RNA_CorrelationHeatMap
2026-01-14
Last updated: 2026-01-22
Checks: 7 0
Knit directory: CrossSpecies_CM_Diff_RNA/
This reproducible R Markdown analysis was created with workflowr (version 1.7.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20251129) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 7299279. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/RNA_CorrelationHeatMap.Rmd) and HTML
(docs/RNA_CorrelationHeatMap.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7299279 | John D. Hurley | 2026-01-22 | wflow_publish("analysis/RNA_CorrelationHeatMap.Rmd") |
| Rmd | b4bc4cd | John D. Hurley | 2026-01-22 | Updating Index HeatMap |
| html | 9aa0597 | John D. Hurley | 2026-01-22 | Website Update HeatMap |
| Rmd | 88bce45 | John D. Hurley | 2026-01-22 | Correcting Eval = FALSE statement |
| Rmd | 59e658c | John D. Hurley | 2026-01-22 | Test |
| html | 59e658c | John D. Hurley | 2026-01-22 | Test |
| html | 9264bd4 | John D. Hurley | 2026-01-21 | Change in RMD organization |
| Rmd | 0725194 | John D. Hurley | 2026-01-21 | Final QC Heatmap |
| Rmd | 5d2c47d | John D. Hurley | 2026-01-21 | Large Changes to Flow and Organization to QC |
####Library Loading####
library("edgeR")Loading required package: limmalibrary("ggplot2")
library("tibble")
library("dplyr")
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionlibrary("ggrepel")
library("readr")
library("org.Hs.eg.db")Loading required package: AnnotationDbiLoading required package: stats4Loading required package: BiocGenericsLoading required package: generics
Attaching package: 'generics'The following object is masked from 'package:dplyr':
explainThe following objects are masked from 'package:base':
as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
setequal, union
Attaching package: 'BiocGenerics'The following object is masked from 'package:dplyr':
combineThe following object is masked from 'package:limma':
