RNA_CorrelationHeatMap
2026-01-14
Last updated: 2026-03-30
Checks: 7 0
Knit directory: CrossSpecies_CM_Diff_RNA/
This reproducible R Markdown analysis was created with workflowr (version 1.7.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20251129) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5a7c251. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/RNA_CorrelationHeatMap_Ensemble.Rmd) and HTML
(docs/RNA_CorrelationHeatMap_Ensemble.html) files. If
you’ve configured a remote Git repository (see
?wflow_git_remote), click on the hyperlinks in the table
below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 5a7c251 | John D. Hurley | 2026-03-30 | Changes to data QC checks |
| html | 0958201 | John D. Hurley | 2026-03-23 | Build site. |
| Rmd | 01611df | John D. Hurley | 2026-03-23 | Data updates |
| html | 8bce9df | John D. Hurley | 2026-02-20 | Build site. |
| html | fcf8fc0 | John D. Hurley | 2026-02-18 | Build site. |
| Rmd | c85837b | John D. Hurley | 2026-02-18 | SiteUpDate_CorrelationHeatMap |
| html | 7789dff | John D. Hurley | 2026-01-28 | Build site. |
| Rmd | 0fcf7c3 | John D. Hurley | 2026-01-28 | HeatMap knit update |
| html | d9f15a6 | John D. Hurley | 2026-01-28 | Build site. |
| Rmd | 19cd9fb | John D. Hurley | 2026-01-28 | Commenting out wokrflowr publish |
| Rmd | 3b54f65 | John D. Hurley | 2026-01-28 | Duplicate Code Labels |
| Rmd | de67e64 | John D. Hurley | 2026-01-28 | Commenting out code not in use. |
| Rmd | da444ed | John D. Hurley | 2026-01-28 | RMarkdown name update |
| Rmd | 085c1db | John D. Hurley | 2026-01-28 | Finalizing CorHeatMap |
RNA_Metadata <- read_excel("~/diff_timeline_tes/RNA/RNA_Metadata.xlsx")
counts_table <- read.delim("~/diff_timeline_tes/RNA/rna_concat/featureCounts/all_counts.txt")
marker_genes <- read.delim("~/diff_timeline_tes/RNA/CM_Diff_PurityPanel.txt")
RNA_fc <- counts_table %>%
filter(Geneid != "#") %>%
filter(Geneid != "Geneid") %>%
column_to_rownames("Geneid")
# #Rename column Names to More Useful Info
col_names <- RNA_Metadata$SampleName
colnames(RNA_fc) <- col_names
dim(RNA_fc)
ensembl_ids_unfilt <- rownames(RNA_fc)
entrez_ids_unfilt <- mapIds(org.Hs.eg.db,
keys = ensembl_ids_unfilt,
column = "ENTREZID",
