863 122
160 0.025 0.60 0.7987013 0.7802198 0.6263059 249 31 871 123
161 0.025 0.61 0.7987013 0.7841444 0.7159693 244 31 876 123
162 0.025 0.62 0.7987013 0.7849294 0.7345742 243 31 877 123
163 0.025 0.63 0.8051948 0.7888540 0.6710996 239 30 881 124
164 0.025 0.64 0.8116883 0.7967033 0.6994709 230 29 890 125
165 0.025 0.65 0.8246753 0.8061224 0.6083481 220 27 900 127
166 0.025 0.66 0.8246753 0.8092622 0.6819088 216 27 904 127
167 0.025 0.67 0.8311688 0.8186813 0.7509079 205 26 915 128
168 0.025 0.68 0.8441558 0.8281005 0.6531980 195 24 925 130
169 0.025 0.69 0.8506494 0.8351648 0.6624428 187 23 933 131
170 0.025 0.70 0.8506494 0.8367347 0.7024542 185 23 935 131
171 0.025 0.71 0.8506494 0.8414443 0.8290641 179 23 941 131
172 0.025 0.72 0.8636364 0.8485086 0.6609532 172 21 948 133
173 0.025 0.73 0.8636364 0.8547881 0.8333300 164 21 956 133
174 0.025 0.74 0.8701299 0.8594976 0.7784900 159 20 961 134
175 0.025 0.75 0.8766234 0.8634223 0.7012304 155 19 965 135
176 0.025 0.76 0.8831169 0.8681319 0.6461094 150 18 970 136
177 0.025 0.77 0.8896104 0.8744113 0.6330983 143 17 977 137
178 0.025 0.78 0.8896104 0.8806907 0.8168264 135 17 985 137
179 0.025 0.79 0.8896104 0.8822606 0.8661953 133 17 987 137
180 0.025 0.80 0.9025974 0.8893250 0.6723274 126 15 994 139
181 0.025 0.81 0.9090909 0.8924647 0.5675824 123 14 997 140
182 0.025 0.82 0.9090909 0.8956044 0.6576012 119 14 1001 140
183 0.025 0.83 0.9090909 0.9018838 0.8601319 111 14 1009 140
184 0.025 0.84 0.9090909 0.9089482 1.0000000 102 14 1018 140
185 0.025 0.85 0.9285714 0.9152276 0.6313866 97 11 1023 143
186 0.025 0.86 0.9285714 0.9207221 0.8216118 90 11 1030 143
187 0.025 0.87 0.9285714 0.9301413 1.0000000 78 11 1042 143
188 0.025 0.88 0.9480519 0.9356358 0.6209248 74 8 1046 146
189 0.025 0.89 0.9480519 0.9379906 0.7084186 71 8 1049 146
190 0.025 0.90 0.9610390 0.9442700 0.4352894 65 6 1055 148
191 0.025 0.91 0.9610390 0.9481947 0.5665503 60 6 1060 148
192 0.025 0.92 0.9610390 0.9521193 0.7250980 55 6 1065 148
193 0.025 0.93 0.9610390 0.9591837 1.0000000 46 6 1074 148
194 0.025 0.94 0.9740260 0.9686028 0.8688029 36 4 1084 150
195 0.025 0.95 0.9805195 0.9740973 0.7913314 30 3 1090 151
196 0.025 0.96 0.9805195 0.9819466 1.0000000 20 3 1100 151
197 0.025 0.97 0.9805195 0.9866562 0.7388667 14 3 1106 151
198 0.025 0.98 0.9935065 0.9937206 1.0000000 7 1 1113 153
199 0.025 0.99 1.0000000 0.9968603 1.0000000 4 0 1116 154
200 0.025 1.00 1.0000000 1.0000000 0.0000000 0 0 1120 154#VCs appear to be better on both, but we'll see in final combining of it--pretty sure it's the second set of doing it that's wthe way I did it for HOMER, but double-check.
#FIGS3 for paper:
pap.enrichment.plotter <- function(df, HiC_col, DE_col, xlab, xmax=0.3, i=c(0.01, 0.025, 0.05, 0.075, 0.1), k=seq(0.01, 1, 0.01), significance=FALSE){
enrich.table <- data.frame(DEFDR = c(rep(i[1], 100), rep(i[2], 100), rep(i[3], 100), rep(i[4], 100), rep(i[5], 100)), DHICFDR=rep(k, 5), prop.obs=NA, prop.exp=NA, chisq.p=NA, Dneither=NA, DE=NA, DHiC=NA, Dboth=NA)
for(de.FDR in i){
for(hic.FDR in k){
enrich.table[which(enrich.table$DEFDR==de.FDR&enrich.table$DHICFDR==hic.FDR), 3:9] <- prop.calculator(df[,DE_col], df[,HiC_col], de.FDR, hic.FDR)
}
}
des.enriched <- ggplot(data=enrich.table, aes(x=DHICFDR, y=prop.obs, group=as.factor(DEFDR), color=as.factor(DEFDR))) +geom_line()+ geom_line(aes(y=prop.exp), linetype="dashed", size=0.5) + ggtitle("Enrichment of DC in DE Genes") + xlab(xlab) + ylab("Proportion of DE genes that are DC") + guides(color=guide_legend(title="FDR for DE Genes"))
dhics.enriched <- ggplot(data=enrich.table, aes(x=DHICFDR, y=Dboth/(Dboth+DHiC), group=as.factor(DEFDR), color=as.factor(DEFDR))) + geom_line() + geom_line(aes(y=(((((DE+Dboth)/(Dneither+DE+DHiC+Dboth))*((DHiC+Dboth)/(Dneither+DE+DHiC+Dboth)))*(Dneither+DE+DHiC+Dboth))/(DHiC+Dboth))), linetype="dashed") + ylab("Proportion of DC genes that are DE") +xlab(xlab) + ggtitle("Enrichment of DE in DC Genes") + coord_cartesian(xlim=c(0, xmax), ylim=c(0.05, 0.32)) + guides(color=FALSE)
joint.enriched <- ggplot(data=enrich.table, aes(x=DHICFDR, y=Dboth/(Dneither+DE+DHiC+Dboth), group=as.factor(DEFDR), color=as.factor(DEFDR))) + geom_line() + ylab("Proportion of ALL Genes both DE & DHi-C") + xlab(xlab) + geom_line(aes(y=((DE+Dboth)/(Dneither+DE+DHiC+Dboth))*((DHiC+Dboth)/(Dneither+DE+DHiC+Dboth))), linetype="dashed") + ggtitle("Enrichment of Joint DE & DHi-C in All Genes")
chisq.p <- ggplot(data=enrich.table, aes(x=DHICFDR, y=-log10(chisq.p), group=as.factor(DEFDR), color=as.factor(DEFDR))) + geom_point() + geom_hline(yintercept=-log10(0.05), color="red") + ggtitle("Chi-squared Test P-values") + xlab(xlab) + ylab("-log10(chi-squared p-values)") + coord_cartesian(xlim=c(0, xmax), ylim=c(0, 3.2)) + guides(color=guide_legend(title="DE FDR"))
if(significance==TRUE){
return(chisq.p)
}
else{
return(dhics.enriched)
}
}
FIGS3A <- pap.enrichment.plotter(gene.hic.VC, "min_FDR.H", "adj.P.Val", "Minimum FDR of Contacts", xmax=1) #FIGS3AWarning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrectFIGS3B <- pap.enrichment.plotter(gene.hic.VC, "min_FDR.H", "adj.P.Val", "Minimum FDR of Contacts", xmax=1, significance = TRUE) #FIGS3BWarning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrectFIGS3C <- pap.enrichment.plotter(gene.hic.VC, "weighted_Z.ALLvar.H", "adj.P.Val", "FDR for Weighted P-val Combination", xmax=1) #FIGS3CWarning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrectFIGS3D <- pap.enrichment.plotter(gene.hic.VC, "weighted_Z.ALLvar.H", "adj.P.Val", "FDR for Weighted P-val Combination", xmax=1, significance = TRUE) #FIGS3DWarning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrect
Warning in chisq.test(mytable): Chi-squared approximation may be incorrectFIGS3 <- plot_grid(FIGS3A, FIGS3B, FIGS3C, FIGS3D, labels=c("A", "B", "C", "D"), align="h", rel_widths=c(1, 1.2))
save_plot("~/Desktop/FIGS3.png", FIGS3, nrow=2, ncol=2)Now that I’ve seen some enrichment of differential contact (DC) in differential expression (DE) based on my linear modeling results from before, I would like to further quantify this effect. In order to do so, I now extract log2 observed/expected homer-normalized contact frequency values and RPKM expression values for each gene/bin set, so I can look at correlations of these values and assess their explanatory power.
###Contact Frequency Extraction In this section, I proceed to create a function in order to extract the Hi-C interaction frequency values for the different types of summaries I’ve made gene overlaps with above. This allows for operations to be performed separately utilizing different summaries of Hi-C contacts.
######Get a df with the H and C coordinates of the hits, and the IF values from homer. This subset df makes things easier to extract. Both for VC and KR.