plotMAThe following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
unsplit, which.max, which.minLoading required package: BiobaseWelcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.Loading required package: IRangesLoading required package: S4Vectors
Attaching package: 'S4Vectors'The following objects are masked from 'package:dplyr':
first, renameThe following object is masked from 'package:utils':
findMatchesThe following objects are masked from 'package:base':
expand.grid, I, unname
Attaching package: 'IRanges'The following objects are masked from 'package:dplyr':
collapse, desc, sliceThe following object is masked from 'package:grDevices':
windows
Attaching package: 'AnnotationDbi'The following object is masked from 'package:dplyr':
selectlibrary("AnnotationDbi")
library("pheatmap")
library("Cormotif")Loading required package: affylibrary("tidyverse")── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ forcats 1.0.1 ✔ stringr 1.5.2
✔ lubridate 1.9.4 ✔ tidyr 1.3.1
✔ purrr 1.1.0 ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ lubridate::%within%() masks IRanges::%within%()
✖ IRanges::collapse() masks dplyr::collapse()
✖ Biobase::combine() masks BiocGenerics::combine(), dplyr::combine()
✖ IRanges::desc() masks dplyr::desc()
✖ tidyr::expand() masks S4Vectors::expand()
✖ dplyr::filter() masks stats::filter()
✖ S4Vectors::first() masks dplyr::first()
✖ dplyr::lag() masks stats::lag()
✖ lubridate::pm() masks affy::pm()
✖ BiocGenerics::Position() masks ggplot2::Position(), base::Position()
✖ purrr::reduce() masks IRanges::reduce()
✖ S4Vectors::rename() masks dplyr::rename()
✖ lubridate::second() masks S4Vectors::second()
✖ lubridate::second<-() masks S4Vectors::second<-()
✖ AnnotationDbi::select() masks dplyr::select()
✖ IRanges::slice() masks dplyr::slice()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errorslibrary("workflowr")
####Loadind R Objects####