keytype = "ENSEMBL",
multiVals = "first")
symbol_ids_unfilt <- mapIds(org.Hs.eg.db,
keys = ensembl_ids_unfilt,
column = "SYMBOL",
keytype = "ENSEMBL",
multiVals = "first")
RNA_fc_df <- as.data.frame(RNA_fc)
RNA_fc_df <- RNA_fc_df %>%
rownames_to_column(var = "Ensemble") %>%
dplyr::mutate(
Entrez_ID = entrez_ids_unfilt,
Symbol = symbol_ids_unfilt
) %>%
dplyr::select(
Ensemble, # 1st column
Entrez_ID, # 2nd column
Symbol, # 3rd column
everything() # rest unchanged
)
rn <- rownames(RNA_fc)
RNA_fc <- as.data.frame(
lapply(RNA_fc, function(x) as.numeric(as.character(x)))
)
rownames(RNA_fc) <- rn
# saveRDS(RNA_fc,"data/QC/concat/RNA_fc.RDS")
# saveRDS(RNA_fc_df,"data/Raw_Data/concat/RNA_fc_df.RDS")
# saveRDS(RNA_Metadata,"data/Raw_Data/concat/RNA_Metadata.RDS")library(tidyverse)
total_reads_df <- RNA_fc %>%
as.data.frame() %>%
mutate(Gene = rownames(.)) %>%
dplyr::select(-Gene) %>%
summarise(across(everything(), \(x) sum(x, na.rm = TRUE))) %>%
pivot_longer(
cols = everything(),
names_to = "Sample",
values_to = "Total_Counts"
) %>%
left_join(RNA_Metadata, by = c("Sample" = "SampleName"))
day_levels <- c("Day0","Day2","Day4","Day5","Day15","Day30")
total_reads_df <- total_reads_df %>%
mutate(
Timepoint = factor(Timepoint, levels = day_levels, ordered = TRUE),
# force species order: chimp first, human second
Species = factor(Species, levels = c("C", "H")),
Individual_Color = ann_colors$individual_cor[Individual]
) %>%
arrange(Species, Individual, Timepoint) %>%
mutate(Sample = factor(Sample, levels = Sample))library(ggplot2)
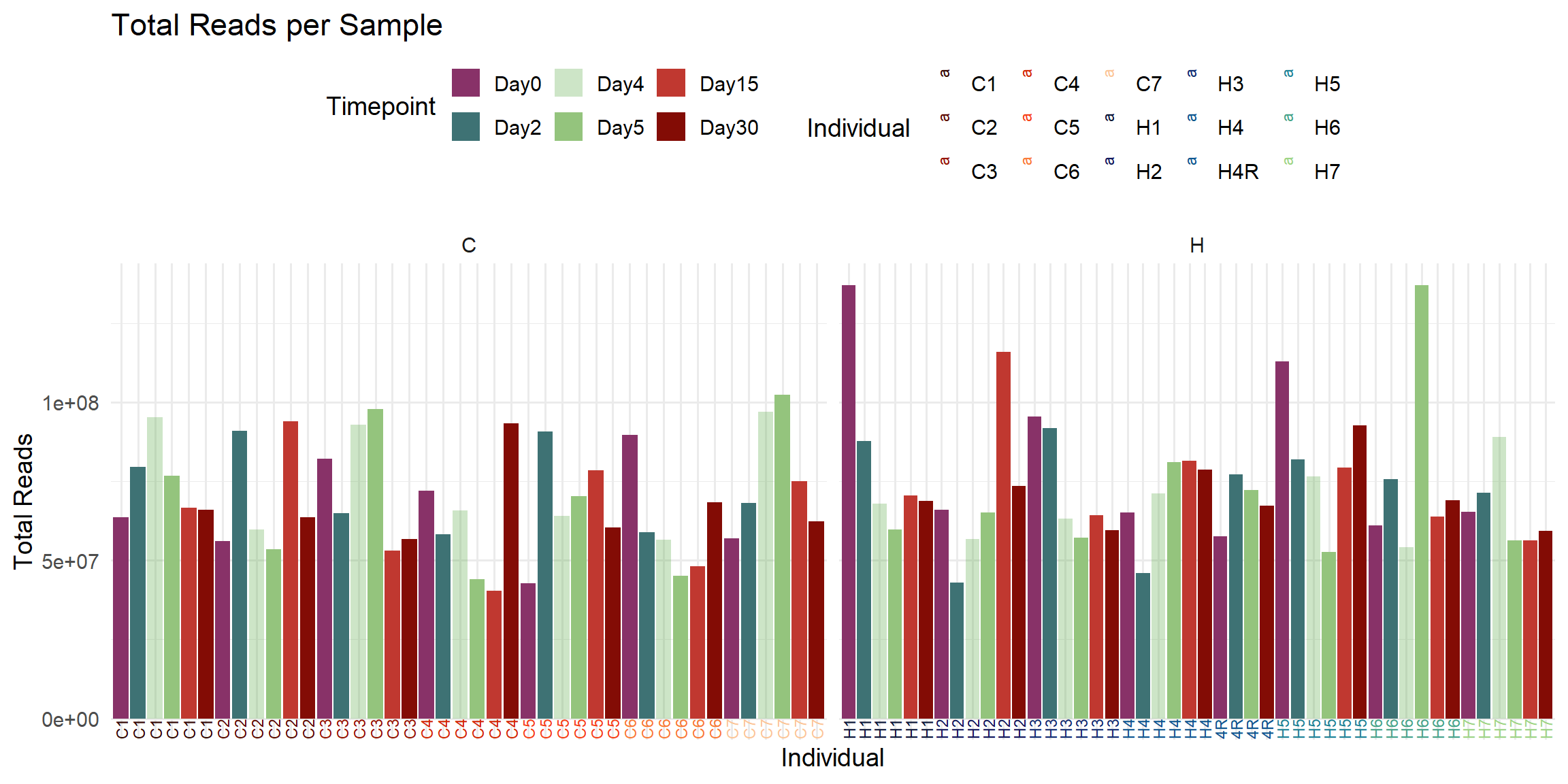
ggplot(total_reads_df, aes(x = Sample, y = Total_Reads, fill = Timepoint)) +
geom_col() +
geom_text(aes(label = Individual, color = Individual),
y = 0, angle = 90, hjust = 1, vjust = 0.5, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
legend.position = "top") +
labs(
title = "Total Reads per Sample",
x = "Individual",
y = "Total Reads",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
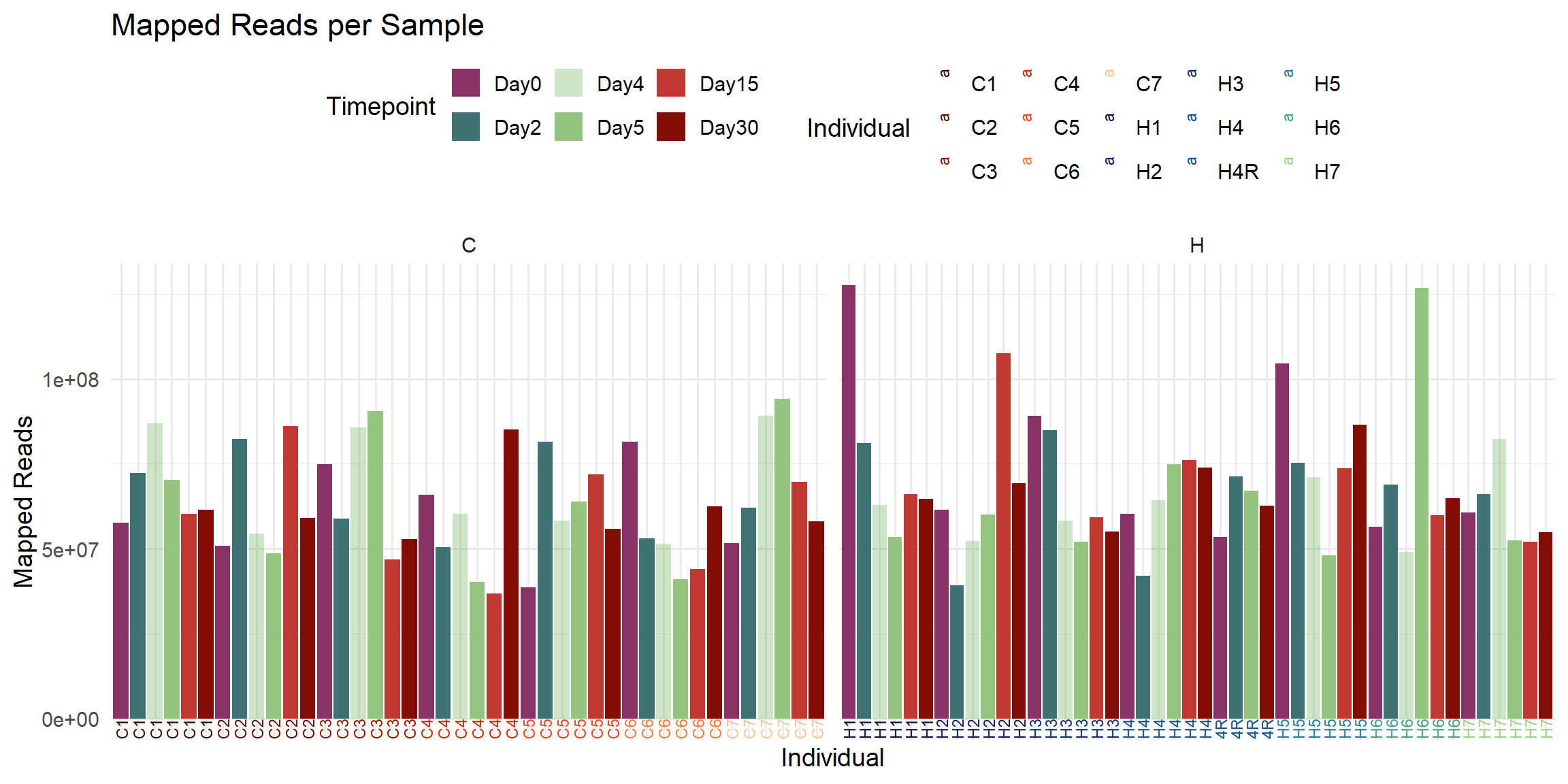
ggplot(total_reads_df, aes(x = Sample, y = Mapped_Reads, fill = Timepoint)) +
geom_col() +
geom_text(aes(label = Individual, color = Individual),
y = 0, angle = 90, hjust = 1, vjust = 0.5, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