###VC
contacts.VC <- data.frame(h1=data.VC$H1, h2=data.VC$H2, c1=data.VC$C1, c2=data.VC$C2, A_21792_HIC=data.VC$`A-21792_VC`, B_28126_HIC=data.VC$`B-28126_VC`, C_3649_HIC=data.VC$`C-3649_VC`, D_40300_HIC=data.VC$`D-40300_VC`, E_28815_HIC=data.VC$`E-28815_VC`, F_28834_HIC=data.VC$`F-28834_VC`, G_3624_HIC=data.VC$`G-3624_VC`, H_3651_HIC=data.VC$`H-3651_VC`, stringsAsFactors = FALSE)
newH1 <- as.numeric(gsub(".*-", "", contacts.VC$h1))
newH2 <- as.numeric(gsub(".*-", "", contacts.VC$h2))
lower.HID <- ifelse(newH1<newH2, contacts.VC$h1, contacts.VC$h2)
higher.HID2 <- ifelse(newH1<newH2, contacts.VC$h2, contacts.VC$h1)
contacts.VC$hpair <- paste(lower.HID, higher.HID2, sep="_")
newC1 <- as.numeric(gsub(".*-", "", contacts.VC$c1))
newC2 <- as.numeric(gsub(".*-", "", contacts.VC$c2))
lower.CID <- ifelse(newC1<newC2, contacts.VC$c1, contacts.VC$c2)
higher.CID2 <- ifelse(newC1<newC2, contacts.VC$c2, contacts.VC$c1)
contacts.VC$cpair <- paste(lower.CID, higher.CID2, sep="_")
###KR
contacts.KR <- data.frame(h1=data.KR$H1, h2=data.KR$H2, c1=data.KR$C1, c2=data.KR$C2, A_21792_HIC=data.KR$`A-21792_KR`, B_28126_HIC=data.KR$`B-28126_KR`, C_3649_HIC=data.KR$`C-3649_KR`, D_40300_HIC=data.KR$`D-40300_KR`, E_28815_HIC=data.KR$`E-28815_KR`, F_28834_HIC=data.KR$`F-28834_KR`, G_3624_HIC=data.KR$`G-3624_KR`, H_3651_HIC=data.KR$`H-3651_KR`, stringsAsFactors = FALSE)
newH1 <- as.numeric(gsub(".*-", "", contacts.KR$h1))
newH2 <- as.numeric(gsub(".*-", "", contacts.KR$h2))
lower.HID <- ifelse(newH1<newH2, contacts.KR$h1, contacts.KR$h2)
higher.HID2 <- ifelse(newH1<newH2, contacts.KR$h2, contacts.KR$h1)
contacts.KR$hpair <- paste(lower.HID, higher.HID2, sep="_")
newC1 <- as.numeric(gsub(".*-", "", contacts.KR$c1))
newC2 <- as.numeric(gsub(".*-", "", contacts.KR$c2))
lower.CID <- ifelse(newC1<newC2, contacts.KR$c1, contacts.KR$c2)
higher.CID2 <- ifelse(newC1<newC2, contacts.KR$c2, contacts.KR$c1)
contacts.KR$cpair <- paste(lower.CID, higher.CID2, sep="_")
#Fix column names for both contacts.VC and contacts.KR
colnames(contacts.KR)[5:12] <- colnames(contacts.VC)[5:12] <- c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC")
#A function that takes a dataframe (like gene.hic.filt) and two columns from the dataframe to create a pair vector for the given interaction. First ensures the first bin in a pair is always lowest to make this easier. Then extracts the IF values for that vector from the contacts df created above. This provides me with the appropriate Hi-C data values for the different bin classes we're examining here, so that I can later test them with linear modeling to quantify their effect on expression.
IF.extractor <- function(dataframe, col1, col2, contacts, species, strand=FALSE){
new1 <- as.numeric(gsub(".*-", "", dataframe[,col1]))
if(strand==FALSE){#In the case where I'm not worried about strand, I just work with the second column selected.
new2 <- as.numeric(gsub(".*-", "", dataframe[,col2]))
lower1 <- ifelse(new1<new2, dataframe[,col1], dataframe[,col2])
higher2 <- ifelse(new1<new2, dataframe[,col2], dataframe[,col1]) #Fix all the columns first
if(species=="H"){ #Then depending on species create the pair column and merge to contact info.
dataframe[,"hpair"] <- paste(lower1, higher2, sep="_")
finaldf <- left_join(dataframe, contacts[,c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC", "hpair")], by="hpair")
}
else if(species=="C"){
dataframe[,"cpair"] <- paste(lower1, higher2, sep="_")
finaldf <- left_join(dataframe, contacts[,c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC", "cpair")], by="cpair")
}
}
else if(strand==TRUE){#If dealing with upstream and downstream hits, need to do things separately for genes on the + and - strand.
if(species=="H"){
US <- ifelse(dataframe[,"Hstrand.H"]=="+", dataframe[,"US_bin.H"], dataframe[,"DS_bin.H"]) #Obtain upstream bins depending on strand.
new2 <- as.numeric(gsub(".*-", "", US)) #Now rearrange the pairs to ensure regardless of stream we can find the pair (first mate lower coordinates than 2nd).
lower1 <- ifelse(new1<new2, dataframe[,col1], US)
higher2 <- ifelse(new1<new2, US, dataframe[,col1])
dataframe[,"hpair"] <- paste(lower1, higher2, sep="_")
dataframe[,"USFDR"] <- ifelse(dataframe[,"Hstrand.H"]=="+", dataframe[,"US_FDR.H"], dataframe[,"DS_FDR.H"])
finaldf <- left_join(dataframe, contacts[,c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC", "hpair")], by="hpair")
}
else if(species=="C"){
US <- ifelse(dataframe[,"Hstrand.C"]=="+", dataframe[,"US_bin.C"], dataframe[,"DS_bin.C"]) #Obtain upstream bins depending on strand.
new2 <- as.numeric(gsub(".*-", "", US)) #Now rearrange the pairs to ensure regardless of stream we can find the pair (first mate lower coordinates than 2nd).
lower1 <- ifelse(new1<new2, dataframe[,col1], US)
higher2 <- ifelse(new1<new2, US, dataframe[,col1])
dataframe[,"cpair"] <- paste(lower1, higher2, sep="_")
dataframe[,"USFDR"] <- ifelse(dataframe[,"Hstrand.C"]=="+", dataframe[,"US_FDR.C"], dataframe[,"DS_FDR.C"])
finaldf <- left_join(dataframe, contacts[,c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC", "cpair")], by="cpair")
}
}
#before finally returning, remove rows where we don't have full Hi-C data.
finaldf <- finaldf[complete.cases(finaldf[,c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC")]),]
return(finaldf)
}
#Now I can use the IF.extractor function to make a number of different dataframes for actually testing different IF values with the RPKM expression values.
h_minFDR.KR <- IF.extractor(gene.hic.KR, "HID", "min_FDR_bin.H", contacts.KR, "H")
#contacts.VC$hpair <- gsub("Chr. ", "chr", contacts.VC$hpair)
h_minFDR.VC <- IF.extractor(gene.hic.VC, "HID", "min_FDR_bin.H", contacts.VC, "H") #The issue here lies in having renamed h1 and h2 in contacts.VC earlier!
#h_maxB <- IF.extractor(gene.hic.filt, "HID", "max_B_bin.H", contacts, "H")
#c_maxB <- IF.extractor(gene.hic.filt, "CID", "max_B_bin.C", contacts, "C")
#h_US <- IF.extractor(gene.hic.filt, "HID", "US_bin.H", contacts, "H", TRUE)
#c_US <- IF.extractor(gene.hic.filt, "CID", "US_bin.C", contacts, "C", TRUE)
#Write these out so they can be permuted upon on midway2.
fwrite(h_minFDR.KR, "data/old_mediation_permutations/HiC_covs/juicer/h_minFDR.KR", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
fwrite(h_minFDR.VC, "data/old_mediation_permutations/HiC_covs/juicer/h_minFDR.VC", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(h_maxB, "data/old_mediation_permutations/HiC_covs/h_maxB", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(c_maxB, "data/old_mediation_permutations/HiC_covs/c_maxB", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(h_US, "data/old_mediation_permutations/HiC_covs/h_US", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(c_US, "data/old_mediation_permutations/HiC_covs/c_US", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#Now the same thing, but subsetting down to ONLY the genes that show evidence for DE at 5% FDR.
h_minFDR_DE.KR <- filter(h_minFDR.KR, adj.P.Val<=0.05)
h_minFDR_DE.VC <- filter(h_minFDR.VC, adj.P.Val<=0.05)
#h_maxB_DE <- filter(h_maxB, adj.P.Val<=0.05)
#c_maxB_DE <- filter(c_maxB, adj.P.Val<=0.05)
#h_US_DE <- filter(h_US, adj.P.Val<=0.05)
#c_US_DE <- filter(c_US, adj.P.Val<=0.05)
fwrite(h_minFDR_DE.KR, "data/old_mediation_permutations/HiC_covs/juicer/h_minFDR_DE.KR", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
fwrite(h_minFDR_DE.VC, "data/old_mediation_permutations/HiC_covs/juicer/h_minFDR_DE.VC", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(h_maxB_DE, "data/old_mediation_permutations/HiC_covs/h_maxB_DE", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(c_maxB_DE, "~/Desktop/Hi-C/HiC_covs/c_maxB_DE", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(h_US_DE, "~/Desktop/Hi-C/HiC_covs/h_US_DE", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)
#fwrite(c_US_DE, "~/Desktop/Hi-C/HiC_covs/c_US_DE", quote = FALSE, sep = "\t", na = "NA", col.names = TRUE)Now I use these data frames and join each again on the RPKM table by genes, in order to obtain RPKM expression values for each gene-Hi-C bin set.