RNA_fc_df <- readRDS("data/QC/RNA_fc_df.RDS")
RNA_fc <- readRDS("data/QC/RNA_fc_Ensemble.RDS")
RNA_log2cpm <- readRDS("data/QC/RNA_log2cpm_Ensemble.RDS")
Filt_RMG0_RNA_fc <- readRDS("data/QC/Filt_RMG0_RNA_fc_Ensemble.RDS")
Filt_RMG0_RNA_log2cpm <- readRDS("data/QC/Filt_RMG0_RNA_log2cpm_Ensemble.RDS")
RNA_Metadata <- readRDS("data/QC/RNA_Metadata.RDS")
cor_Filt_RMG0_RNA_log2cpm <- readRDS("data/QC/Cor_Filt_RMG0_RNA_log2cpm.RDS")
Cor_metadata <- readRDS("data/QC/Cor_metadata.RDS")
ann_colors <- readRDS("data/QC/ann_colors.RDS")
RNA_Metadata_No4 <- readRDS("data/QC/RNA_Metatdata_No4.RDS")
Filt_RMG0_RNA_fc_NoD4 <- readRDS("data/QC/Filt_RMG0_RNA_fc_NoD4_Ensemble.RDS")
Filt_RMG0_RNA_log2cpm_NoD4 <- readRDS("data/QC/Filt_RMG0_RNA_log2cpm_NoD4_Ensemble.RDS")
Cor_metadata_No4 <- readRDS("data/QC/Cor_metadata_No4.RDS")
cor_Filt_RMG0_RNA_log2cpm_NoD4 <- readRDS("data/QC/cor_Filt_RMG0_RNA_log2cpm_NoD4.RDS")
ann_colors_No4 <- readRDS("data/QC/ann_colors_no4.RDS")chimp_RNA_pantro5_fc <- read.delim("~/diff_timeline_tes/RNA/Run1_Run2_Concat/featurecounts/c_samples_counts.txt", comment.char="#",row.names=1)
human_RNA_hg38_fc <- read.delim("~/diff_timeline_tes/RNA/Run1_Run2_Concat/featurecounts/h_samples_counts.txt", comment.char="#",row.names=1)
human_df <- human_RNA_hg38_fc %>%
tibble::rownames_to_column("gene")
chimp_df <- chimp_RNA_pantro5_fc %>%
tibble::rownames_to_column("gene")
RNA_joined_fc <- left_join(human_df, chimp_df, by = "gene")
#To keep only OrthoGenes and sample columns
RNA_fc <- RNA_joined_fc[ , !(names(RNA_joined_fc) %in% c("gene","Chr.x","Start.x","End.x","Strand.x","Length.x","Geneid.x","Chr.y","Start.y","End.y","Strand.y","Length.y","Geneid.y"))]
# 89 columns; 7 x 6 = 42 Human Exp, 7 x 6 = 42 Chimp Exp, 4 Human Replicate
rownames(RNA_fc) <- RNA_joined_fc$gene
#Rename column Names to More Useful Info
col_names <- c("H28126_D0",
"H28126_D2",
"H28126_D4",
"H28126_D5",
"H28126_D15",
"H28126_D30",