legend.position = "top") +
labs(
title = "Mapped Reads per Sample",
x = "Individual",
y = "Mapped Reads",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
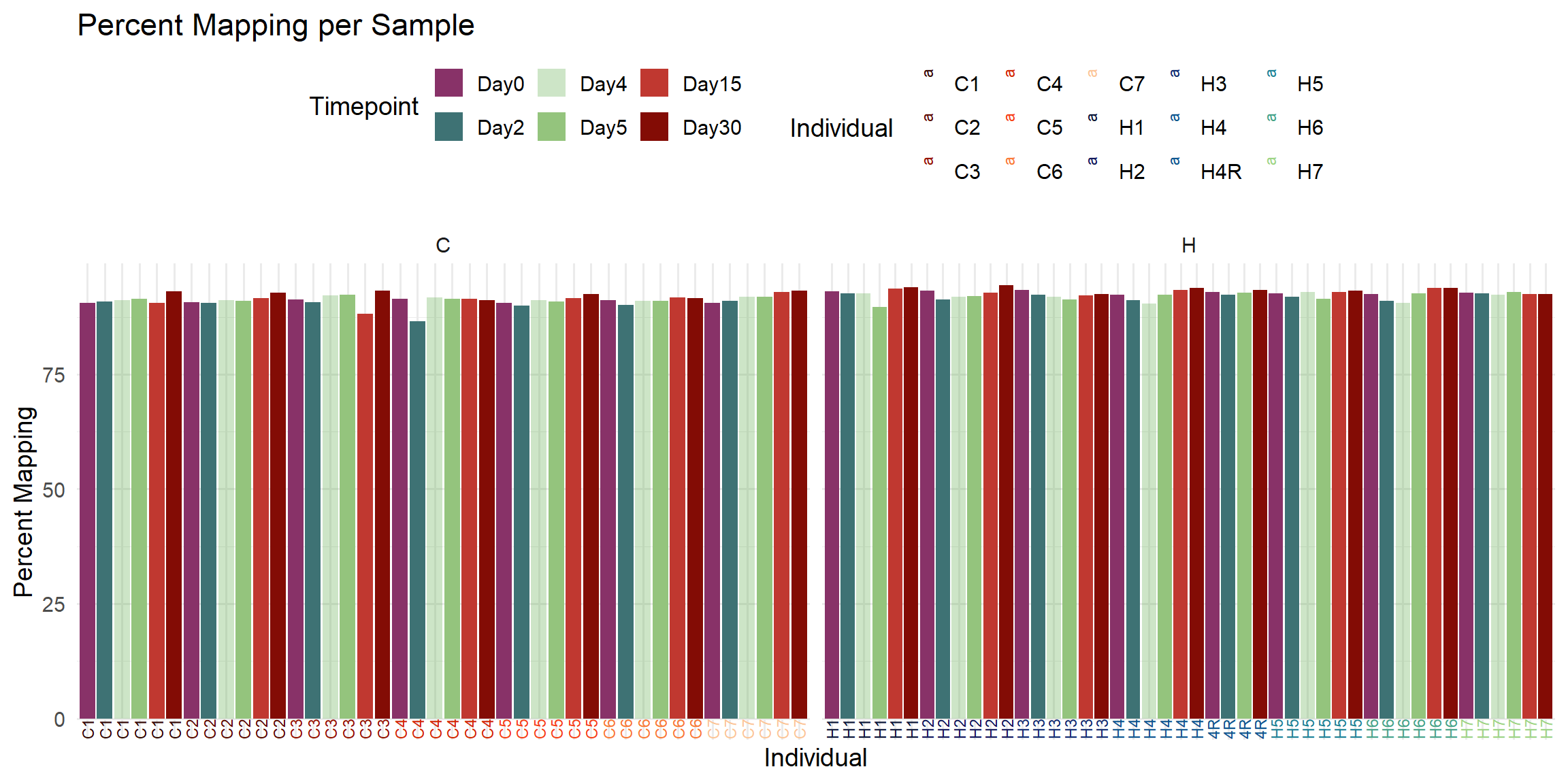
ggplot(total_reads_df, aes(x = Sample, y = Percent_Mapped, fill = Timepoint)) +
geom_col() +
geom_text(aes(label = Individual, color = Individual),
y = 0, angle = 90, hjust = 1, vjust = 0.5, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
legend.position = "top") +
labs(
title = "Percent Mapping per Sample",
x = "Individual",
y = "Percent Mapping",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
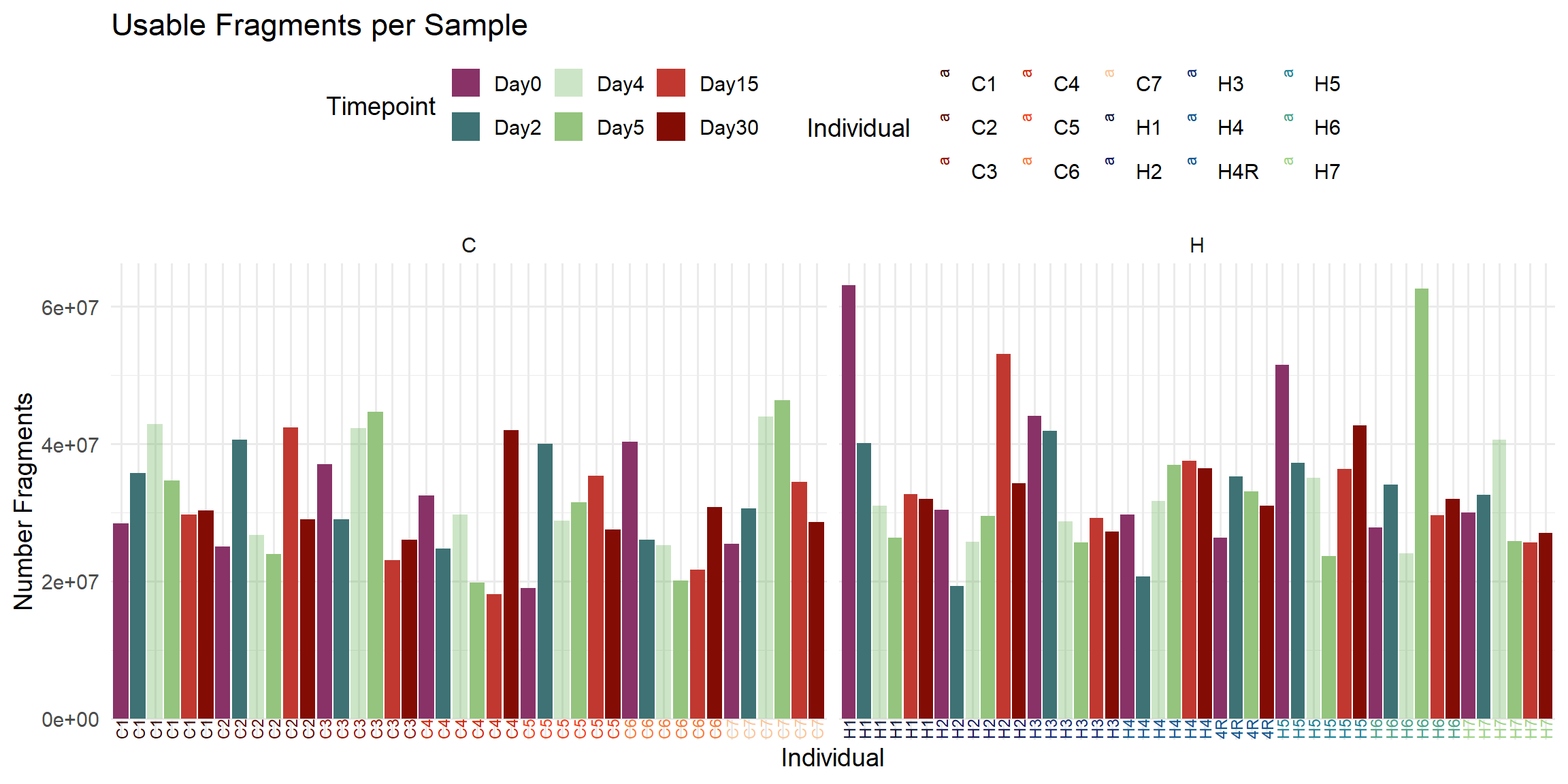
ggplot(total_reads_df, aes(x = Sample, y = Usable_Fragments, fill = Timepoint)) +
geom_col() +
geom_text(aes(label = Individual, color = Individual),
y = 0, angle = 90, hjust = 1, vjust = 0.5, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
legend.position = "top") +
labs(
title = "Usable Fragments per Sample",
x = "Individual",
y = "Number Fragments",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
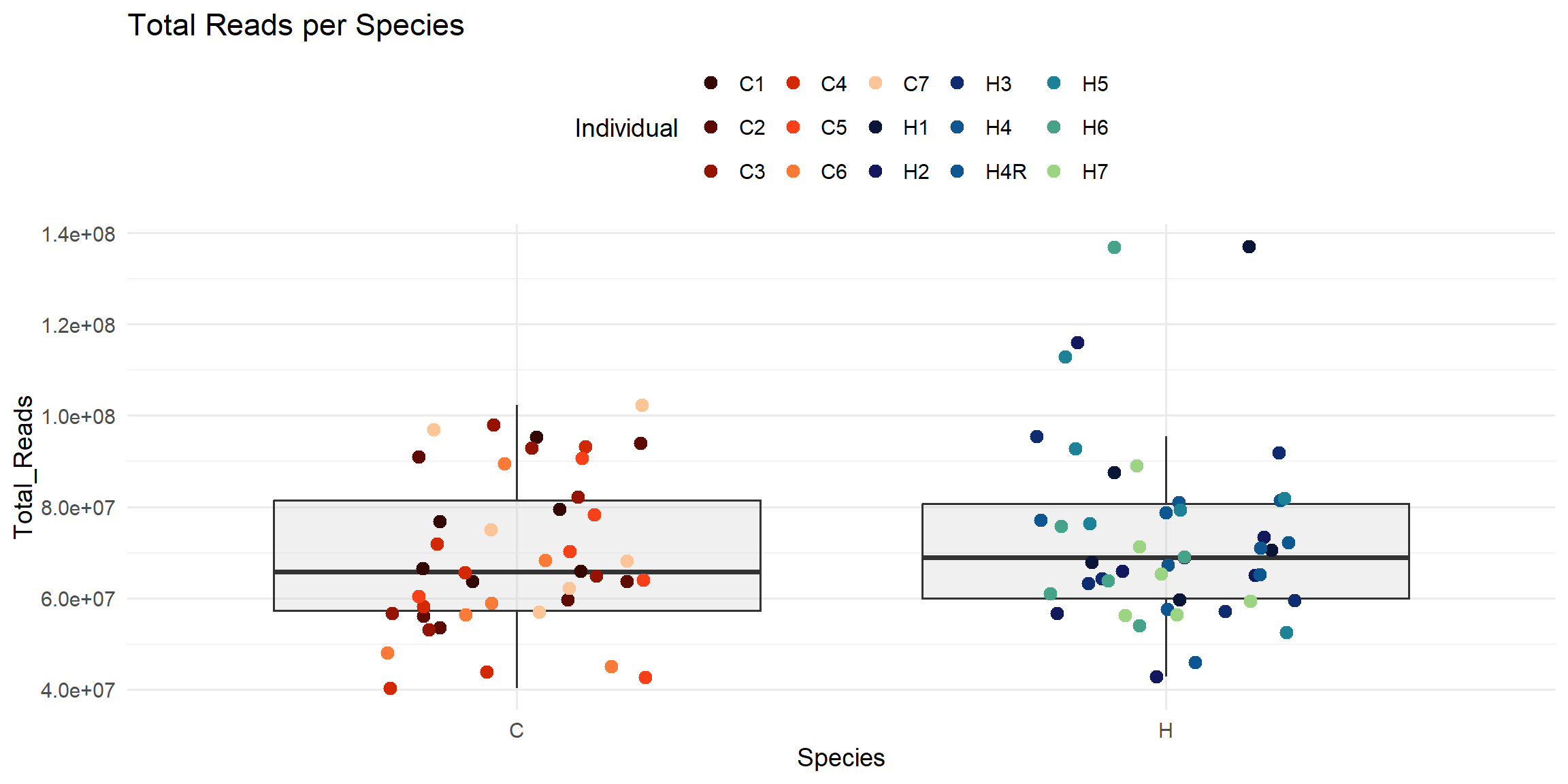
ggplot(total_reads_df, aes(x = Species, y = Total_Reads)) +
geom_boxplot(fill = "#CCCCCC", alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.2, size = 3) +
scale_color_manual(values = ann_colors$individual_cor) +
theme_minimal(base_size = 14) +
theme(legend.position = "top") +
labs(
title = "Total Reads per Species",
x = "Species",
y = "Total_Reads",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
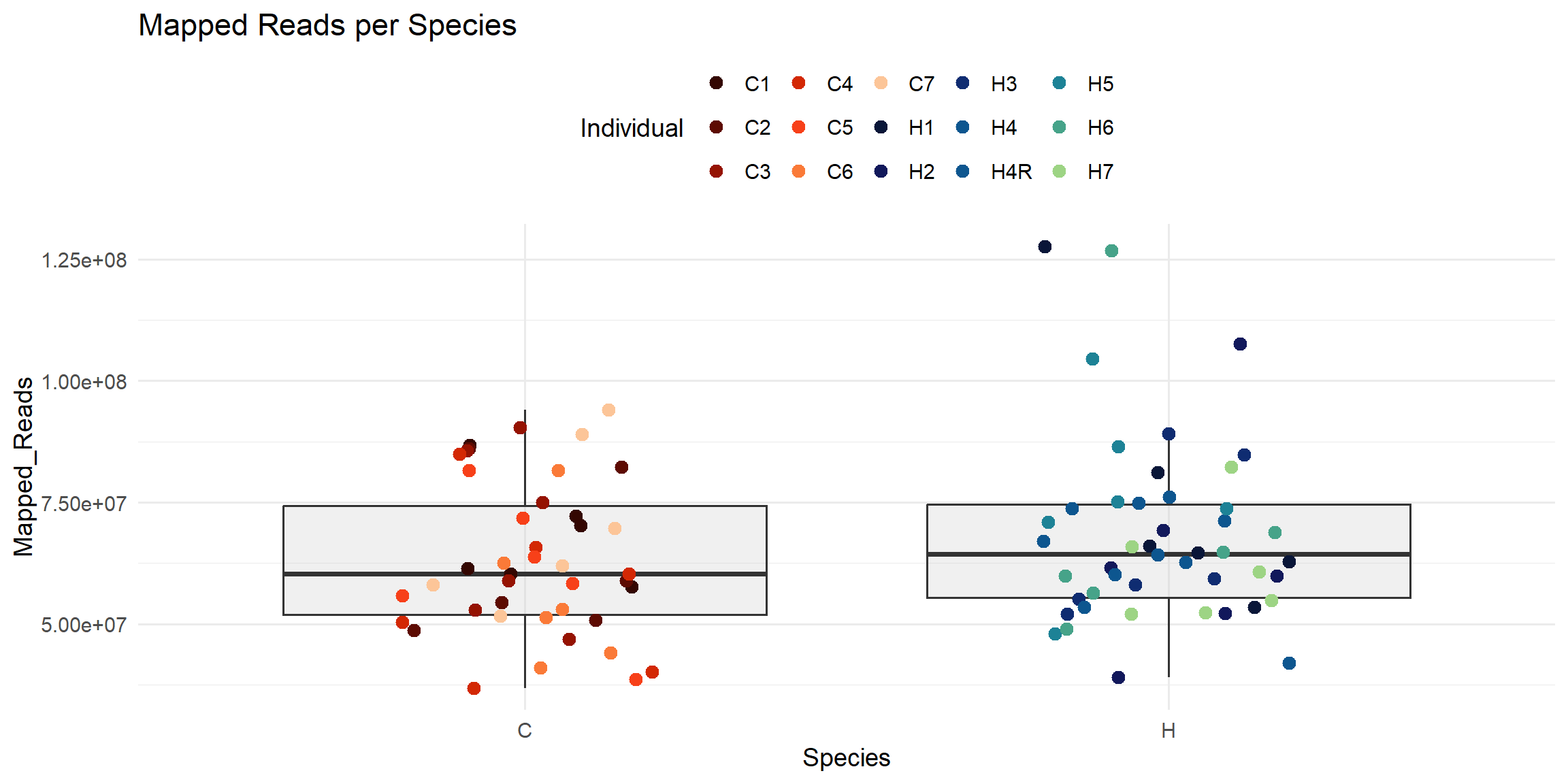
ggplot(total_reads_df, aes(x = Species, y = Mapped_Reads)) +
geom_boxplot(fill = "#CCCCCC", alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.2, size = 3) +
scale_color_manual(values = ann_colors$individual_cor) +
theme_minimal(base_size = 14) +
theme(legend.position = "top") +
labs(
title = "Mapped Reads per Species",
x = "Species",
y = "Mapped_Reads",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
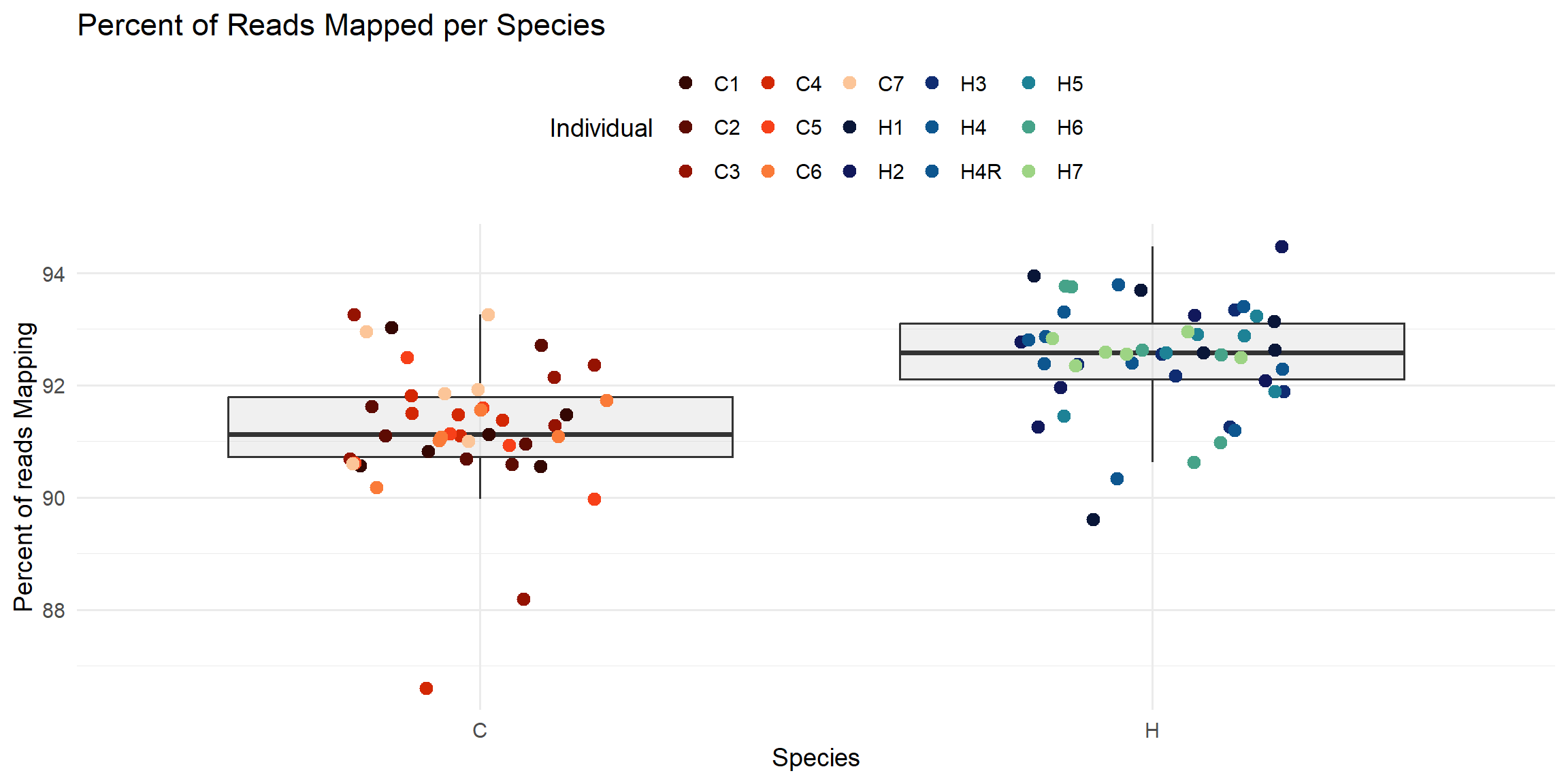
ggplot(total_reads_df, aes(x = Species, y = Percent_Mapped)) +
geom_boxplot(fill = "#CCCCCC", alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.2, size = 3) +