###Merging contact frequency values with expression values. In this next section I join each of the data frames created above on the RPKM table by genes, in order to have a merged table with expression values and contact frequencies for each gene-Hi-C bin set. I move then to explore correlations between the values.
#Join all of the previously-made Hi-C interaction frequency tables to the RPKM table by genes, to obtain RPKM values in concert with contact frequency values.
RPKM <- as.data.frame(weighted.data$E)
RPKM$genes <- rownames(RPKM)
hmin.KR <- left_join(h_minFDR.KR, RPKM, by="genes")
hmin.VC <- left_join(h_minFDR.VC, RPKM, by="genes")
#hmaxB <- left_join(h_maxB, RPKM, by="genes")
#cmaxB <- left_join(c_maxB, RPKM, by="genes")
#hUS <- left_join(h_US, RPKM, by="genes")
#cUS <- left_join(c_US, RPKM, by="genes")
#Get the same thing filtered for only DE genes.
hminDE.KR <- filter(hmin.KR, adj.P.Val<=0.05)
hminDE.VC <- filter(hmin.VC, adj.P.Val<=0.05)
#hmaxBDE <- filter(hmaxB, adj.P.Val<=0.05)
#cmaxBDE <- filter(cmaxB, adj.P.Val <=0.05)
#hUSDE <- filter(hUS, adj.P.Val<=0.05)
#cUSDE <- filter(cUS, adj.P.Val<=0.05)
hmin_noDE.KR <- filter(hmin.KR, adj.P.Val>0.05) #Pull out specific set of non-DE hits.
hmin_noDE.VC <- filter(hmin.VC, adj.P.Val>0.05)
#Extract contacts and expression for the contact with the lowest FDR from linear modeling.
KRcontacts <- hmin.KR[,61:68]
KRexprs <- hmin.KR[,69:76]
KRcontactsDE <- hminDE.KR[,61:68]
KRexprsDE <- hminDE.KR[,69:76]
nonDEcontactsKR <- hmin_noDE.KR[,61:68]
nonDEexprsKR <- hmin_noDE.KR[,69:76]
VCcontacts <- hmin.VC[,61:68]
VCexprs <- hmin.VC[,69:76]
VCcontactsDE <- hminDE.VC[,61:68]
VCexprsDE <- hminDE.VC[,69:76]
nonDEcontactsVC <- hmin_noDE.VC[,61:68]
nonDEexprsVC <- hmin_noDE.VC[,69:76]
#Function for calculating gene-wise correlations.
cor.calc <- function(hicdata, exprdata){
cor.exp <- vector(length=nrow(hicdata))
for(i in 1:nrow(hicdata)){
cor.exp[i] <- cor(as.numeric(hicdata[i,]), as.numeric(exprdata[i,]))
}
return(cor.exp)
}
#Calculate correlations for different sets! I've commented out the species-specific calculations here because they just aren't that interesting. If you look at DE dynamics within each species you do NOT get a gorgeous bimodal as you do for across--looks more uniform and messy.
fullcorsKR <- data.frame(cor=cor.calc(KRcontacts, KRexprs), type="all")
fullDEcorsKR <- data.frame(cor=cor.calc(KRcontactsDE, KRexprsDE), type="DE")
fullnoDEcorsKR <- data.frame(cor=cor.calc(nonDEcontactsKR, nonDEexprsKR), type="non-DE")
#VC now
fullcorsVC <- data.frame(cor=cor.calc(VCcontacts, VCexprs), type="all")
fullDEcorsVC <- data.frame(cor=cor.calc(VCcontactsDE, VCexprsDE), type="DE")
fullnoDEcorsVC <- data.frame(cor=cor.calc(nonDEcontactsVC, nonDEexprsVC), type="non-DE")
#Combine these dfs to plot in one gorgeous ggplot!
ggcorsKR <- rbind(fullcorsKR, fullDEcorsKR, fullnoDEcorsKR)
ggcorsVC <- rbind(fullcorsVC, fullDEcorsVC, fullnoDEcorsVC)
#VC then KR
ggplot(data=filter(ggcorsVC, type=="all"|type=="DE"|type=="non-DE")) + stat_density(aes(x=cor, group=type, color=type, y=..scaled..), position="identity", geom="line") + ggtitle("Correlation b/t RPKM Expression and VC Hi-C Contact Frequency") + xlab("Pearson Correlations b/t RPKM Expression and VC Hi-C Contact Frequency") + ylab("Density") + scale_color_manual("Gene Set", values=c("red", "blue", "green"), labels=c("All genes", "DE genes", "non-DE genes"))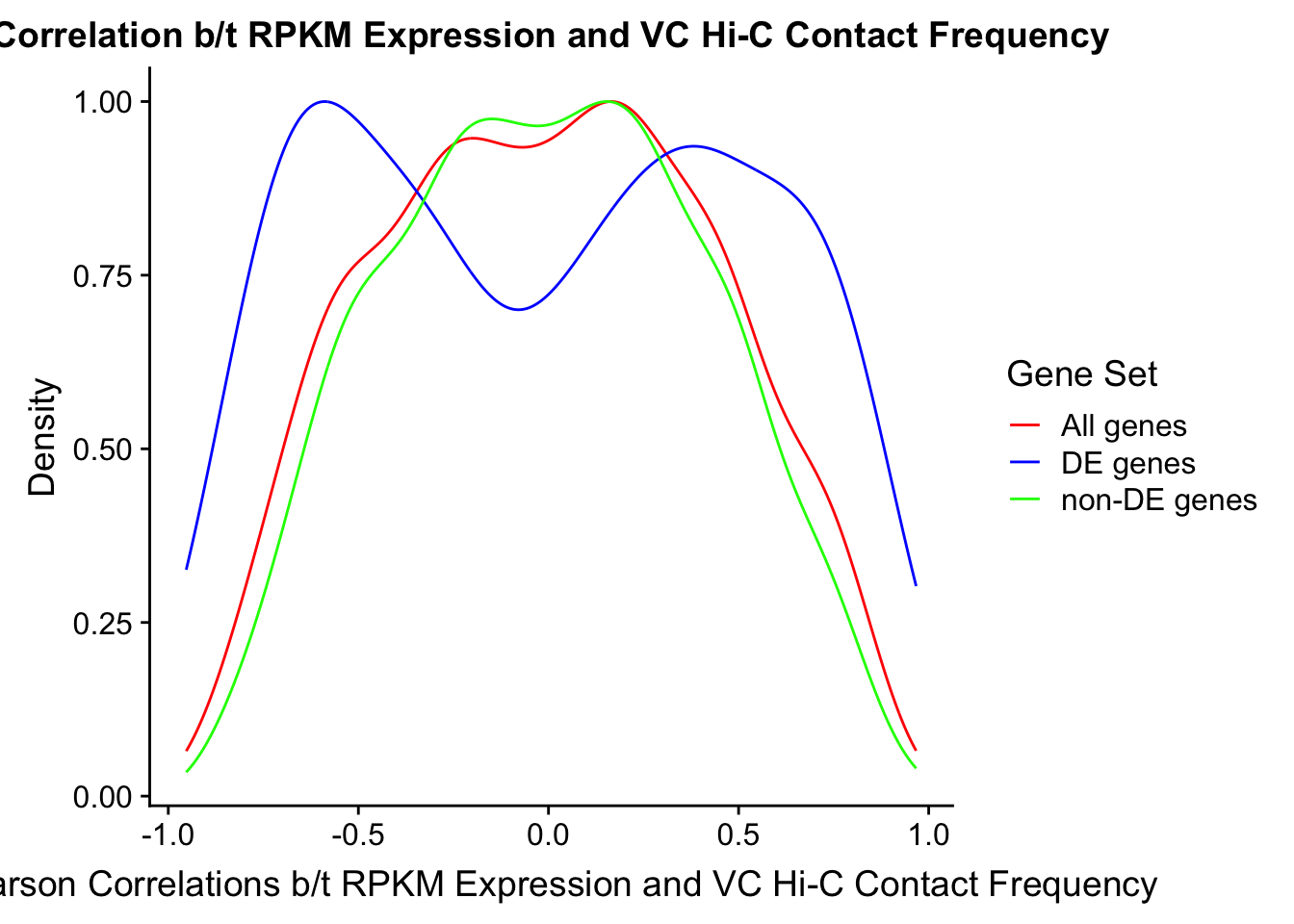