"H17_D0",
"H17_D2",
"H17_D4",
"H17_D5",
"H17_D15",
"H17_D30",
"H78_D0",
"H78_D2",
"H78_D4",
"H78_D5",
"H78_D15",
"H78_D30",
"H20682_D0",
"H20682_D2",
"H20682_D4",
"H20682_D5",
"H20682_D15",
"H20682_D30",
"H22422_D0",
"H22422_D2",
"H22422_D4",
"H22422_D5",
"H22422_D15",
"H22422_D30",
"H21792_D0",
"H21792_D2",
"H21792_D4",
"H21792_D5",
"H21792_D15",
"H21792_D30",
"H24280_D0",
"H24280_D2",
"H24280_D4",
"H24280_D5",
"H24280_D15",
"H24280_D30",
"H20682R_D0",
"H20682R_D2",
"H20682R_D5",
"H20682R_D30",
"C3649_D0",
"C3649_D2",
"C3649_D4",
"C3649_D5",
"C3649_D15",
"C3649_D30",
"C4955_D0",
"C4955_D2",
"C4955_D4",
"C4955_D5",
"C4955_D15",
"C4955_D30",
"C3651_D0",
"C3651_D2",
"C3651_D4",
"C3651_D5",
"C3651_D15",
"C3651_D30",
"C40210_D0",
"C40210_D2",
"C40210_D4",
"C40210_D5",
"C40210_D15",
"C40210_D30",
"C8861_D0",
"C8861_D2",
"C8861_D4",
"C8861_D5",
"C8861_D15",
"C8861_D30",
"C40280_D0",
"C40280_D2",
"C40280_D4",
"C40280_D5",
"C40280_D15",
"C40280_D30",
"C3647_D0",
"C3647_D2",
"C3647_D4",
"C3647_D5",
"C3647_D15",
"C3647_D30"
)
colnames(RNA_fc) <- col_names
dim(RNA_fc)[1] 44125 88# saveRDS(RNA_fc,"data/QC/RNA_fc_Ensemble.RDS")
sum(duplicated(rownames(RNA_fc)))[1] 0ensembl_ids_unfilt <- rownames(RNA_fc)
entrez_ids_unfilt <- mapIds(org.Hs.eg.db,
keys = ensembl_ids_unfilt,
column = "ENTREZID",
keytype = "ENSEMBL",
multiVals = "first")'select()' returned 1:many mapping between keys and columnssymbol_ids_unfilt <- mapIds(org.Hs.eg.db,
keys = ensembl_ids_unfilt,
column = "SYMBOL",
keytype = "ENSEMBL",
multiVals = "first")'select()' returned 1:many mapping between keys and columnsRNA_fc_df <- as.data.frame(RNA_fc)
RNA_fc_df <- RNA_fc_df %>%
rownames_to_column(var = "Ensemble") %>%
dplyr::mutate(
Entrez_ID = entrez_ids_unfilt,
Symbol = symbol_ids_unfilt
) %>%
dplyr::select(
Ensemble, # 1st column
Entrez_ID, # 2nd column
Symbol, # 3rd column
everything() # rest unchanged
)