scale_color_manual(values = ann_colors$individual_cor) +
theme_minimal(base_size = 14) +
theme(legend.position = "top") +
labs(
title = "Percent of Reads Mapped per Species",
x = "Species",
y = "Percent of reads Mapping",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
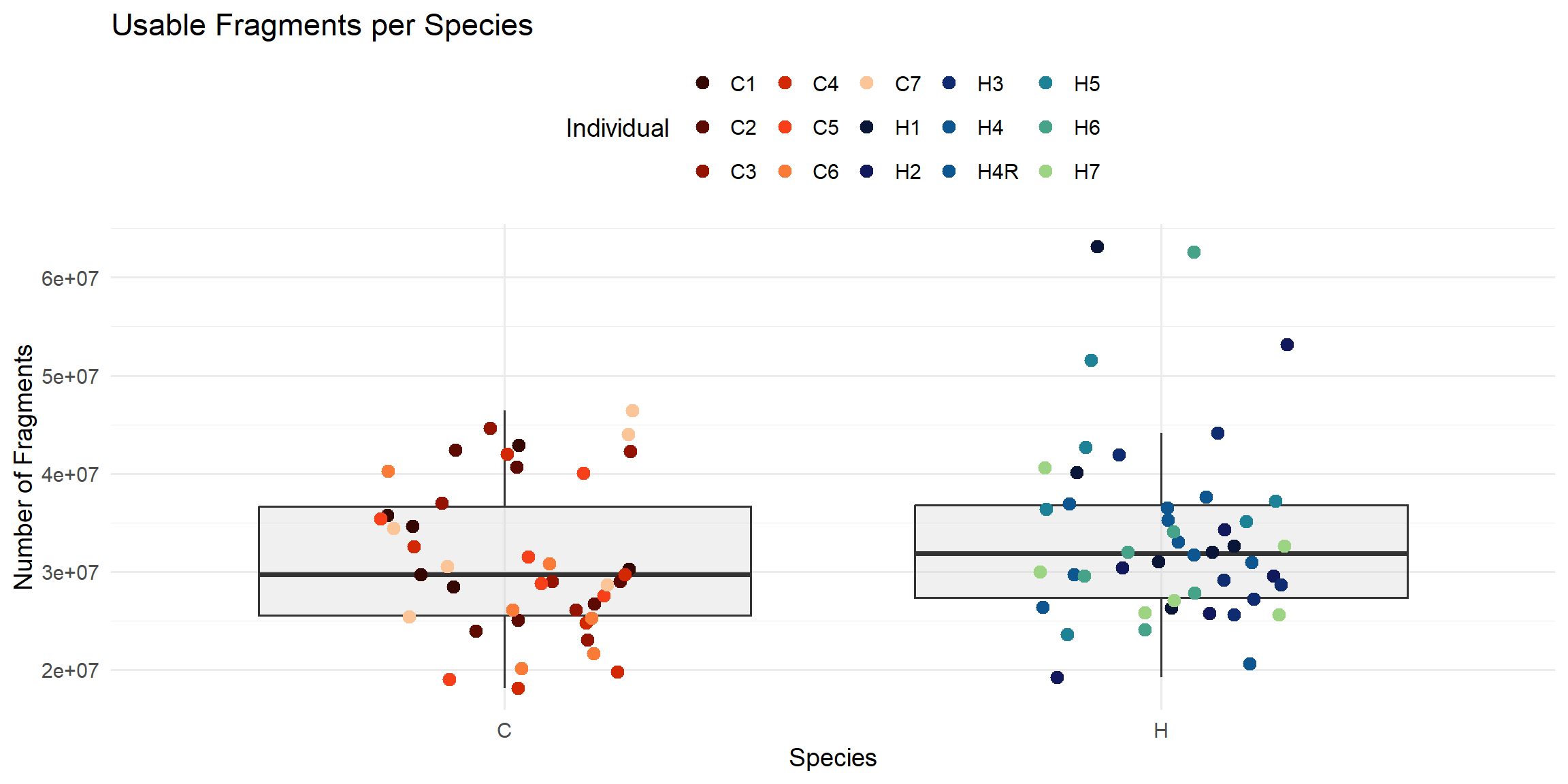
ggplot(total_reads_df, aes(x = Species, y = Usable_Fragments)) +
geom_boxplot(fill = "#CCCCCC", alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.2, size = 3) +
scale_color_manual(values = ann_colors$individual_cor) +
theme_minimal(base_size = 14) +
theme(legend.position = "top") +
labs(
title = "Usable Fragments per Species",
x = "Species",
y = "Number of Fragments",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
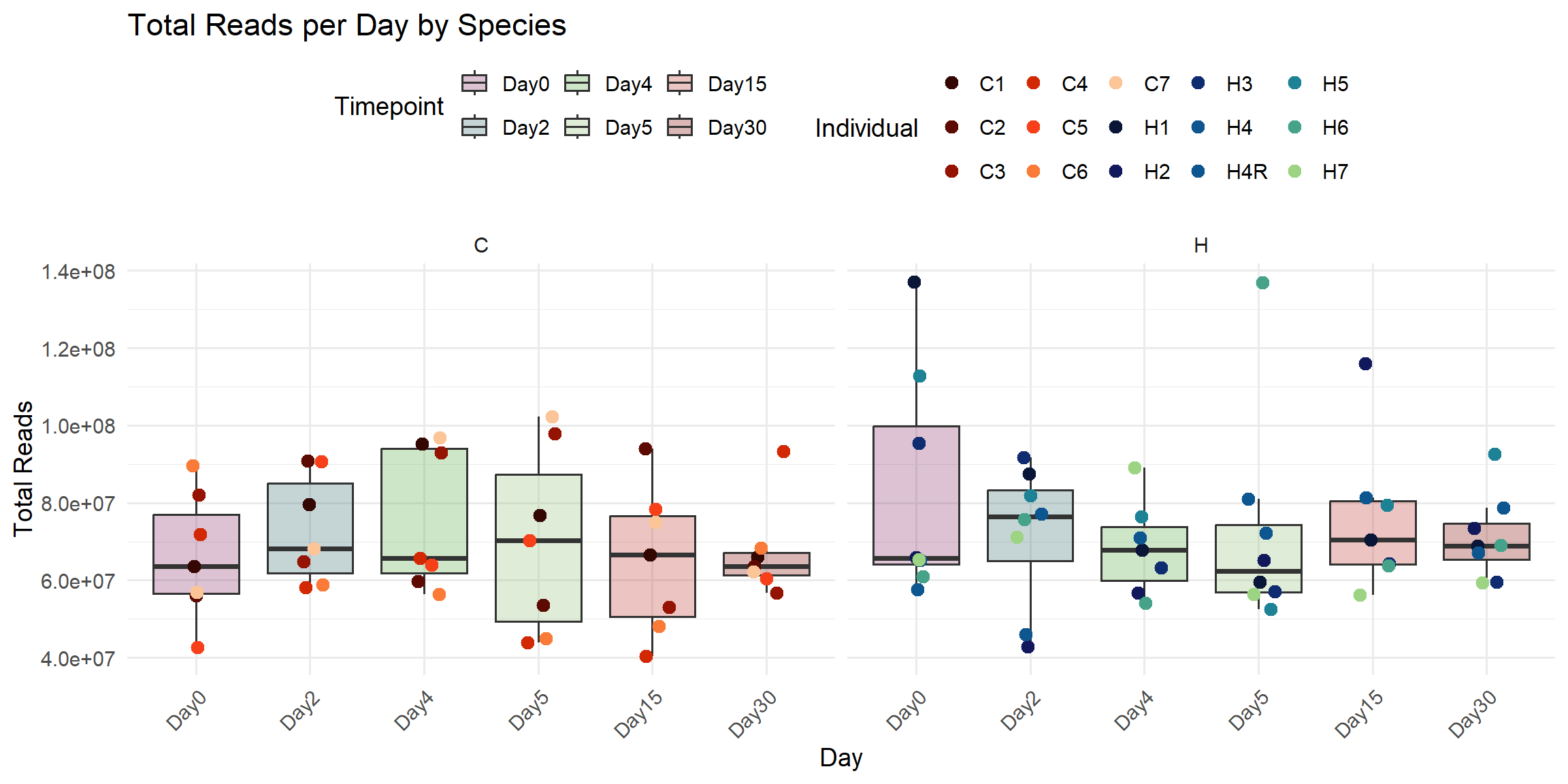
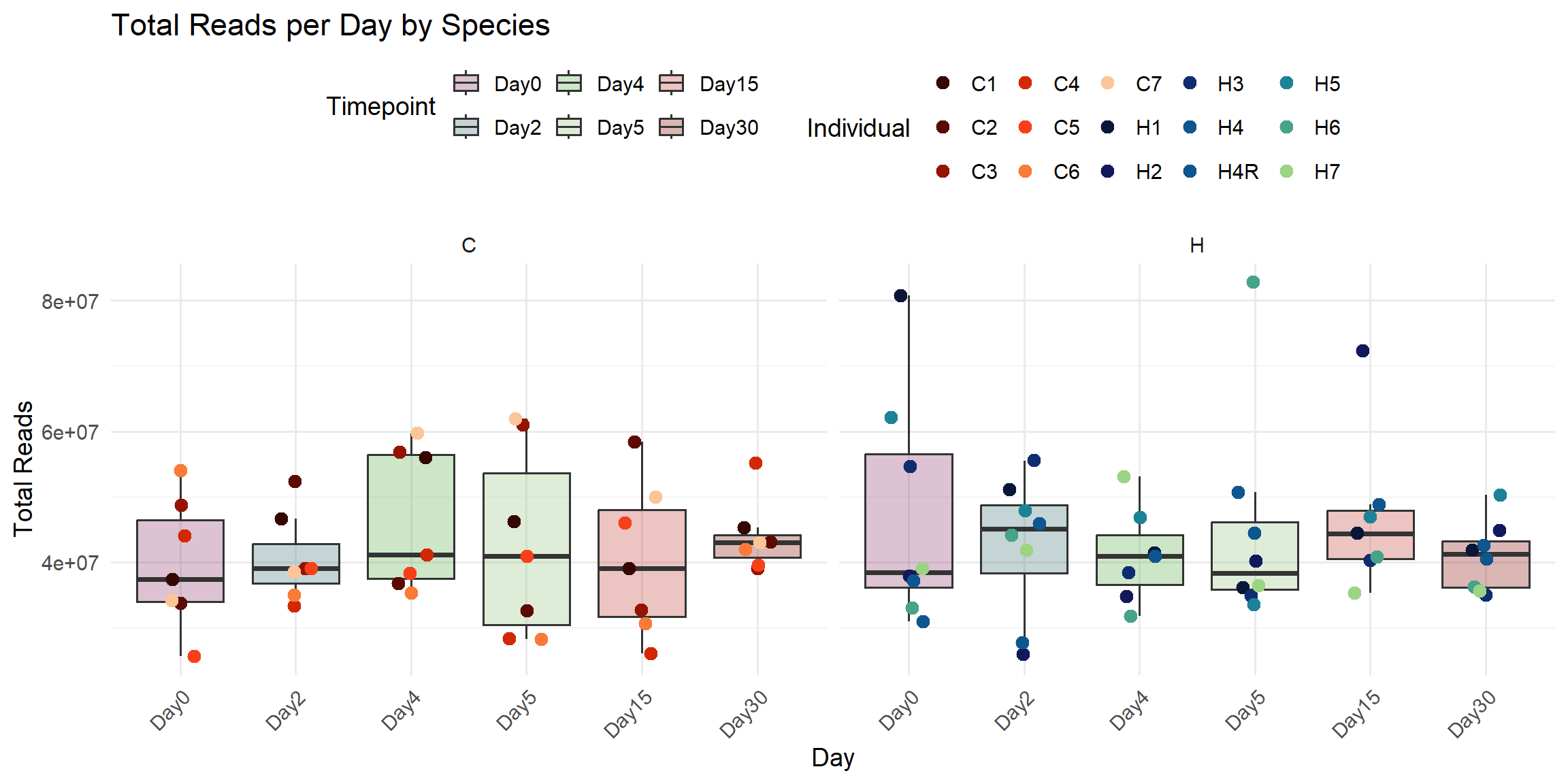
ggplot(total_reads_df, aes(x = Timepoint, y = Total_Reads)) +
geom_boxplot(aes(fill = Timepoint), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.15, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Total Reads per Day by Species",
x = "Day",
y = "Total Reads",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
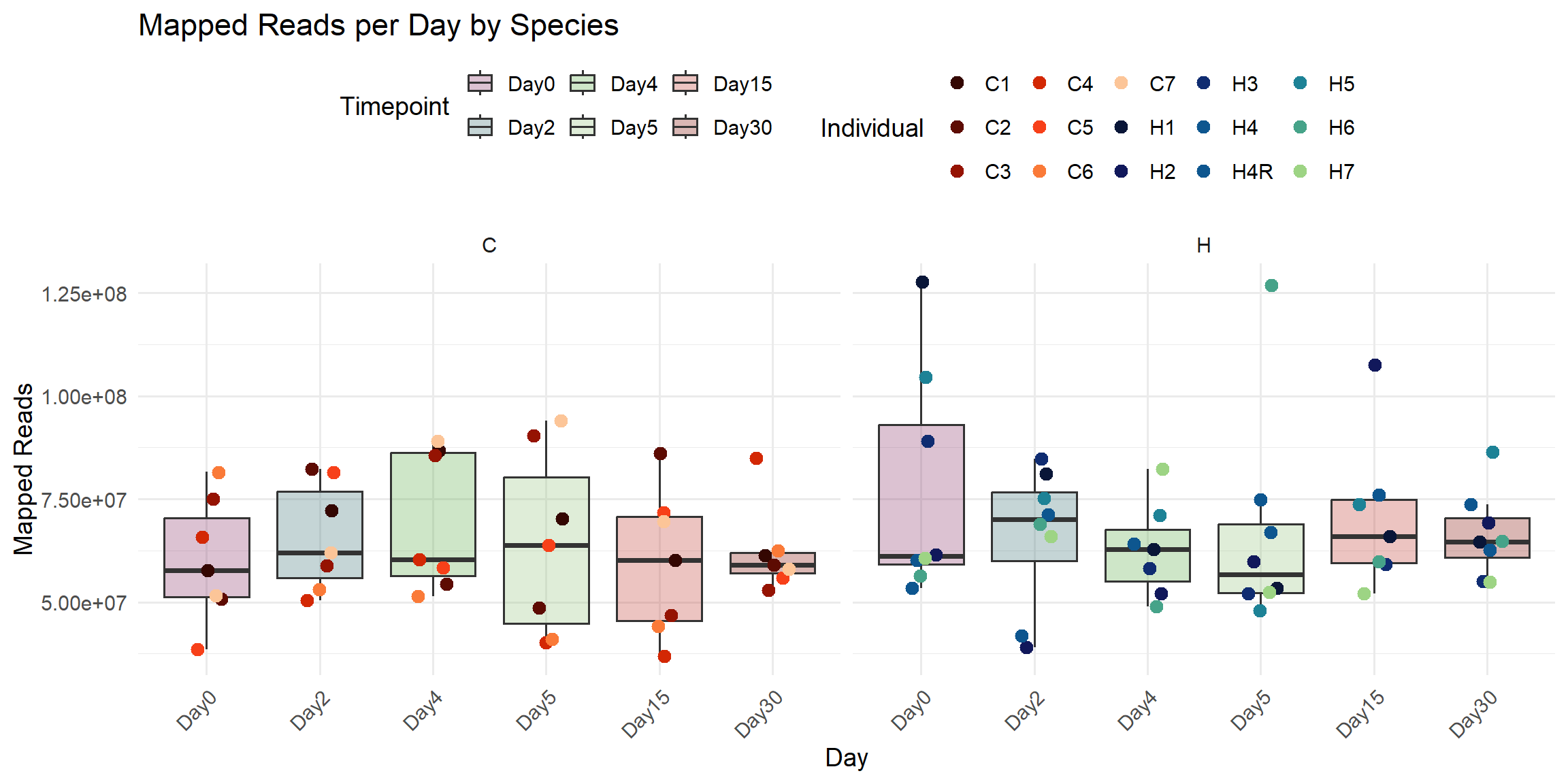
ggplot(total_reads_df, aes(x = Timepoint, y = Mapped_Reads)) +
geom_boxplot(aes(fill = Timepoint), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.15, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Mapped Reads per Day by Species",
x = "Day",
y = "Mapped Reads",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
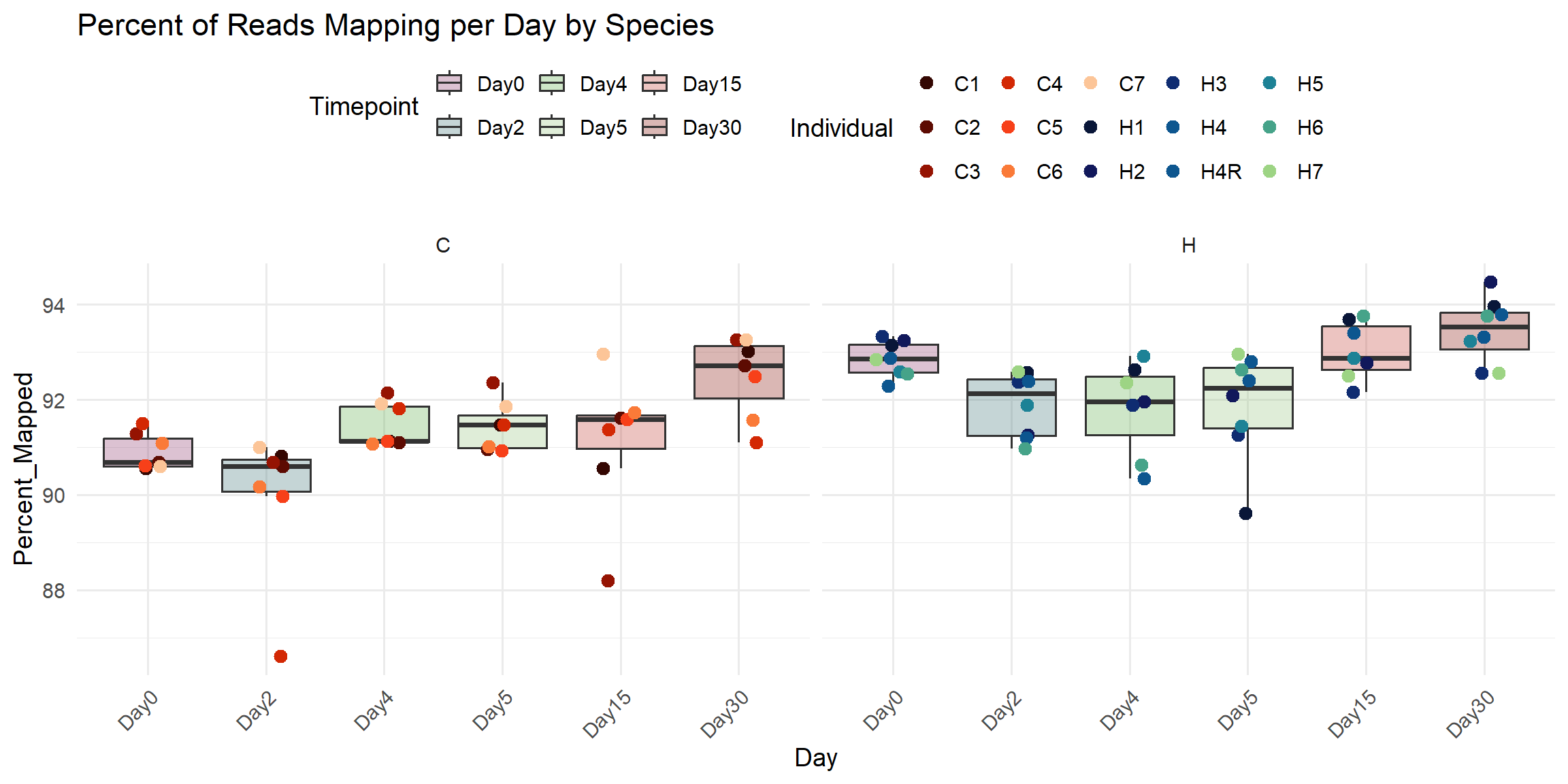
ggplot(total_reads_df, aes(x = Timepoint, y = Percent_Mapped)) +
geom_boxplot(aes(fill = Timepoint), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.15, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Percent of Reads Mapping per Day by Species",
x = "Day",
y = "Percent_Mapped",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
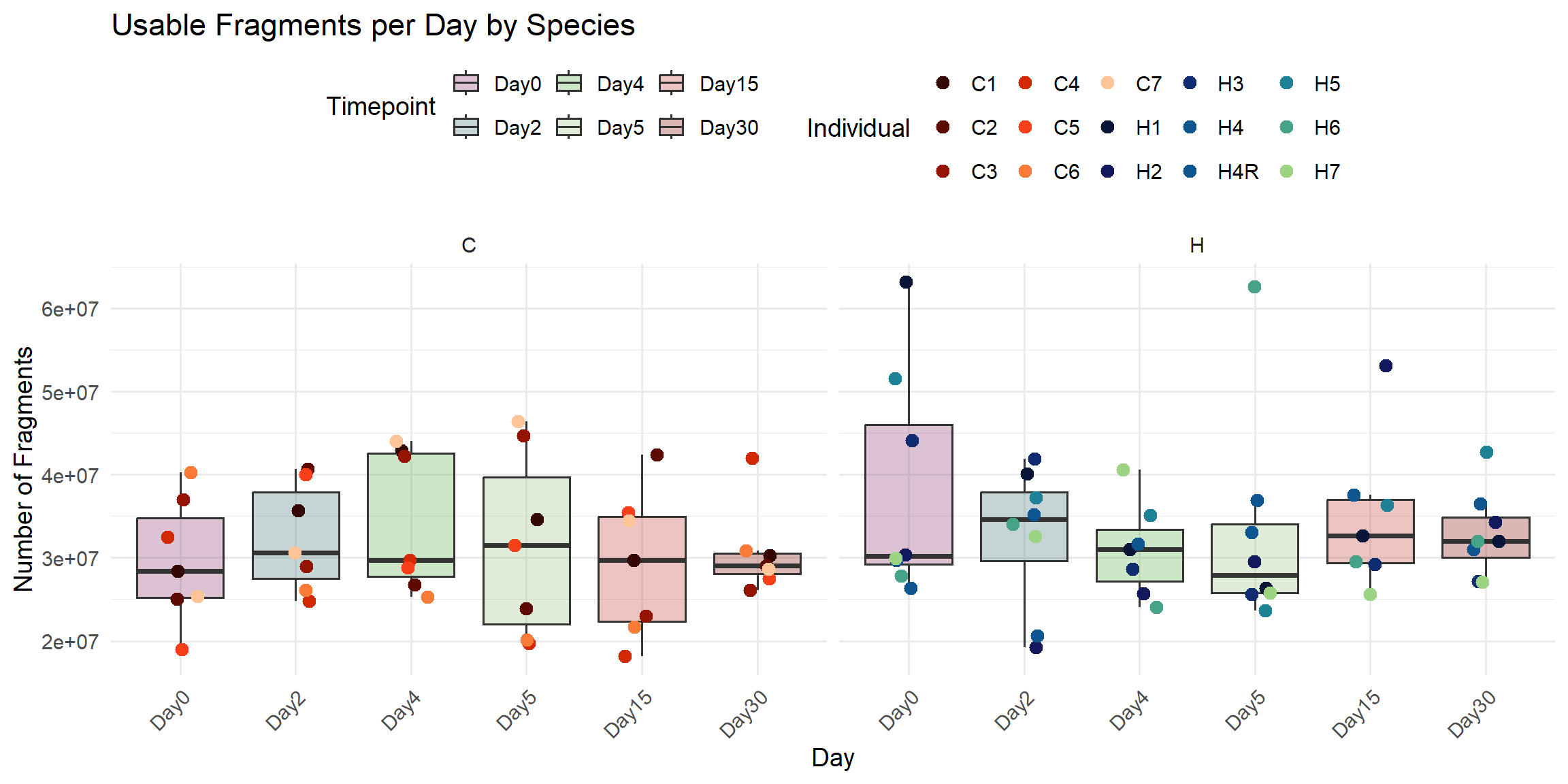
ggplot(total_reads_df, aes(x = Timepoint, y = Usable_Fragments)) +
geom_boxplot(aes(fill = Timepoint), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Individual), width = 0.15, size = 3) +
scale_fill_manual(values = ann_colors$timepoint_cor) +