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
ggplot(data=filter(ggcorsKR, type=="all"|type=="DE"|type=="non-DE")) + stat_density(aes(x=cor, group=type, color=type, y=..scaled..), position="identity", geom="line") + ggtitle("Correlation b/t RPKM Expression and KR Hi-C Contact Frequency") + xlab("Pearson Correlations b/t RPKM Expression and KR Hi-C Contact Frequency") + ylab("Density") + scale_color_manual("Gene Set", values=c("red", "blue", "green"), labels=c("All genes", "DE genes", "non-DE genes"))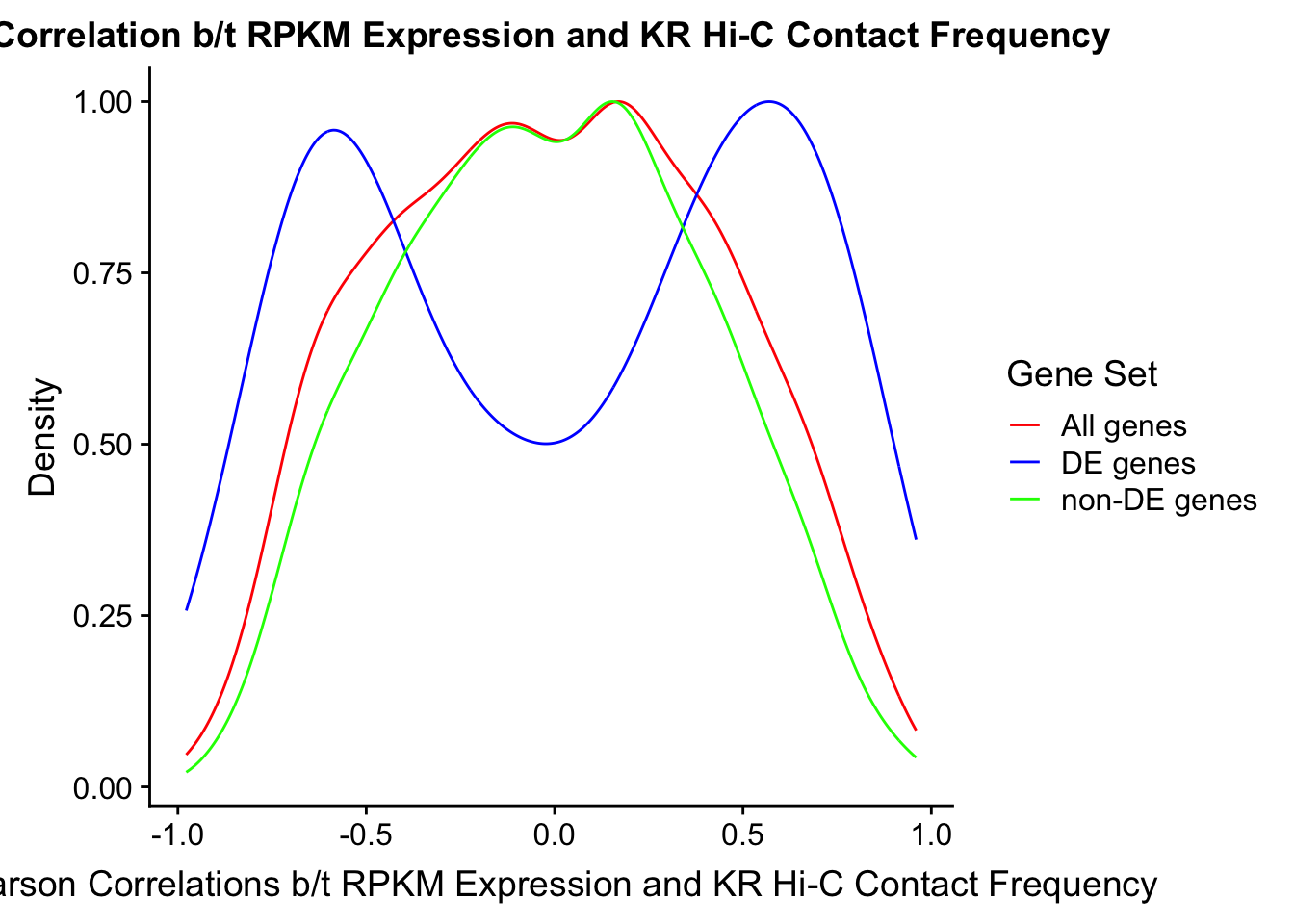
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
###This looks great, showing strong bimodal distribution for the DE genes and broader distributions with a peak at 0 for the non-DE set and the full set. To assess if this is legit at all, I now re-run this correlation analysis after permuting the Hi-C values. I shuffle sample IDs on a gene-by-gene basis to accomplish this:
cor.permuter <- function(hicdata, exprdata, nperm){
result <- data.frame(cor=NA, type=rep(1:nperm, each=nrow(exprdata)))
for(perm in 1:nperm){
permute <- hicdata
for(row in 1:nrow(hicdata)){
permute[row,] <- sample(hicdata[row,])
}
myindices <- which(result$type==perm)
result[myindices,1] <- cor.calc(permute, exprdata)
}
return(result)
}
#Just do it with 5 permutations to see the general effect quickly:
full.perm.KR <- cor.permuter(KRcontacts, KRexprs, 5)
DE.perm.KR <- cor.permuter(KRcontactsDE, KRexprsDE, 5)
nonDE.perm.KR <- cor.permuter(nonDEcontactsKR, nonDEexprsKR, 5)
full.perm.VC <- cor.permuter(VCcontacts, VCexprs, 5)
DE.perm.VC <- cor.permuter(VCcontactsDE, VCexprsDE, 5)
nonDE.perm.VC <- cor.permuter(nonDEcontactsVC, nonDEexprsVC, 5)
#Now visualize.
ggplot(data=filter(ggcorsKR, type=="all"|type=="DE"|type=="non-DE")) + stat_density(aes(x=cor, group=type, color=type, y=..scaled..), position="identity", geom="line") + stat_density(data=full.perm.KR, aes(x=cor, group=type), geom="line", linetype="dotted", position="identity") + stat_density(data=DE.perm.KR, aes(x=cor, group=type), geom="line", linetype="dashed", position="identity") + stat_density(data=nonDE.perm.KR, aes(x=cor, group=type), geom="line", linetype="twodash", position="identity") + ggtitle("Correlation b/t RPKM Expression and KR Hi-C Contact Frequency") + xlab("Pearson Correlations b/t RPKM Expression and KR Hi-C Contact Frequency") + ylab("Density") + scale_color_manual("Gene Set", values=c("red", "blue", "green"), labels=c("All genes", "DE genes", "non-DE genes"))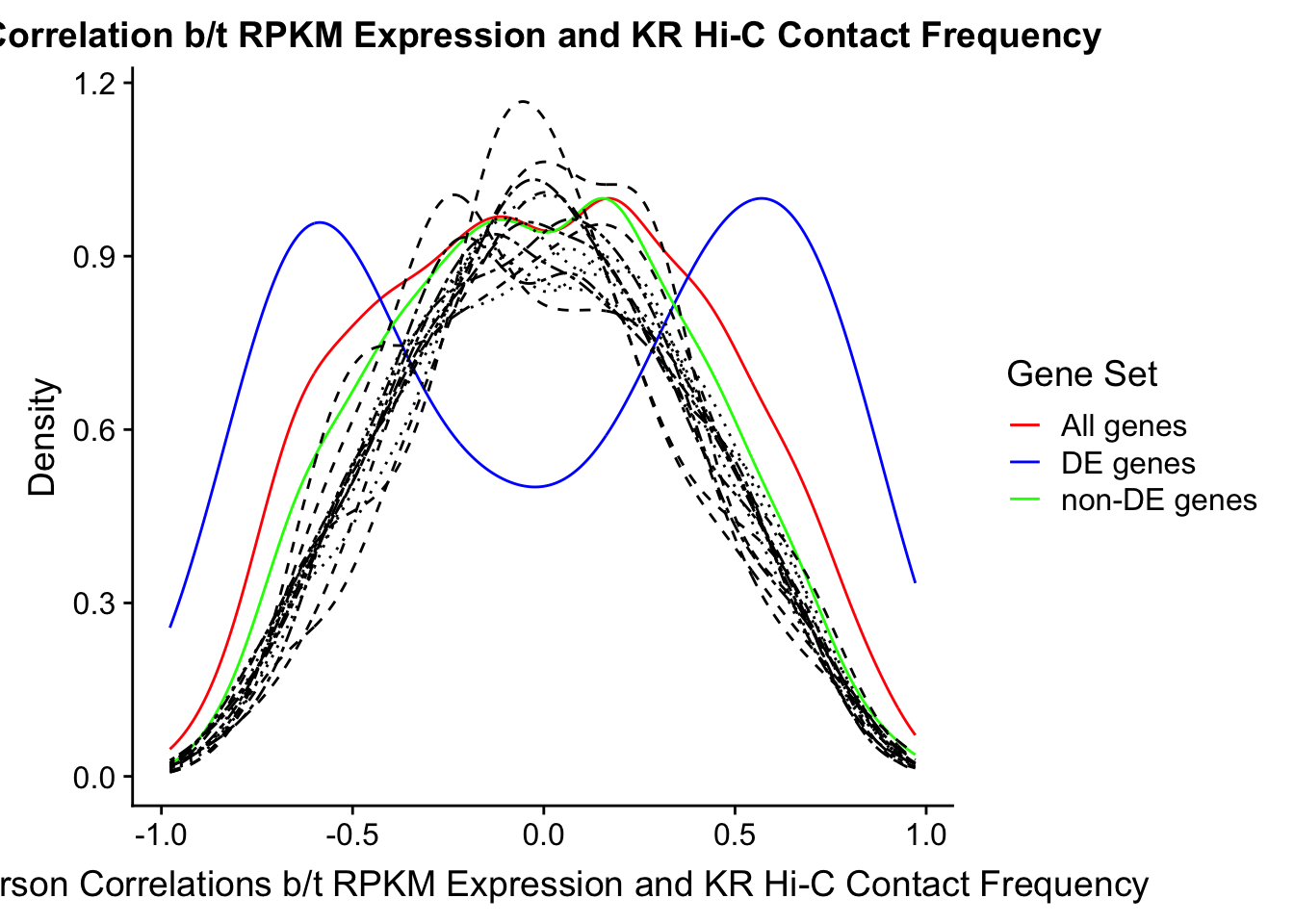
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
ggplot(data=filter(ggcorsVC, type=="all"|type=="DE"|type=="non-DE")) + stat_density(aes(x=cor, group=type, color=type, y=..scaled..), position="identity", geom="line") + stat_density(data=full.perm.VC, aes(x=cor, group=type), geom="line", linetype="dotted", position="identity") + stat_density(data=DE.perm.VC, aes(x=cor, group=type), geom="line", linetype="dashed", position="identity") + stat_density(data=nonDE.perm.VC, aes(x=cor, group=type), geom="line", linetype="twodash", position="identity") + ggtitle("Correlation b/t RPKM Expression and VC Hi-C Contact Frequency") + xlab("Pearson Correlations b/t RPKM Expression and VC Hi-C Contact Frequency") + ylab("Density") + scale_color_manual("Gene Set", values=c("red", "blue", "green"), labels=c("All genes", "DE genes", "non-DE genes"))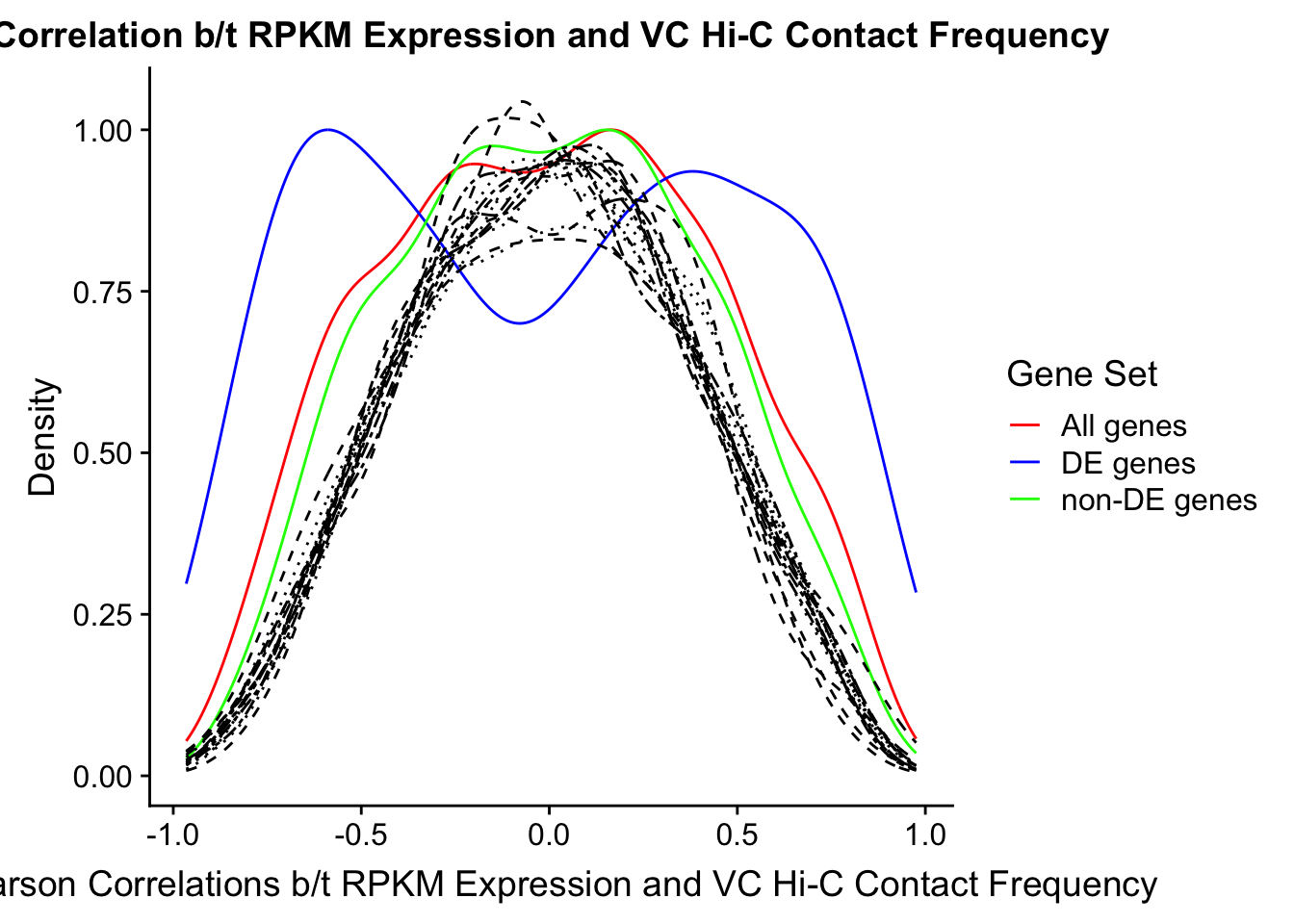