# saveRDS(RNA_fc_df,"data/QC/RNA_fc_df.RDS")#####Unfiltered####
RNA_log2cpm <- cpm(RNA_fc,log=TRUE)
print(hist(RNA_log2cpm, main = "Histogram of all counts (unfiltered)",
xlab =expression("Log"[2]*" counts-per-million"), col =4 ))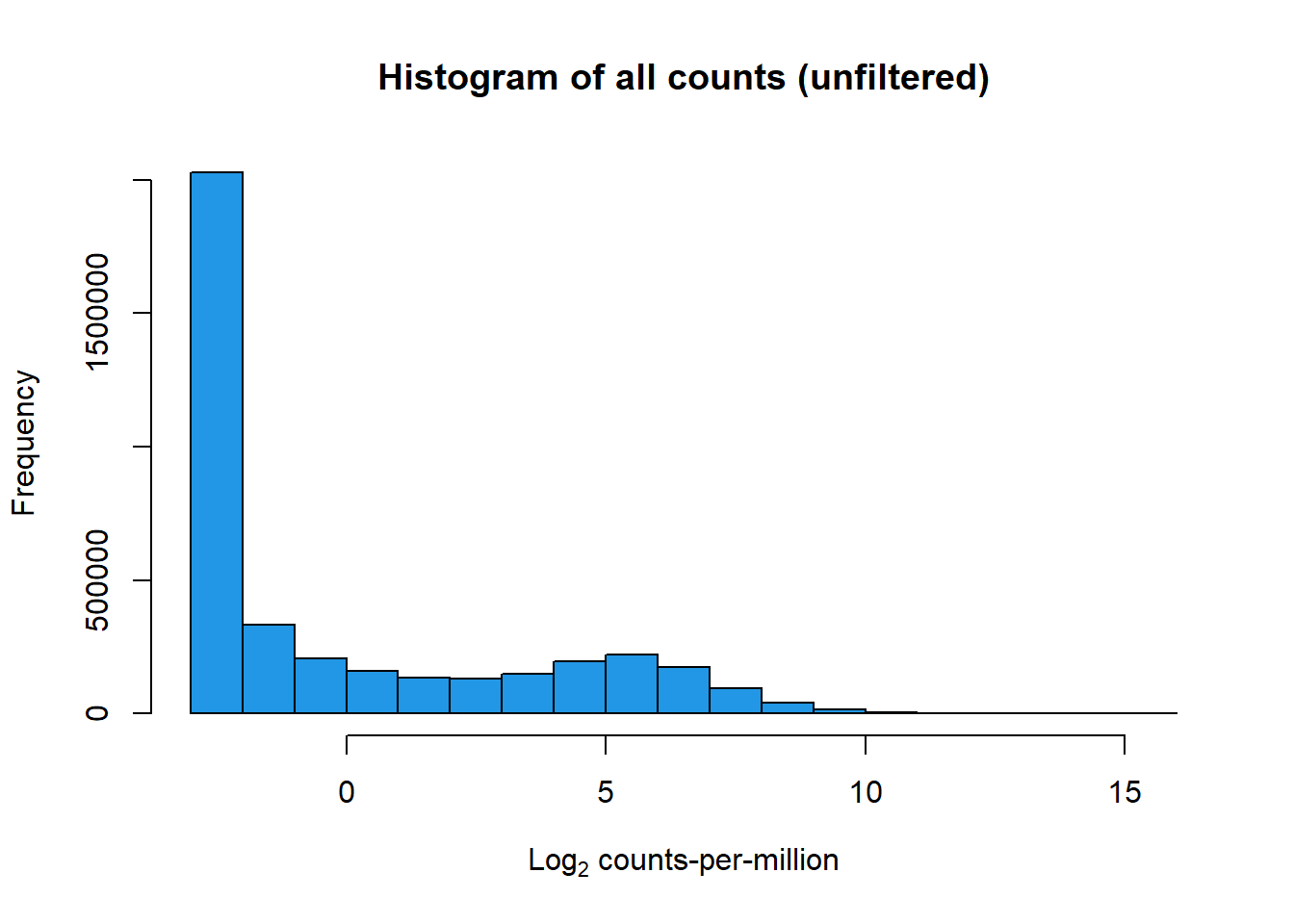
| Version | Author | Date |
|---|---|---|
| 9aa0597 | John D. Hurley | 2026-01-22 |
$breaks
[1] -3 -2 -1 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
$counts
[1] 2026734 332340 206543 159084 134510 130097 148151 195453 219757
[10] 174507 94391 39658 15034 5070 1267 310 70 21
[19] 3
$density
[1] 5.219506e-01 8.558846e-02 5.319160e-02 4.096935e-02 3.464074e-02
[6] 3.350425e-02 3.815375e-02 5.033557e-02 5.659464e-02 4.494128e-02
[11] 2.430878e-02 1.021324e-02 3.871749e-03 1.305691e-03 3.262941e-04
[16] 7.983518e-05 1.802730e-05 5.408190e-06 7.725985e-07
$mids
[1] -2.5 -1.5 -0.5 0.5 1.5 2.5 3.5 4.5 5.5 6.5 7.5 8.5 9.5 10.5 11.5
[16] 12.5 13.5 14.5 15.5
$xname
[1] "RNA_log2cpm"
$equidist
[1] TRUE
attr(,"class")
[1] "histogram"boxplot(RNA_log2cpm, main = "Boxplots of log cpm per sample
(unfiltered)", xaxt = "n", xlab= "")
axis(1,
at = 1:length(col_names), # positions (one per sample)
labels = col_names, # your labels vector
las = 2, # rotate text vertically (like srt=90)
cex.axis = 0.3) # shrink label size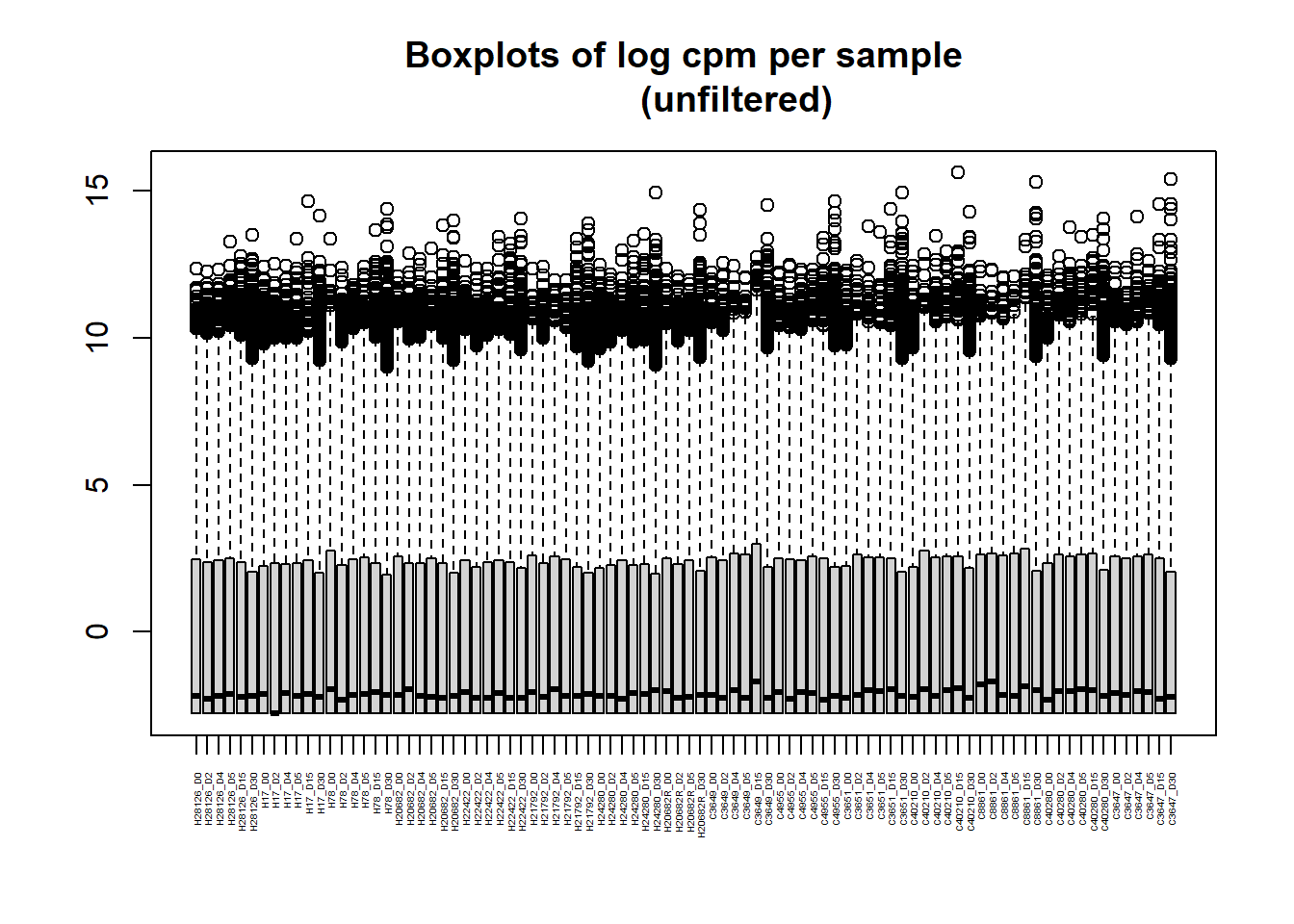
| Version | Author | Date |
|---|---|---|
| 9aa0597 | John D. Hurley | 2026-01-22 |
# saveRDS(RNA_log2cpm,"data/QC/RNA_log2cpm_Ensemble.RDS")#####RowMu>0####
row_means <- rowMeans(RNA_log2cpm)
Filt_RMG0_RNA_fc <- RNA_fc[row_means >0,]
Filt_RMG0_RNA_log2cpm <- cpm(Filt_RMG0_RNA_fc,log=TRUE)
hist(Filt_RMG0_RNA_log2cpm, main = "Histogram of filtered counts using rowMeans > 0 method",
xlab =expression("Log"[2]*" counts-per-million"), col =5 )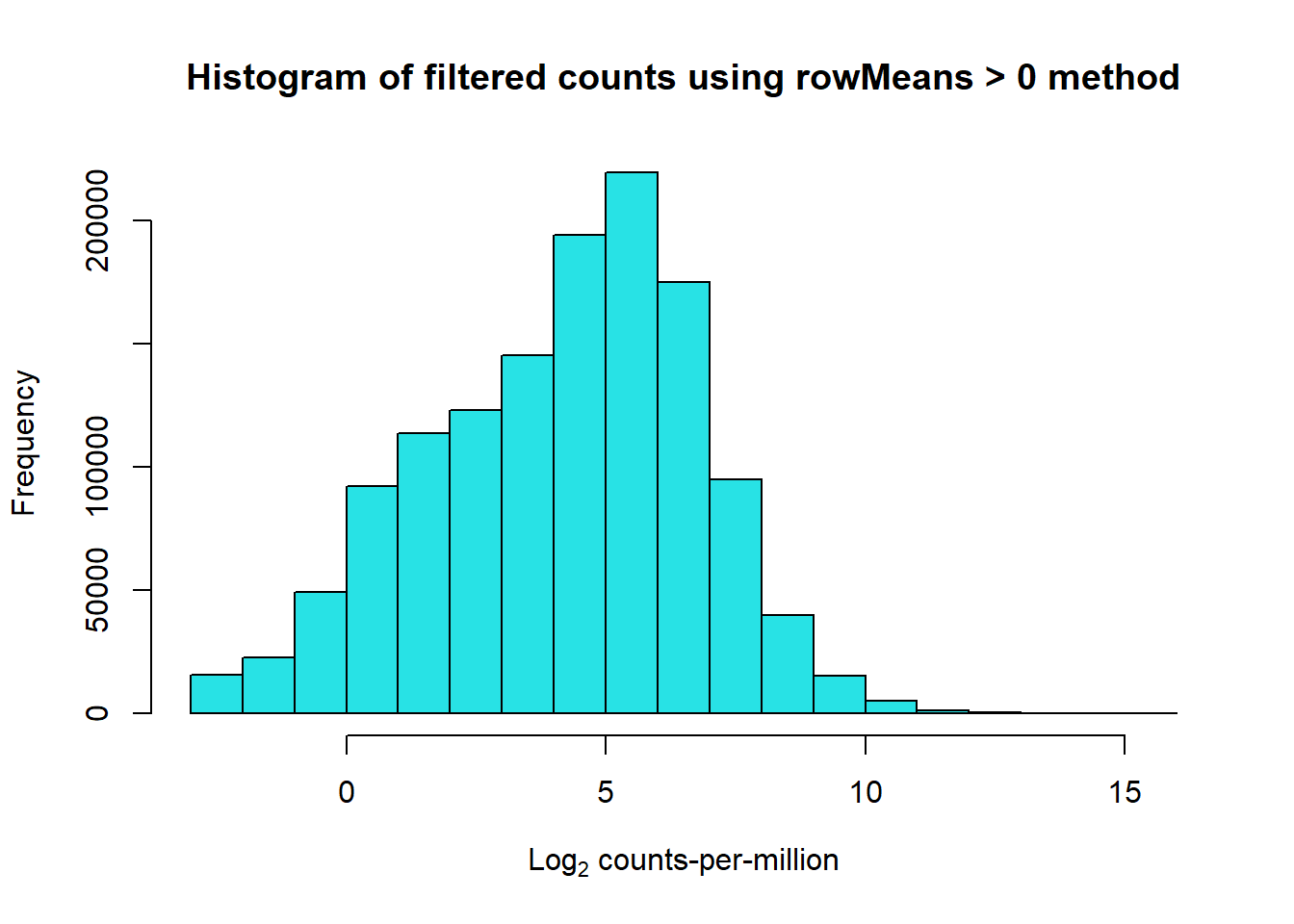
| Version | Author | Date |
|---|---|---|
| 9aa0597 | John D. Hurley | 2026-01-22 |