scale_color_manual(values = ann_colors$individual_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Usable Fragments per Day by Species",
x = "Day",
y = "Number of Fragments",
fill = "Timepoint",
color = "Individual"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
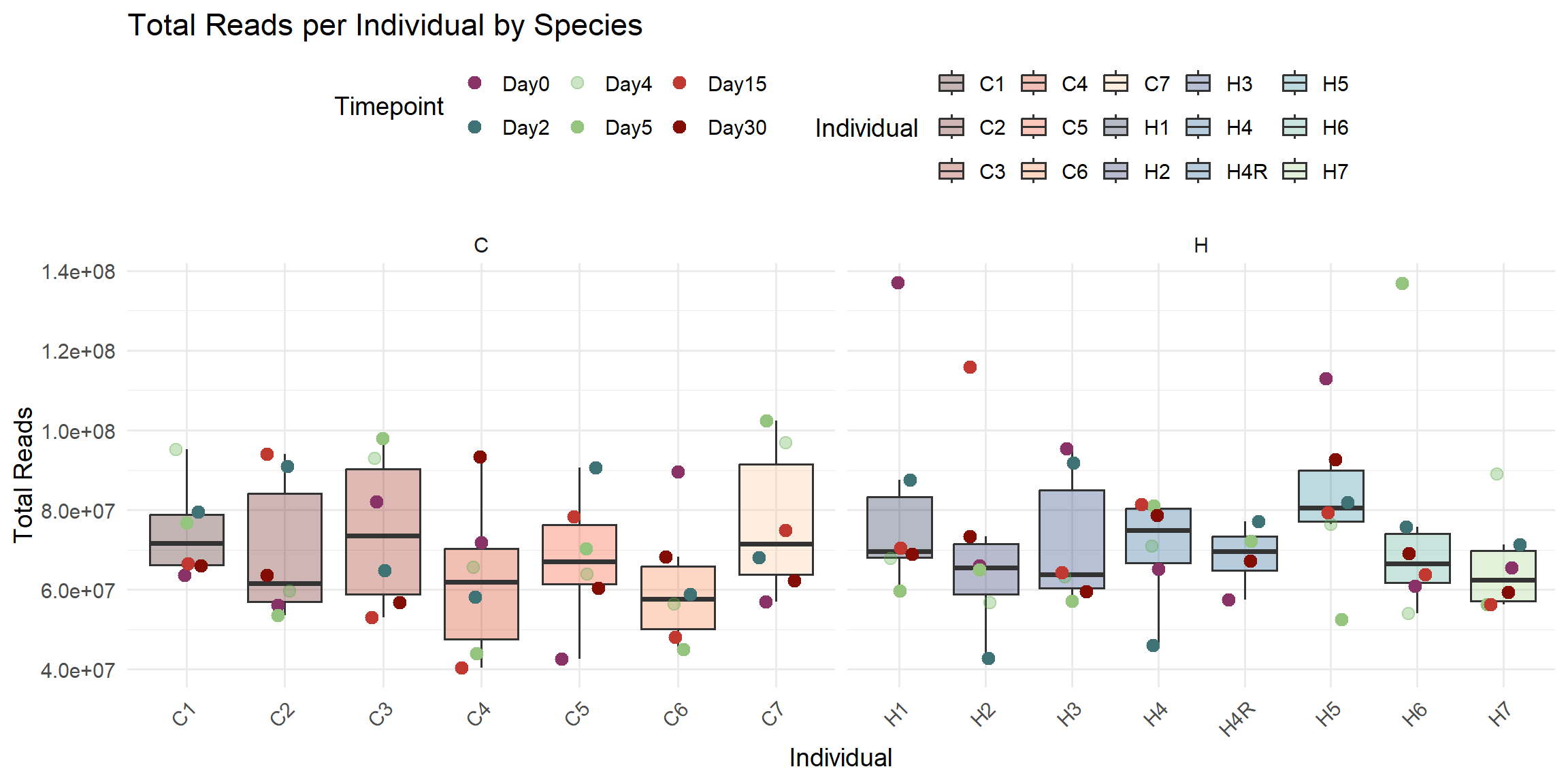
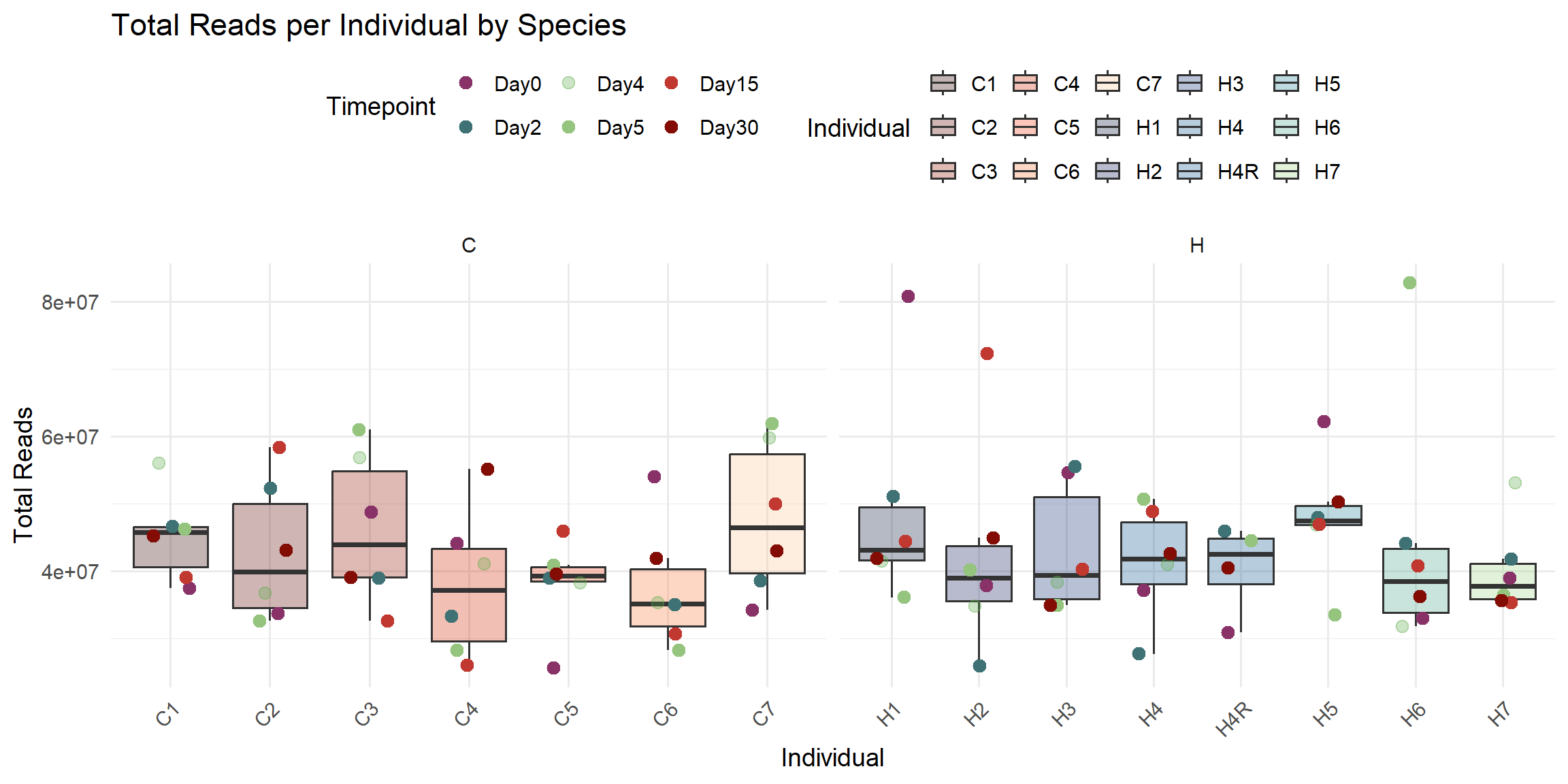
ggplot(total_reads_df, aes(x = Individual, y = Total_Reads)) +
geom_boxplot(aes(fill = Individual), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Timepoint), width = 0.2, size = 3) +
scale_fill_manual(values = ann_colors$individual_cor) +
scale_color_manual(values = ann_colors$timepoint_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Total Reads per Individual by Species",
x = "Individual",
y = "Total Reads",
fill = "Individual",
color = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
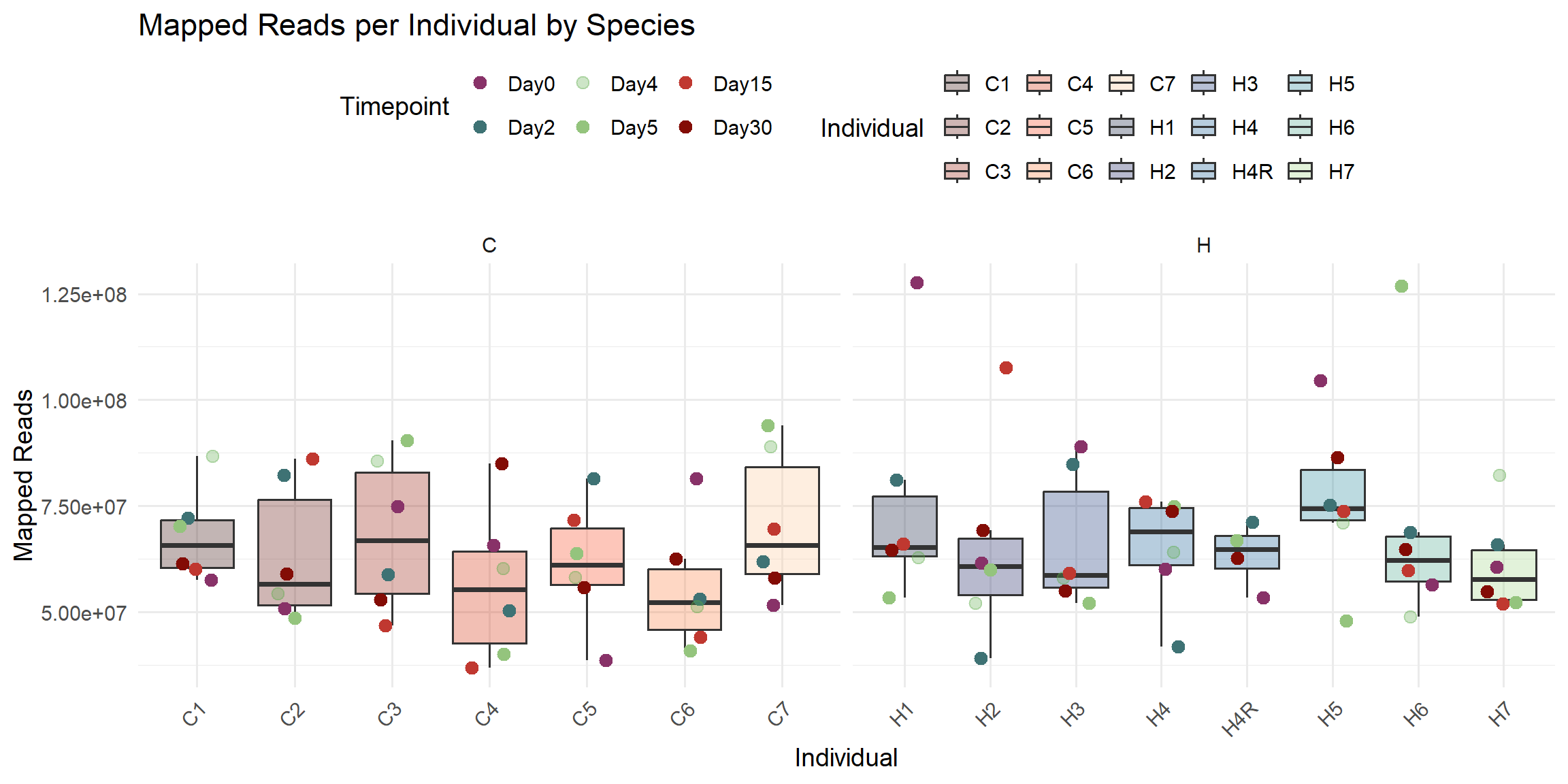
ggplot(total_reads_df, aes(x = Individual, y = Mapped_Reads)) +
geom_boxplot(aes(fill = Individual), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Timepoint), width = 0.2, size = 3) +
scale_fill_manual(values = ann_colors$individual_cor) +
scale_color_manual(values = ann_colors$timepoint_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Mapped Reads per Individual by Species",
x = "Individual",
y = "Mapped Reads",
fill = "Individual",
color = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
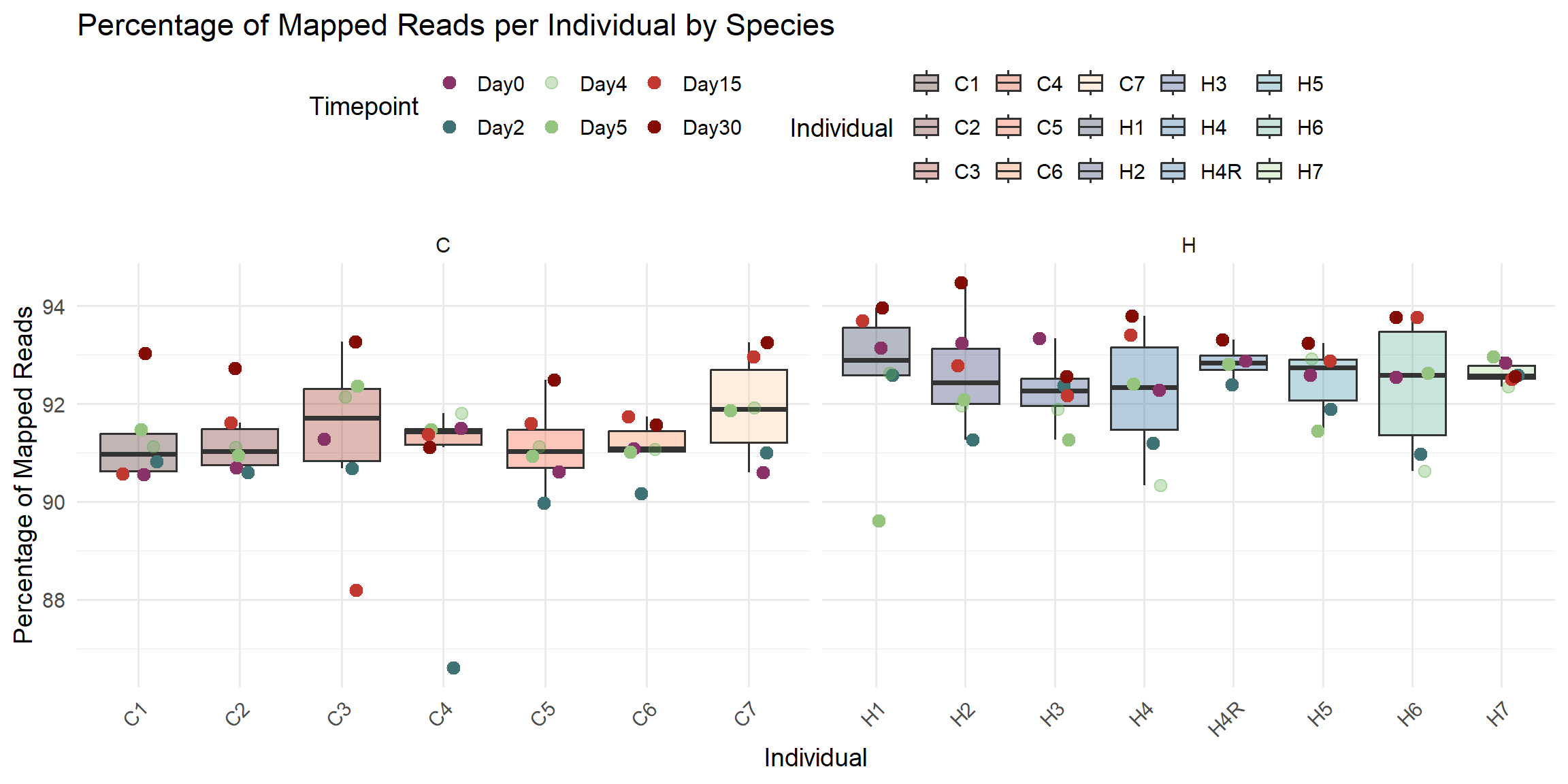
ggplot(total_reads_df, aes(x = Individual, y = Percent_Mapped)) +
geom_boxplot(aes(fill = Individual), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Timepoint), width = 0.2, size = 3) +
scale_fill_manual(values = ann_colors$individual_cor) +
scale_color_manual(values = ann_colors$timepoint_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Percentage of Mapped Reads per Individual by Species",
x = "Individual",
y = "Percentage of Mapped Reads",
fill = "Individual",
color = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
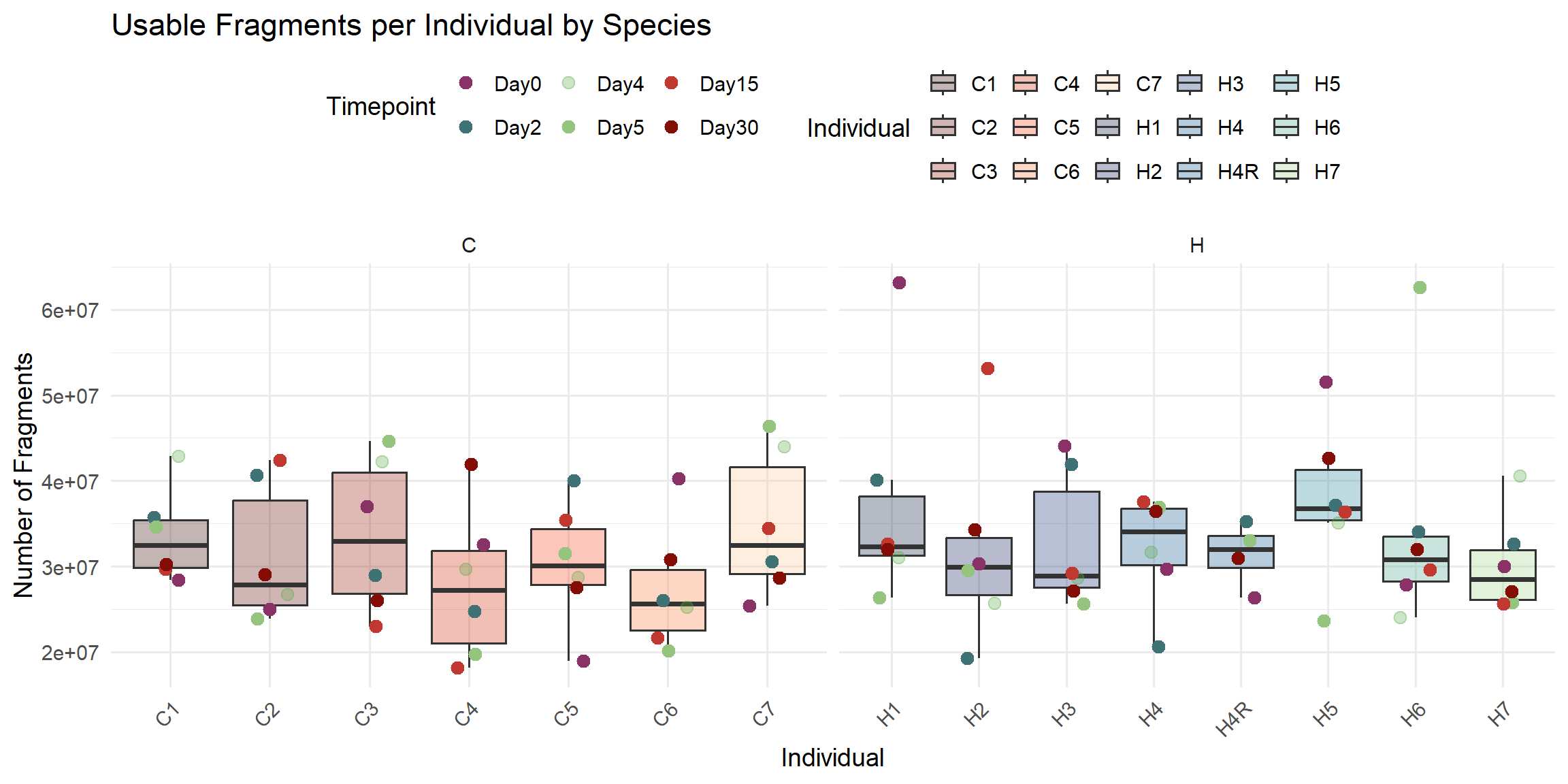
ggplot(total_reads_df, aes(x = Individual, y = Usable_Fragments)) +
geom_boxplot(aes(fill = Individual), alpha = 0.3, outlier.shape = NA) +
geom_jitter(aes(color = Timepoint), width = 0.2, size = 3) +