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
#See that permuted datasets have a tighter correlation distribution with a strong peak at 0. Reassuring. In the future I will repeat this analysis on Midway2, running 10000 permutations to see the full range of permuted data possible.Now that I’ve seen some nice effects in the correlation between expression and contact for different sets of genes, and for permutations on those sets, I move to see what kind of quantitiative explanatory power differential contact (DC) might actually have for differential expression (DE).
###How well does Hi-C data explain expression data? In this next section I “regress out” the effect of Hi-C contacts from their overlapping genes’ RPKM expression values, comparing a linear model run on the base values to one run on the residuals of expression after regressing out Hi-C data. Comparing the p-values before and after this regression can give some sense of whether the DE is being driven by differential Hi-C contacts (DC).
###A function to calculate gene-wise correlations between Hi-C data and expression data, but for spearman correlations. Pearson correlation calculator function is in the chunk above.
cor.calc.spear <- function(hicdata, exprdata){
cor.exp <- vector(length=nrow(hicdata))
for(i in 1:nrow(hicdata)){
cor.exp[i] <- cor(as.numeric(hicdata[i,]), as.numeric(exprdata[i,]), method="spearman")
}
return(cor.exp)
}
###A function to permute a Hi-C df by going gene-by-gene (row-by-row) and shuffling all sample IDs.
shuffler <- function(hicdata){
for(row in 1:nrow(hicdata)){
hicdata[row,] <- sample(hicdata[row,])
}
return(hicdata)
}
###A function that runs a linear model, both with and without Hi-C corrected expression values, and returns a dataframe of hit classes (DE or not before and after correction). Also spits back out p-values before and after correction, as well as correlations between Hi-C data and expression data. Since the former is a one-row data frame of 4 points and the latter is a 3-column data frame with the number of genes rows, returns a list.
lmcorrect <- function(voom.obj, exprs, cov_matrix, meta_df){
mygenes <- cov_matrix$genes #Pull out the relevant genes here; used for subsetting the voom.obj in a bit.
cov_matrix <- cov_matrix[, -9] #Remove genes from the cov matrix
hic_present <- sapply(1:nrow(cov_matrix), function(i) !any(is.na(cov_matrix[i,]))) #First, remove any rows w/ missing Hi-C data.
exprs <- data.matrix(exprs[hic_present,]) #Filter expression with this
cov_matrix <- data.matrix(cov_matrix[hic_present,]) #Filter Hi-C data with this
#Now, prepare to run the actual models. First run a model w/ Hi-C as a covariate to evaluate
SP <- factor(meta_df$SP,levels = c("H","C"))
design <- model.matrix(~0+SP)
colnames(design) <- c("Human", "Chimp")
resid_hic <- array(0, dim=c(nrow(exprs), ncol(cov_matrix))) #Initialize a dataframe for storing the residuals.
for(i in 1:nrow(exprs)){#Loop through rows of the expression df, running linear modeling w/ Hi-C to obtain residuals.
resid_hic[i,] <- lm(exprs[i,]~cov_matrix[i,])$resid
}
mycon <- makeContrasts(HvC = Human-Chimp, levels = design)
#Filter the voom object to only contain genes that had Hi-C information here.
good.indices <- which(rownames(voom.obj$E) %in% mygenes)
voom.obj <- voom.obj[good.indices,]
#Now, replace the RPKM values in the voom object for after linear modeling with the residuals.