# saveRDS(Filt_RMG0_RNA_fc,"data/QC/Filt_RMG0_RNA_fc_Ensemble.RDS")
# saveRDS(Filt_RMG0_RNA_log2cpm,"data/QC/Filt_RMG0_RNA_log2cpm_Ensemble.RDS")boxplot(Filt_RMG0_RNA_log2cpm, main = "Boxplots of log cpm per sample (RowMeans>0)",xaxt = "n", xlab= "")
axis(1,
at = 1:length(col_names), # positions (one per sample)
labels = col_names, # your labels vector
las = 2, # rotate text vertically (like srt=90)
cex.axis = 0.3) # shrink label size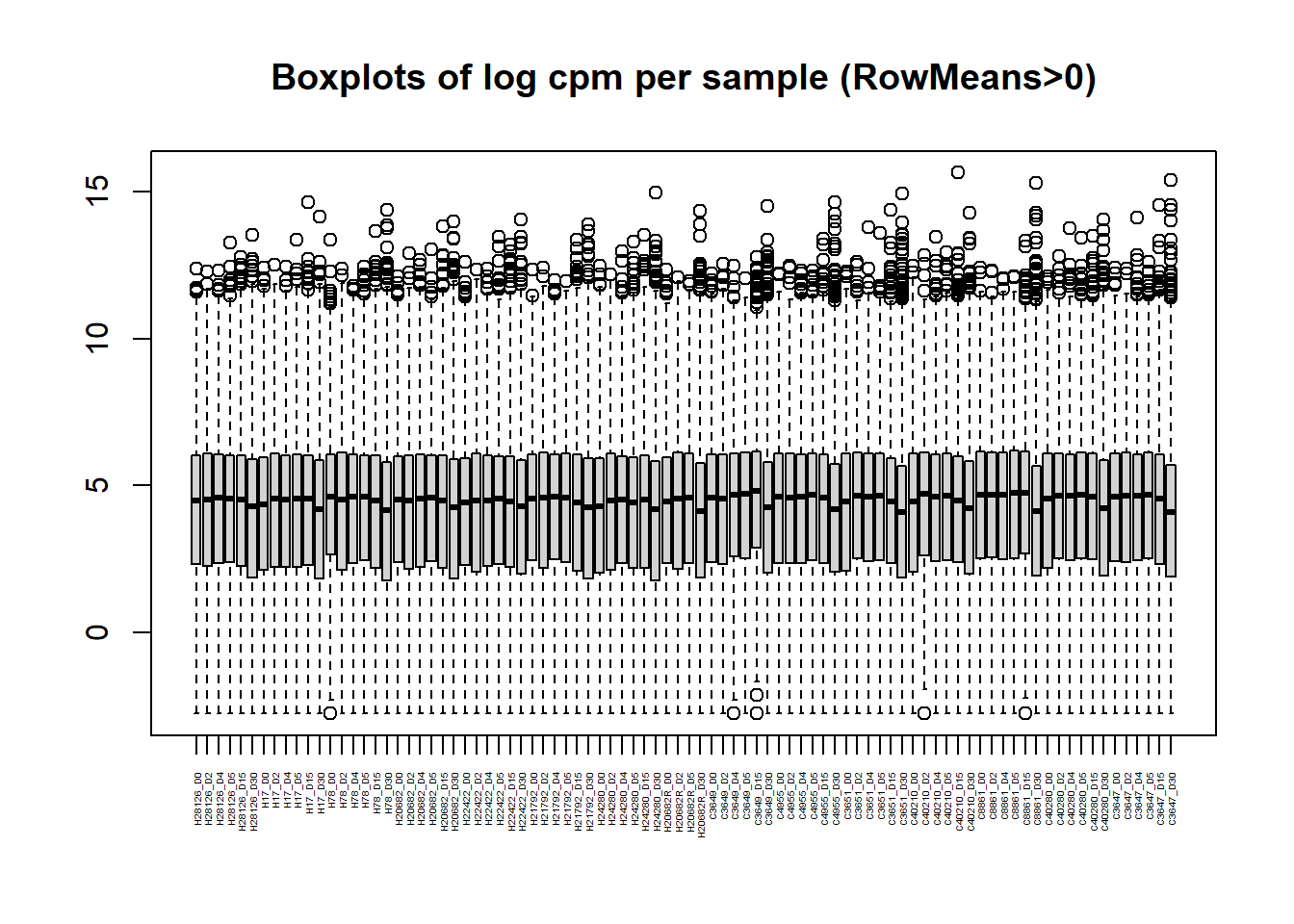
| Version | Author | Date |
|---|---|---|
| 9aa0597 | John D. Hurley | 2026-01-22 |
######Cor_HeatMap####
cor_Filt_RMG0_RNA_log2cpm <- cor(Filt_RMG0_RNA_log2cpm, method = "spearman")
individual <- RNA_Metadata$Individual
species <- RNA_Metadata$Species
timepoint <- RNA_Metadata$Timepoint
timepoint <- factor(timepoint,levels = c("Day0","Day2","Day4","Day5","Day15","Day30"))
Cor_metadata <- data.frame(
sample_cor = colnames(Filt_RMG0_RNA_log2cpm),
species_cor = species,
timepoint_cor = timepoint,
individual_cor = individual
)
ann_colors <- list(
timepoint_cor = c(
"Day0" = "#883268", # Purple
"Day2" = "#3E7274", # blue
"Day4" = "#5AAA464D", # light green
"Day5" = "#94C47D", # Green
"Day15" = "#C03830", # red
"Day30" = "#830C05" # dark red
),
species_cor = c(
"H" = "#171717", # black
"C" = "#17171717" # light grey
),
individual_cor = c(
H1 = "#091638", #Blue-Green Darkest
H2 = "#11185B",
H3 = "#0F2C71",
H4 = "#0D568F",
H5 = "#1D8296",
H6 = "#46A389",
H7 = "#9DD484", #Blue-Green Lightest
C1 = "#340702", #Brown-Orange darkest
C2 = "#5D0B02",
C3 = "#951302",
C4 = "#D32804",
C5 = "#F74019",
C6 = "#FA7A38",
C7 = "#FCC598"
)
)
rownames(Cor_metadata) <- Cor_metadata$sample_cor
# saveRDS(cor_Filt_RMG0_RNA_log2cpm, "data/QC/Cor_Filt_RMG0_RNA_log2cpm.RDS")
# saveRDS(Cor_metadata, "data/QC/Cor_metadata.RDS")
# saveRDS(ann_colors,"data/QC/ann_colors.RDS")print(
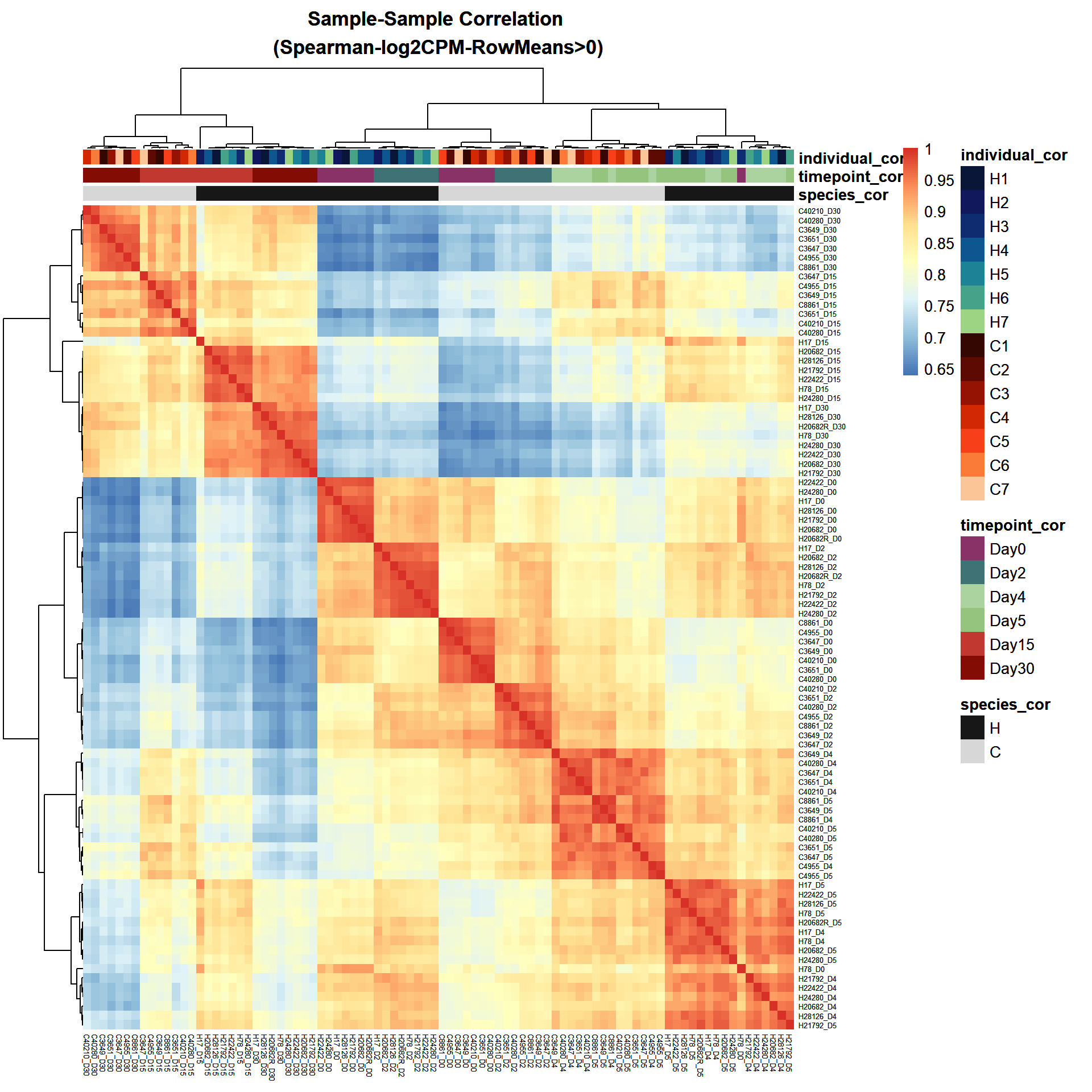
pheatmap(cor_Filt_RMG0_RNA_log2cpm,
fontsize_row = 5,
fontsize_col = 5,