scale_fill_manual(values = ann_colors$individual_cor) +
scale_color_manual(values = ann_colors$timepoint_cor) +
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "top") +
labs(
title = "Usable Fragments per Individual by Species",
x = "Individual",
y = "Number of Fragments",
fill = "Individual",
color = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
# -----------------------------
# 1. Prepare long-format data
# -----------------------------
day_levels <- c("Day0","Day2","Day4","Day5","Day15","Day30")
marker_levels <- c("OCT3/4", "Brachy", "ISL-1", "TNNT-2")
RNA_Metadata_NoRep <- RNA_Metadata %>%
dplyr::filter(!SampleName %in% c(
"H20682R_Day0",
"H20682R_Day2",
"H20682R_Day5",
"H20682R_Day30"
))
df_long_all <- RNA_Metadata_NoRep %>%
dplyr::select(Individual,Cond, `OCT3/4`, Brachy, `ISL-1`, `TNNT-2`) %>%
mutate(
`OCT3/4` = as.numeric(`OCT3/4`),
Brachy = as.numeric(Brachy),
`ISL-1` = as.numeric(`ISL-1`),
`TNNT-2` = as.numeric(`TNNT-2`),
Group = ifelse(grepl("^H_", Cond), "H", "C")
) %>%
pivot_longer(
cols = c(`OCT3/4`, Brachy, `ISL-1`, `TNNT-2`),
names_to = "Marker",
values_to = "Percent_Positive"
) %>%
mutate(
Day = gsub(".*_(Day\\d+)$", "\\1", Cond),
Day = factor(Day, levels = day_levels, ordered = TRUE),
Marker = factor(Marker, levels = marker_levels, ordered = TRUE)
)
# -----------------------------
# 2. Compute summary with SEM
# -----------------------------
summary_df <- df_long_all %>%
group_by(Marker, Day, Group) %>%
summarise(
Mean = mean(Percent_Positive, na.rm = TRUE),
SD = sd(Percent_Positive, na.rm = TRUE),
N = sum(!is.na(Percent_Positive)),
SEM = SD / sqrt(N),
.groups = "drop"
)# -----------------------------
# 3. Plot lines over time with SEM error bars
# -----------------------------
library(dplyr)
library(scales)
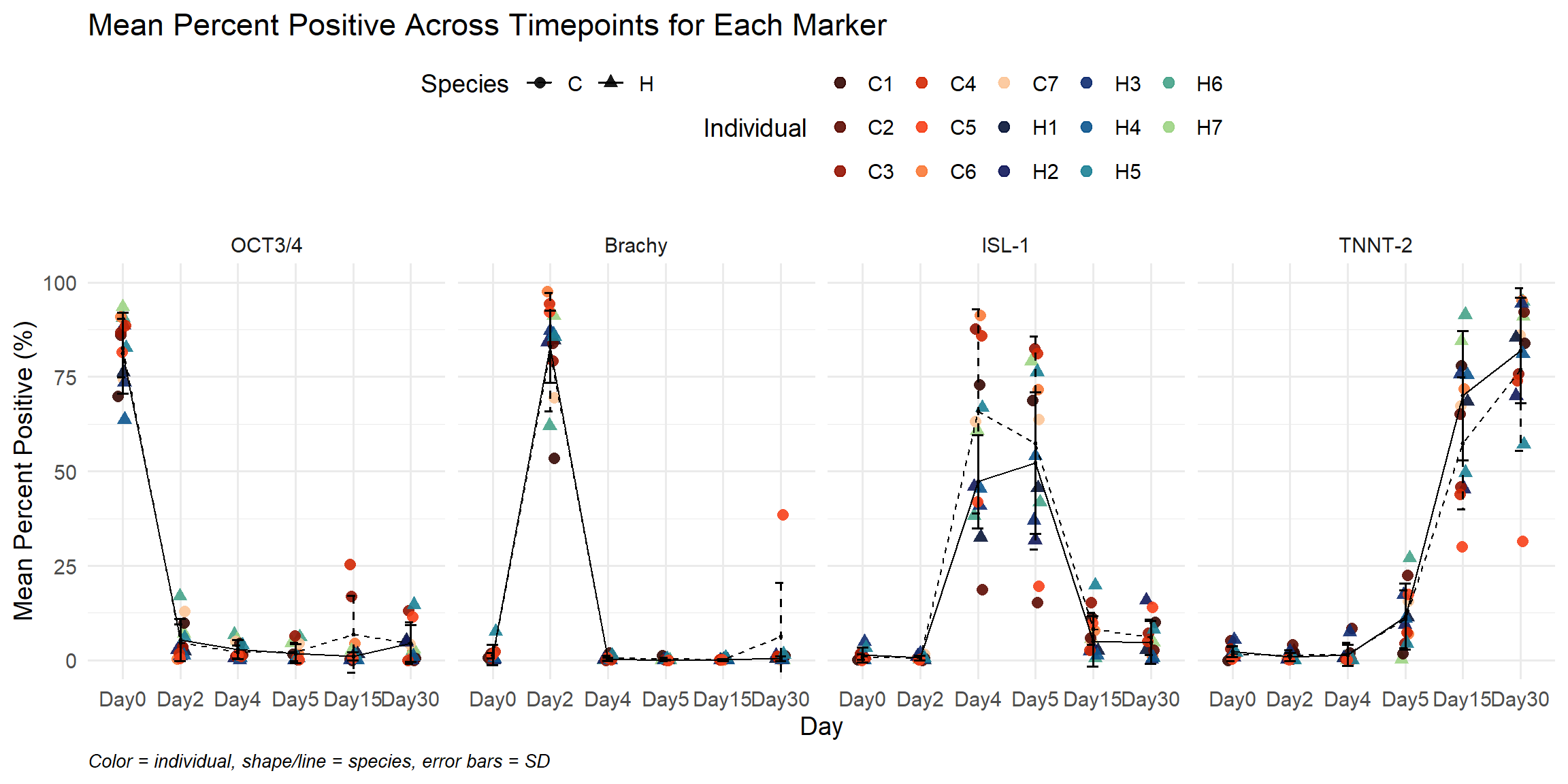
ggplot(summary_df,
aes(x = Day, y = Mean,
linetype = Group,
group = Group)) +
# Individual points
geom_point(
data = df_long_all,
aes(
x = Day,
y = Percent_Positive,
color = Individual,
shape = Group
),
size = 2.5,
alpha = 0.9,
position = position_jitter(width = 0.08, height = 0)
) +
# Mean line (black, texture = species)
geom_line(
linewidth = 0.5,
color = "black"
) +
# Mean point (black)
geom_point(
size = 0.5,
color = "black"