voom.obj.after <- voom.obj
voom.obj.after$E <- resid_hic
lmFit(voom.obj, design=design) %>% eBayes(.) %>% contrasts.fit(., mycon) %>% eBayes(.) %>% topTable(., coef = 1, adjust.method = "BH", number = Inf, sort.by="none") -> fit_before
lmFit(voom.obj.after, design=design) %>% eBayes(.) %>% contrasts.fit(., mycon) %>% eBayes(.) %>% topTable(., coef = 1, adjust.method = "BH", number = Inf, sort.by="none") -> fit_after
result.DE.cats <- data.frame(DEneither=sum(fit_before$adj.P.Val>0.05&fit_after$adj.P.Val>0.05), DEbefore=sum(fit_before$adj.P.Val<=0.05&fit_after$adj.P.Val>0.05), DEafter=sum(fit_before$adj.P.Val>0.05&fit_after$adj.P.Val<=0.05), DEboth=sum(fit_before$adj.P.Val<=0.05&fit_after$adj.P.Val<=0.05))
result.DE.stats <- data.frame(cor.pear=cor.calc(cov_matrix, exprs), cor.spear=cor.calc.spear(cov_matrix, exprs), pval.before=fit_before$adj.P.Val, pval.after=fit_after$adj.P.Val)
result <- list("categories"=result.DE.cats, "stats"=result.DE.stats)
return(result)
}
#Proceed to use the function on the different dataframes I've created with different sets of overlapping Hi-C contacts:
h_minFDR_pvals_KR <- lmcorrect(weighted.data, hmin.KR[,c("A-21792", "B-28126", "C-3649", "D-40300", "E-28815", "F-28834", "G-3624", "H-3651")], hmin.KR[,c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC", "genes")], meta.data)
h_minFDR_pvals_VC <- lmcorrect(weighted.data, hmin.VC[,c("A-21792", "B-28126", "C-3649", "D-40300", "E-28815", "F-28834", "G-3624", "H-3651")], hmin.VC[,c("A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC", "genes")], meta.data)
#I now proceed to visualize the difference in p-values for expression before and after "correcting" for the Hi-C data. I am hoping to see many hits in the bottom right quadrant of the following plots, indicating genes that showed up as DE before Hi-C correction, but not after. I also expect that p-values falling farther away from the null line of expectation for p-values being identical between models will have higher correlations, which I color here for the pearson correlation (results look similar for spearman).
#ggplot(data=h_minFDR_pvals_KR$stats, aes(x=-log10(pval.before), y=-log10(pval.after), color=abs(cor.pear))) + geom_point(size=0.01) + geom_hline(yintercept=-log10(0.05), color="red") + geom_vline(xintercept=-log10(0.05), color="red") + geom_abline(slope=1, intercept=0, color="green", linetype="dashed") + ggtitle("Evidence for DE before vs. after regressing out Hi-C min. FDR, KR") + xlab("-log10(p-value of DE before Hi-C regression)") + ylab("-log10(p-value of DE after Hi-C regression")
ggplot(data=h_minFDR_pvals_KR$stats, aes(x=-log10(pval.before), y=-log10(pval.after))) + geom_point(size=0.01) + geom_hline(yintercept=-log10(0.05), color="red") + geom_vline(xintercept=-log10(0.05), color="red") + geom_abline(slope=1, intercept=0, color="green", linetype="dashed") + ggtitle("Evidence for DE Before vs. After Regressing out Hi-C Contact Frequency") + xlab("-log10(p-value of DE before KR Hi-C regression)") + ylab("-log10(p-value of DE after KR Hi-C regression)") + theme(plot.title=element_text(hjust=1))<img src=“figure/juicer_gene_expression.Rmd/Mediation effect by”regressing out" Hi-C effects from Expression Data-1.png" width=“672” style=“display: block; margin: auto;” />
| Version | Author | Date |
|---|---|---|
| <a href=“https://github.com/IttaiEres/HiCiPSC/blob/b7d82fcedada8542a39964036d316c305ed7bf1a/docs/figure/juicer_gene_expression.Rmd/Mediation effect by”regressing out" Hi-C effects from Expression Data-1.png" target="_blank">b7d82fc | Ittai Eres | 2019-04-30 |
ggplot(data=h_minFDR_pvals_VC$stats, aes(x=-log10(pval.before), y=-log10(pval.after))) + geom_point(size=0.01) + geom_hline(yintercept=-log10(0.05), color="red") + geom_vline(xintercept=-log10(0.05), color="red") + geom_abline(slope=1, intercept=0, color="green", linetype="dashed") + ggtitle("Evidence for DE Before vs. After Regressing out Hi-C Contact Frequency") + xlab("-log10(p-value of DE before VC Hi-C regression)") + ylab("-log10(p-value of DE after VC Hi-C regression)") + theme(plot.title=element_text(hjust=1))<img src=“figure/juicer_gene_expression.Rmd/Mediation effect by”regressing out" Hi-C effects from Expression Data-2.png" width=“672” style=“display: block; margin: auto;” />
| Version | Author | Date |
|---|---|---|
| <a href=“https://github.com/IttaiEres/HiCiPSC/blob/b7d82fcedada8542a39964036d316c305ed7bf1a/docs/figure/juicer_gene_expression.Rmd/Mediation effect by”regressing out" Hi-C effects from Expression Data-2.png" target="_blank">b7d82fc | Ittai Eres | 2019-04-30 |
Critically, here I see that “regressing out” Hi-C data and trying to model expression again moves many genes from being significantly differentially expressed (at FDR of 5%) to no longer showing differential expression. Seeing most of the hits in the bottom right corner of these visualizations is what confirms this. Reassuringly, I also see stronger correlations between RPKM expression values and normalized Hi-C interaction frequency values for points farther away from the diagonal green line of expectation. However, since this is not a statistical test, I have no assessment of significance. To accomplish this, I compare these results to running the same kind of analysis on permuted data in the next section.
###Differences in effect size/sign changes This next section would be to look at differences in effect size and sign changes when doing the two separate lms (one before and one after regressing out Hi-C data from RPKM expression). Unfortunately the ash results I get have all their svalues as 1 so this is boring/not interesting right now. Hence it is not included. But let’s give it another go!
#######FRESH CODE 6-21-18#########
design <- model.matrix(~meta.data$SP)
exprs <- select(hmin.VC, "A-21792", "B-28126", "C-3649", "D-40300", "E-28815", "F-28834", "G-3624", "H-3651", genes) #Should be done on just DE genes, filter with adj.P.Val<=0.05
hic <- select(hmin.VC,"A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC")
present <- sapply(1:nrow(hic), function(i) !any(is.na(hic[i,])))
genes <- exprs$genes
DEgenes <- filter(hmin.VC, adj.P.Val<=0.05) %>% select(., genes)
myDEindices <- which(genes %in% DEgenes[,1])
exprs <- exprs[,-9]
exprs <- data.matrix(exprs[present,])
hic <- data.matrix(hic[present,])
designy <- as.factor(c("human", "human", "chimp", "chimp", "human", "human", "chimp", "chimp"))
colnames(hic) <- colnames(exprs)
hic <- as.data.table(hic)
exprs <- as.data.table(exprs)
source("code/mediate.test.regressing.R")
test <- mediate.test.regressing(exprs, designy, hic, cov=as.factor(meta.data$SX))
Attaching package: 'assertthat'The following object is masked from 'package:tibble':
has_namevshrDE <- vash(test$d_se[myDEindices], df=4)
vshrnonDE <- vash(test$d_se[-myDEindices], df=4)
vshrink <- vash(test$d_se, df=4)
DEtest <- test[myDEindices,]
nonDEtest <- test[-myDEindices,]
ash_reg <- ash(as.vector(DEtest$d), as.vector(vshrDE$sd.post), mixcompdist = "normal") #Now it works with this design coding. Use 6 df, as 2-group comparison and 8 individual samples (samples - 2)
summary(ash_reg$result$svalue<=0.05) #Incredible result, looks too good to be true. Mode FALSE TRUE
logical 281 18 ###### START EXAMPLE
#Figure out where some of these are to pull out good examples, try to find a dense region of them
# myhits <- which(ash_reg$result$svalue<=0.05) #These SHOULD line up with DEtest indices...
# realhits <- myDEindices[myhits] #Now go back to exprs original with these indices to get the gene names!
# exprs <- select(hmin.KR, "A-21792", "B-28126", "C-3649", "D-40300", "E-28815", "F-28834", "G-3624", "H-3651", genes) #Should be done on just DE genes, filter with adj.P.Val<=0.05
# demgenes <- exprs[realhits,9]
# testcases <- filter(gene.hic.KR, genes %in% demgenes)
# table(testcases$genechr.H)
# testcases$sval <- ash_reg$result$svalue[myhits]
# testcases <- testcases[order(testcases$sval),]
#
# #Now, pull out expression values for those suckers:
# options(scipen = 999)
# mean.species.expression <- as.data.frame(weighted.data$E) #Now, need to pull out the mean expression values for all those genes!