annotation_col = Cor_metadata[, c("species_cor", "timepoint_cor","individual_cor")],
annotation_colors = ann_colors,
clustering_distance_rows = "correlation",
clustering_distance_cols = "correlation",
main = "Sample-Sample Correlation \n(Spearman-log2CPM-RowMeans>0)")
)
| Version | Author | Date |
|---|---|---|
| 9aa0597 | John D. Hurley | 2026-01-22 |
####Subset####
RNA_Metadata_No4 <- RNA_Metadata %>%
filter(timepoint != "Day4")
RNA_fc_NoD4 <- RNA_fc %>%
dplyr::select(-ends_with("_D4"))
RNA_log2cpm_NoD4 <- cpm(RNA_fc_NoD4,log=TRUE)
dim(RNA_log2cpm_NoD4)[1] 44125 74dim(RNA_fc)[1] 44125 88row_means_NoD4 <- rowMeans(RNA_log2cpm_NoD4)
Filt_RMG0_RNA_fc_NoD4 <- RNA_fc_NoD4[row_means_NoD4 >0,]
dim(Filt_RMG0_RNA_fc_NoD4)[1] 14838 74Filt_RMG0_RNA_log2cpm_NoD4 <- cpm(Filt_RMG0_RNA_fc_NoD4,log=TRUE)
# saveRDS(RNA_Metadata_No4,"data/QC/RNA_Metatdata_No4.RDS")
# saveRDS(Filt_RMG0_RNA_fc_NoD4,"data/QC/Filt_RMG0_RNA_fc_NoD4_Ensemble.RDS")
# saveRDS(Filt_RMG0_RNA_log2cpm_NoD4, "data/QC/Filt_RMG0_RNA_log2cpm_NoD4_Ensemble.RDS")######Cor_HeatMap####
cor_Filt_RMG0_RNA_log2cpm_NoD4 <- cor(Filt_RMG0_RNA_log2cpm_NoD4, method = "spearman")
Cor_metadata_No4 <- Cor_metadata %>%
dplyr::filter(timepoint_cor !="Day4")
ann_colors_No4 <- ann_colors
ann_colors_No4$timepoint_cor <- ann_colors$timepoint_cor[
names(ann_colors$timepoint_cor) != "Day4"
]
# saveRDS(Cor_metadata_No4, "data/QC/Cor_metadata_No4.RDS")
# saveRDS(cor_Filt_RMG0_RNA_log2cpm_NoD4, "data/QC/cor_Filt_RMG0_RNA_log2cpm_NoD4.RDS")
# saveRDS(ann_colors_No4,"data/QC/ann_colors_no4.RDS")print(
pheatmap(cor_Filt_RMG0_RNA_log2cpm_NoD4,
fontsize_row = 5,
fontsize_col = 5,
annotation_col = Cor_metadata_No4[, c("species_cor", "timepoint_cor","individual_cor")],
annotation_colors = ann_colors_No4,
clustering_distance_rows = "correlation",
clustering_distance_cols = "correlation",
main = "Sample-Sample Correlation (Spearman) \n (log2CPM-RowMeans>0-NoDay4)")
)