) +
# Error bars (black)
geom_errorbar(
aes(ymin = Mean - SD, ymax = Mean + SD),
width = 0.2,
color = "black"
) +
facet_wrap(~ Marker, nrow = 1) +
# species as line texture
scale_linetype_manual(
values = c(
"H" = "solid",
"C" = "dashed"
),
name = "Species"
) +
# species as shape
scale_shape_manual(
values = c(
"H" = 17, # triangle
"C" = 16 # circle
),
name = "Species"
) +
# individual colors from your manual palette
scale_color_manual(
values = ann_colors$individual_cor,
name = "Individual"
) +
# Use coord_cartesian instead of scale_y_continuous to avoid dropped points
coord_cartesian(ylim = c(0, 100)) +
labs(
title = "Mean Percent Positive Across Timepoints for Each Marker",
x = "Day",
y = "Mean Percent Positive (%)",
caption = "Color = individual, shape/line = species, error bars = SD"
) +
theme_minimal(base_size = 14) +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 0, hjust = 0.5),
plot.caption = element_text(hjust = 0, face = "italic", size = 10)
)
# -----------------------------
# 1. Prepare long-format data
# -----------------------------
day_levels <- c("Day0","Day2","Day4","Day5","Day15","Day30")
marker_levels <- c("OCT3/4", "Brachy", "ISL-1", "TNNT-2")
RNA_Metadata_NoD4_NoRep <- RNA_Metadata %>%
dplyr::filter(Timepoint != "Day4") %>%
dplyr::filter(!SampleName %in% c(
"H20682R_Day0",
"H20682R_Day2",
"H20682R_Day5",
"H20682R_Day30"
))
df_long_all <- RNA_Metadata_NoD4_NoRep %>%
dplyr::select(Individual,Cond, `OCT3/4`, Brachy, `ISL-1`, `TNNT-2`) %>%
mutate(
`OCT3/4` = as.numeric(`OCT3/4`),
Brachy = as.numeric(Brachy),
`ISL-1` = as.numeric(`ISL-1`),
`TNNT-2` = as.numeric(`TNNT-2`),
Group = ifelse(grepl("^H_", Cond), "H", "C")
) %>%
pivot_longer(
cols = c(`OCT3/4`, Brachy, `ISL-1`, `TNNT-2`),
names_to = "Marker",
values_to = "Percent_Positive"
) %>%
mutate(
Day = gsub(".*_(Day\\d+)$", "\\1", Cond),
Day = factor(Day, levels = day_levels, ordered = TRUE),
Marker = factor(Marker, levels = marker_levels, ordered = TRUE)
)
# -----------------------------
# 2. Compute summary with SEM
# -----------------------------
summary_df <- df_long_all %>%
group_by(Marker, Day, Group) %>%
summarise(
Mean = mean(Percent_Positive, na.rm = TRUE),
SD = sd(Percent_Positive, na.rm = TRUE),