# mean.species.expression$genes <- rownames(mean.species.expression)
# mean.species.expression$Hmean <- rowMeans(mean.species.expression[,c(1:2,5:6)])
# mean.species.expression$Cmean <- rowMeans(mean.species.expression[,c(3:4,7:8)])
# mean.species.expression <- mean.species.expression[,(-1:-8)]
#
# left_join(testcases, mean.species.expression, by="genes") -> filt.example
# filter(filt.example, min_FDR.H<=0.05|min_FDR.C<=0.05) -> final.example
# final.example$Hmean-final.example$Cmean
# #gonna end up pulling 48th row example from final.example. Pull out its contact frequency vals for arc diagram creation:
# final.example[48,]
# which((data.filtered$H1=="chr17-64470000"|data.filtered$H2=="chr17-64470000")&(data.filtered$C1=="chr17-63970000"|data.filtered$C2=="chr17-63970000"))
# which((data.filtered$H1=="chr17-64470000"&data.filtered$H2=="chr17-64430000")|(data.filtered$H2=="chr17-64470000"&data.filtered$H1=="chr17-64430000"))
#
# #Check on what other contacts might be significant in the region
# QC <- filter(data.filtered, Hchr=="Chr. 17"&sp_BH_pval<=0.05)
# QC$H1 <- gsub(".*-", "", QC$H1)
# QC$H2 <- gsub(".*-", "", QC$H2)
# which((QC$H1>=64400000&QC$H1<=64500000)&(QC$H2>=64400000&QC$H2<=64500000))
# QC[c(218,323,397),]
######END EXAMPLE
ind_sig <- (ash_reg$result$svalue < 0.05)
ind_sig[ind_sig == TRUE] <- "red"
ind_sig[ind_sig == FALSE] <- "black"
plot(x=ash_reg$result$betahat, y=ash_reg$result$sebetahat, ylab="Standard error of the difference", xlab="Difference in Effect Size from 2 Linear Models, VC-Norm", main="Difference of Effect Size in DE genes", pch=16, cex=0.6, col=ind_sig, xlim=c(-2.5, 2.5), ylim=c(0, 2.6))
legend("topleft", legend=c("s < 0.05", "s >= 0.05"), col=c("red", "black"), pch=16:16, cex=0.8) #FIGS6A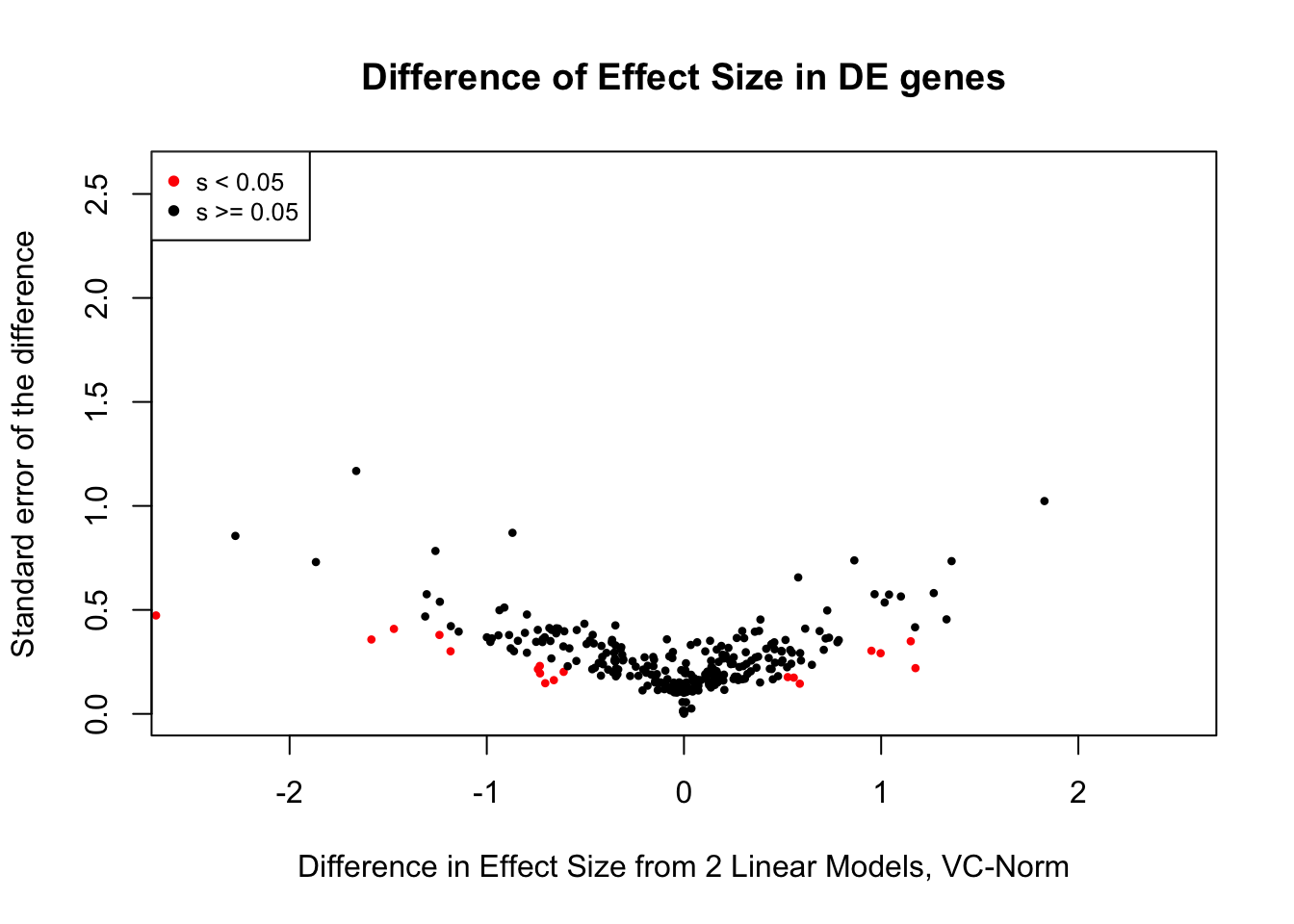
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
ash_reg_nonDE <- ash(as.vector(nonDEtest$d), as.vector(vshrnonDE$sd.post), mixcompdist = "normal") #Now it works with this design coding. Use 6 df, as 2-group comparison and 8 individual samples (samples - 2)?
summary(ash_reg_nonDE$result$svalue<=0.05) Mode FALSE
logical 1331 ind_sig <- (ash_reg_nonDE$result$svalue < 0.05)
ind_sig[ind_sig == TRUE] <- "red"
ind_sig[ind_sig == FALSE] <- "black"
plot(x=ash_reg_nonDE$result$betahat, y=ash_reg_nonDE$result$sebetahat, ylab="Standard error of the difference", xlab="Difference in Effect Size from 2 Linear Models, VC-Norm", main="Difference of Effect Size in non-DE genes", pch=16, cex=0.6, col=ind_sig, xlim=c(-2.5, 2.5), ylim=c(0, 2.6))
legend("topleft", legend=c("s < 0.05", "s >= 0.05"), col=c("red", "black"), pch=16:16, cex=0.8) #FIGS6B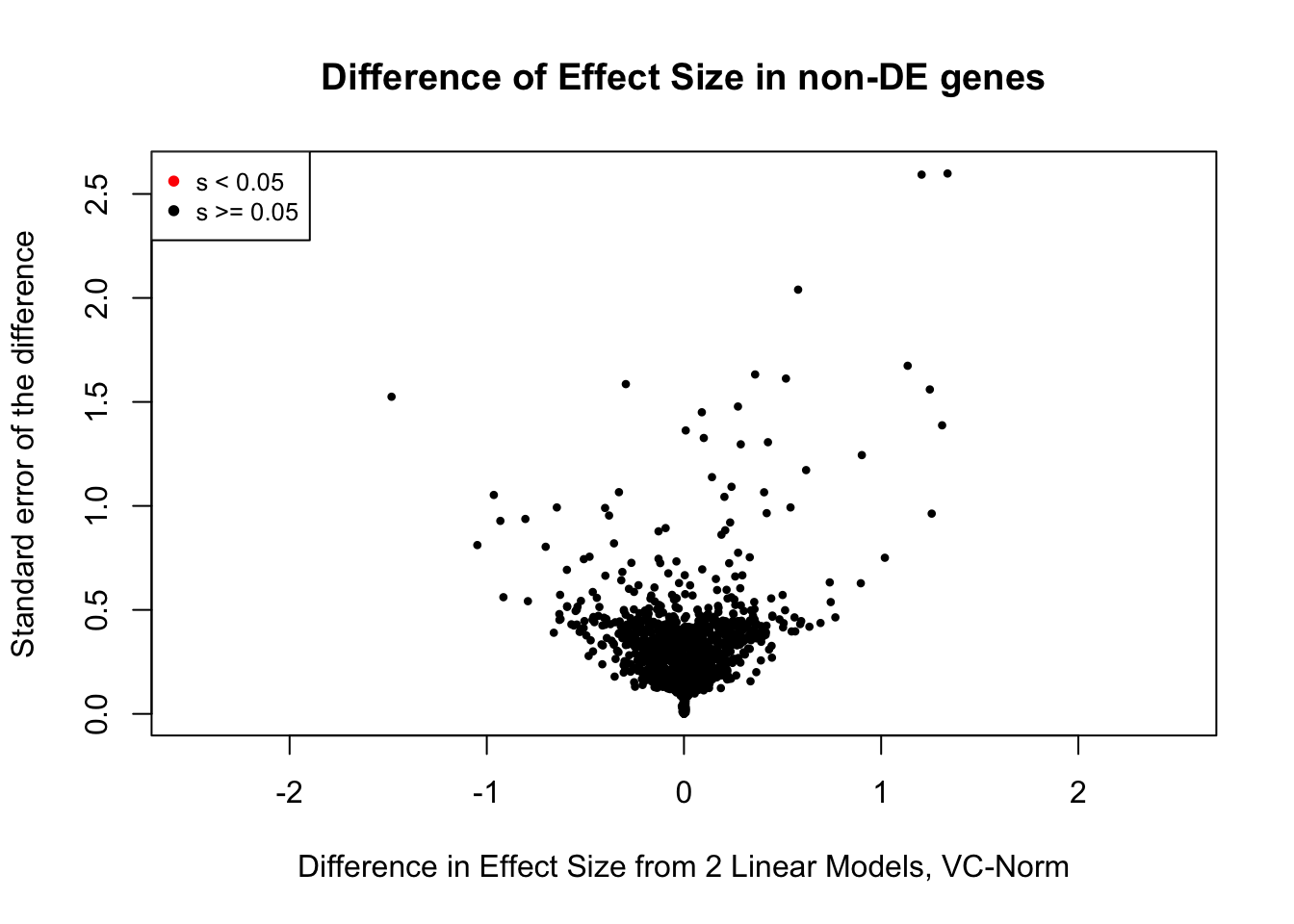
| Version | Author | Date |
|---|---|---|
| b7d82fc | Ittai Eres | 2019-04-30 |
# exprs <- select(hmin.VC, "A-21792", "B-28126", "C-3649", "D-40300", "E-28815", "F-28834", "G-3624", "H-3651", genes) #Should be done on just DE genes, filter with adj.P.Val<=0.05
# hic <- select(hmin.VC,"A_21792_HIC", "B_28126_HIC", "C_3649_HIC", "D_40300_HIC", "E_28815_HIC", "F_28834_HIC", "G_3624_HIC", "H_3651_HIC")
# present <- sapply(1:nrow(hic), function(i) !any(is.na(hic[i,])))
# genes <- exprs$genes
# DEgenes <- filter(hmin.KR, adj.P.Val<=0.05) %>% select(., genes)
# myDEindices <- which(genes %in% DEgenes[,1])
# exprs <- exprs[,-9]
# exprs <- data.matrix(exprs[present,])
# hic <- data.matrix(hic[present,])
# designy <- as.factor(c("human", "human", "chimp", "chimp", "human", "human", "chimp", "chimp"))
# colnames(hic) <- colnames(exprs)
# hic <- as.data.table(hic)
# exprs <- as.data.table(exprs)
# test <- mediate.test.regressing(exprs, designy, hic)
# vshrDE <- vash(test$d_se[myDEindices], df=6)
# vshrnonDE <- vash(test$d_se[-myDEindices], df=6)
# vshrink <- vash(test$d_se, df=6)
# DEtest <- test[myDEindices,]
# nonDEtest <- test[-myDEindices,]
# ash_reg <- ash(as.vector(DEtest$d), as.vector(vshrDE$sd.post), mixcompdist = "normal") #Now it works with this design coding. Use 6 df, as 2-group comparison and 8 individual samples (samples - 2)
# summary(ash_reg$result$svalue<=0.05) #Incredible result, looks too good to be true.