| Version | Author | Date |
|---|---|---|
| 9aa0597 | John D. Hurley | 2026-01-22 |
# #git -> commit all changes
# #git -> push
wflow_publish("analysis/RNA_CorrelationHeatMap.Rmd")
sessionInfo()R version 4.5.1 (2025-06-13 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 11 x64 (build 26100)
Matrix products: default
LAPACK version 3.12.1
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] lubridate_1.9.4 forcats_1.0.1 stringr_1.5.2
[4] purrr_1.1.0 tidyr_1.3.1 tidyverse_2.0.0
[7] Cormotif_1.54.0 affy_1.86.0 pheatmap_1.0.13
[10] org.Hs.eg.db_3.21.0 AnnotationDbi_1.70.0 IRanges_2.42.0
[13] S4Vectors_0.46.0 Biobase_2.68.0 BiocGenerics_0.54.0
[16] generics_0.1.4 readr_2.1.5 ggrepel_0.9.6
[19] dplyr_1.1.4 tibble_3.3.0 ggplot2_4.0.0
[22] edgeR_4.6.3 limma_3.64.3 workflowr_1.7.2
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 farver_2.1.2 blob_1.3.0
[4] Biostrings_2.76.0 S7_0.2.0 fastmap_1.2.0
[7] promises_1.3.3 digest_0.6.37 timechange_0.3.0
[10] lifecycle_1.0.5 statmod_1.5.0 processx_3.8.6
[13] KEGGREST_1.48.1 RSQLite_2.4.3 magrittr_2.0.3
[16] compiler_4.5.1 rlang_1.1.6 sass_0.4.10
[19] tools_4.5.1 yaml_2.3.10 knitr_1.51
[22] bit_4.6.0 RColorBrewer_1.1-3 withr_3.0.2
[25] grid_4.5.1 preprocessCore_1.70.0 git2r_0.36.2
[28] scales_1.4.0 cli_3.6.5 rmarkdown_2.30
[31] crayon_1.5.3 otel_0.2.0 rstudioapi_0.17.1
[34] httr_1.4.7 tzdb_0.5.0 DBI_1.2.3
[37] cachem_1.1.0 BiocManager_1.30.27 XVector_0.48.0
[40] vctrs_0.6.5 jsonlite_2.0.0 callr_3.7.6
[43] hms_1.1.4 bit64_4.6.0-1 locfit_1.5-9.12
[46] jquerylib_0.1.4 affyio_1.78.0 glue_1.8.0
[49] ps_1.9.1 stringi_1.8.7 gtable_0.3.6
[52] later_1.4.4 GenomeInfoDb_1.44.3 UCSC.utils_1.4.0
[55] pillar_1.11.1 htmltools_0.5.8.1 GenomeInfoDbData_1.2.14
[58] R6_2.6.1 rprojroot_2.1.1 evaluate_1.0.5
[61] lattice_0.22-7 png_0.1-8 memoise_2.0.1
[64] httpuv_1.6.16 bslib_0.9.0 Rcpp_1.1.0
[67] whisker_0.4.1 xfun_0.53 fs_1.6.6
[70] getPass_0.2-4 pkgconfig_2.0.3