# ind_sig <- (ash_reg$result$svalue < 0.05)
# ind_sig[ind_sig == TRUE] <- "red"
# ind_sig[ind_sig == FALSE] <- "black"
# plot(x=ash_reg$result$betahat, y=ash_reg$result$sebetahat, ylab="Standard error of the difference", xlab="Difference in Effect Size from 2 Linear Models, VC-Norm", main="Difference of Effect Size in DE Genes", pch=16, cex=0.6, col=ind_sig, xlim=c(-2.5, 2.5), ylim=c(0, 2.5)) #FIGS15A
# legend("topleft", legend=c("s < 0.05", "s >= 0.05"), col=c("red", "black"), pch=16:16, cex=0.8)
# ash_reg_nonDE <- ash(as.vector(nonDEtest$d), as.vector(vshrnonDE$sd.post), mixcompdist = "normal") #Now it works with this design coding. Use 6 df, as 2-group comparison and 8 individual samples (samples - 2)
# summary(ash_reg_nonDE$result$svalue<=0.05) #Incredible result, looks too good to be true.
# ind_sig <- (ash_reg_nonDE$result$svalue < 0.05)
# ind_sig[ind_sig == TRUE] <- "red"
# ind_sig[ind_sig == FALSE] <- "black"
# plot(x=ash_reg_nonDE$result$betahat, y=ash_reg_nonDE$result$sebetahat, ylab="Standard error of the difference", xlab="Difference in Effect Size from 2 Linear Models, VC-Norm", main="Difference of Effect Size in non-DE Genes", pch=16, cex=0.6, col=ind_sig, xlim=c(-2.5, 2.5), ylim=c(0, 2.5))
# legend("topleft", legend=c("s < 0.05", "s >= 0.05"), col=c("red", "black"), pch=16:16, cex=0.8) #FIGS15B
sessionInfo()R version 3.4.0 (2017-04-21)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: OS X El Capitan 10.11.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] compiler stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] assertthat_0.2.1 glmnet_2.0-16 foreach_1.4.4
[4] Matrix_1.2-15 medinome_0.0.1 vashr_0.99.1
[7] qvalue_2.10.0 SQUAREM_2017.10-1 ashr_2.2-32
[10] bedr_1.0.6 forcats_0.4.0 purrr_0.3.2
[13] readr_1.3.1 tibble_2.1.1 tidyverse_1.2.1
[16] edgeR_3.20.9 RColorBrewer_1.1-2 heatmaply_0.15.2
[19] viridis_0.5.1 viridisLite_0.3.0 stringr_1.4.0
[22] gplots_3.0.1.1 Hmisc_4.2-0 Formula_1.2-3
[25] survival_2.44-1 lattice_0.20-38 dplyr_0.8.0.1
[28] plotly_4.8.0 cowplot_0.9.4 ggplot2_3.1.0
[31] reshape2_1.4.3 data.table_1.12.0 tidyr_0.8.3
[34] plyr_1.8.4 limma_3.34.9
loaded via a namespace (and not attached):
[1] colorspace_1.4-1 class_7.3-15 modeltools_0.2-22
[4] mclust_5.4.3 rprojroot_1.3-2 htmlTable_1.13.1
[7] futile.logger_1.4.3 base64enc_0.1-3 fs_1.2.7
[10] rstudioapi_0.10 flexmix_2.3-15 mvtnorm_1.0-8
[13] lubridate_1.7.4 xml2_1.2.0 R.methodsS3_1.7.1
[16] codetools_0.2-16 splines_3.4.0 pscl_1.5.2
[19] doParallel_1.0.14 robustbase_0.92-8 knitr_1.22
[22] jsonlite_1.6 workflowr_1.2.0 broom_0.5.1
[25] cluster_2.0.7-1 kernlab_0.9-27 R.oo_1.22.0
[28] httr_1.4.0 backports_1.1.3 lazyeval_0.2.2
[31] cli_1.1.0 formatR_1.6 acepack_1.4.1
[34] htmltools_0.3.6 tools_3.4.0 gtable_0.3.0
[37] glue_1.3.1 Rcpp_1.0.1 cellranger_1.1.0
[40] trimcluster_0.1-2.1 gdata_2.18.0 nlme_3.1-137
[43] iterators_1.0.10 fpc_2.1-11.1 xfun_0.5
[46] testthat_2.0.1 rvest_0.3.2 gtools_3.8.1
[49] dendextend_1.10.0 DEoptimR_1.0-8 MASS_7.3-51.1
[52] scales_1.0.0 TSP_1.1-6 hms_0.4.2
[55] parallel_3.4.0 lambda.r_1.2.3 yaml_2.2.0
[58] gridExtra_2.3 rpart_4.1-13 latticeExtra_0.6-28
[61] stringi_1.4.3 gclus_1.3.2 checkmate_1.9.1
[64] seriation_1.2-3 caTools_1.17.1.2 truncnorm_1.0-8
[67] rlang_0.3.3 pkgconfig_2.0.2 prabclus_2.2-7
[70] bitops_1.0-6 evaluate_0.13 labeling_0.3
[73] htmlwidgets_1.3 tidyselect_0.2.5 magrittr_1.5
[76] R6_2.4.0 generics_0.0.2 pillar_1.3.1
[79] haven_2.1.0 whisker_0.3-2 foreign_0.8-71
[82] withr_2.1.2 mixsqp_0.1-97 nnet_7.3-12
[85] modelr_0.1.4 crayon_1.3.4 futile.options_1.0.1
[88] KernSmooth_2.23-15 rmarkdown_1.12 locfit_1.5-9.1
[91] grid_3.4.0 readxl_1.3.1 git2r_0.25.2
[94] digest_0.6.18 diptest_0.75-7 webshot_0.5.1
[97] VennDiagram_1.6.20 R.utils_2.8.0 stats4_3.4.0
[100] munsell_0.5.0 registry_0.5-1