N = sum(!is.na(Percent_Positive)),
SEM = SD / sqrt(N),
.groups = "drop"
)# -----------------------------
# 3. Plot lines over time with SEM error bars
# -----------------------------
library(dplyr)
library(scales)
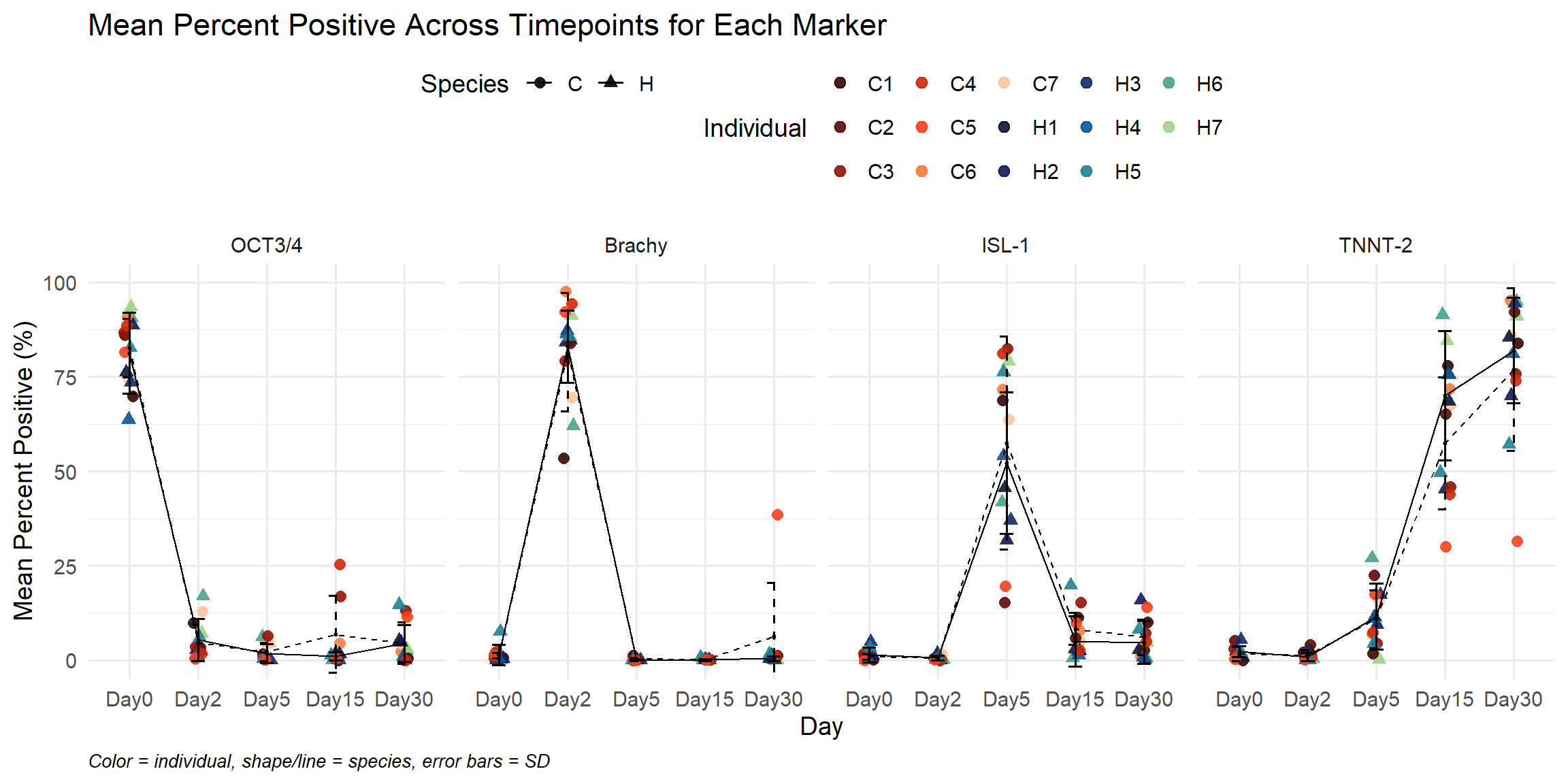
ggplot(summary_df,
aes(x = Day, y = Mean,
linetype = Group,
group = Group)) +
# Individual points
geom_point(
data = df_long_all,
aes(
x = Day,
y = Percent_Positive,
color = Individual,
shape = Group
),
size = 2.5,
alpha = 0.9,
position = position_jitter(width = 0.08, height = 0)
) +
# Mean line (black, texture = species)
geom_line(
linewidth = 0.5,
color = "black"
) +
# Mean point (black)
geom_point(
size = 0.5,
color = "black"
) +
# Error bars (black)
geom_errorbar(
aes(ymin = Mean - SD, ymax = Mean + SD),
width = 0.2,
color = "black"
) +
facet_wrap(~ Marker, nrow = 1) +
# species as line texture
scale_linetype_manual(
values = c(
"H" = "solid",
"C" = "dashed"
),
name = "Species"
) +
# species as shape
scale_shape_manual(
values = c(
"H" = 17, # triangle
"C" = 16 # circle
),
name = "Species"
) +
# individual colors from your manual palette
scale_color_manual(
values = ann_colors$individual_cor,
name = "Individual"
) +
# Use coord_cartesian instead of scale_y_continuous to avoid dropped points
coord_cartesian(ylim = c(0, 100)) +
labs(
title = "Mean Percent Positive Across Timepoints for Each Marker",
x = "Day",
y = "Mean Percent Positive (%)",
caption = "Color = individual, shape/line = species, error bars = SD"
) +
theme_minimal(base_size = 14) +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 0, hjust = 0.5),
plot.caption = element_text(hjust = 0, face = "italic", size = 10)
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
#####Unfiltered####
RNA_log2cpm <- cpm(RNA_fc, log = TRUE)
hist(RNA_log2cpm, main = "Histogram of all counts (unfiltered)",
xlab =expression("Log"[2]*" counts-per-million"), col =4 )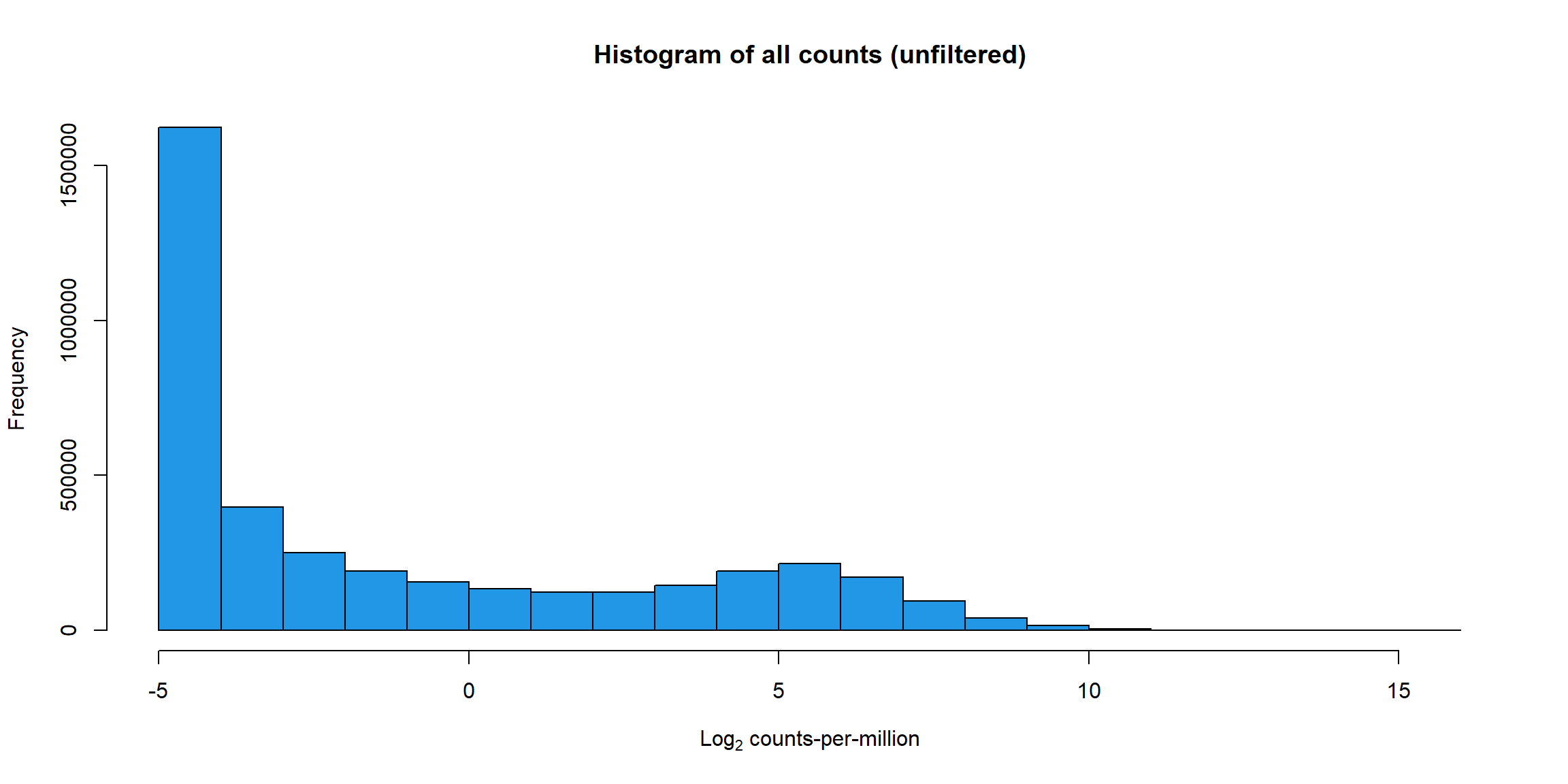
boxplot(RNA_log2cpm, main = "Boxplots of log cpm per sample
(unfiltered)", xaxt = "n", xlab= "")
axis(1,
at = 1:length(col_names), # positions (one per sample)
labels = col_names, # your labels vector
las = 2, # rotate text vertically (like srt=90)
cex.axis = 0.6) # shrink label size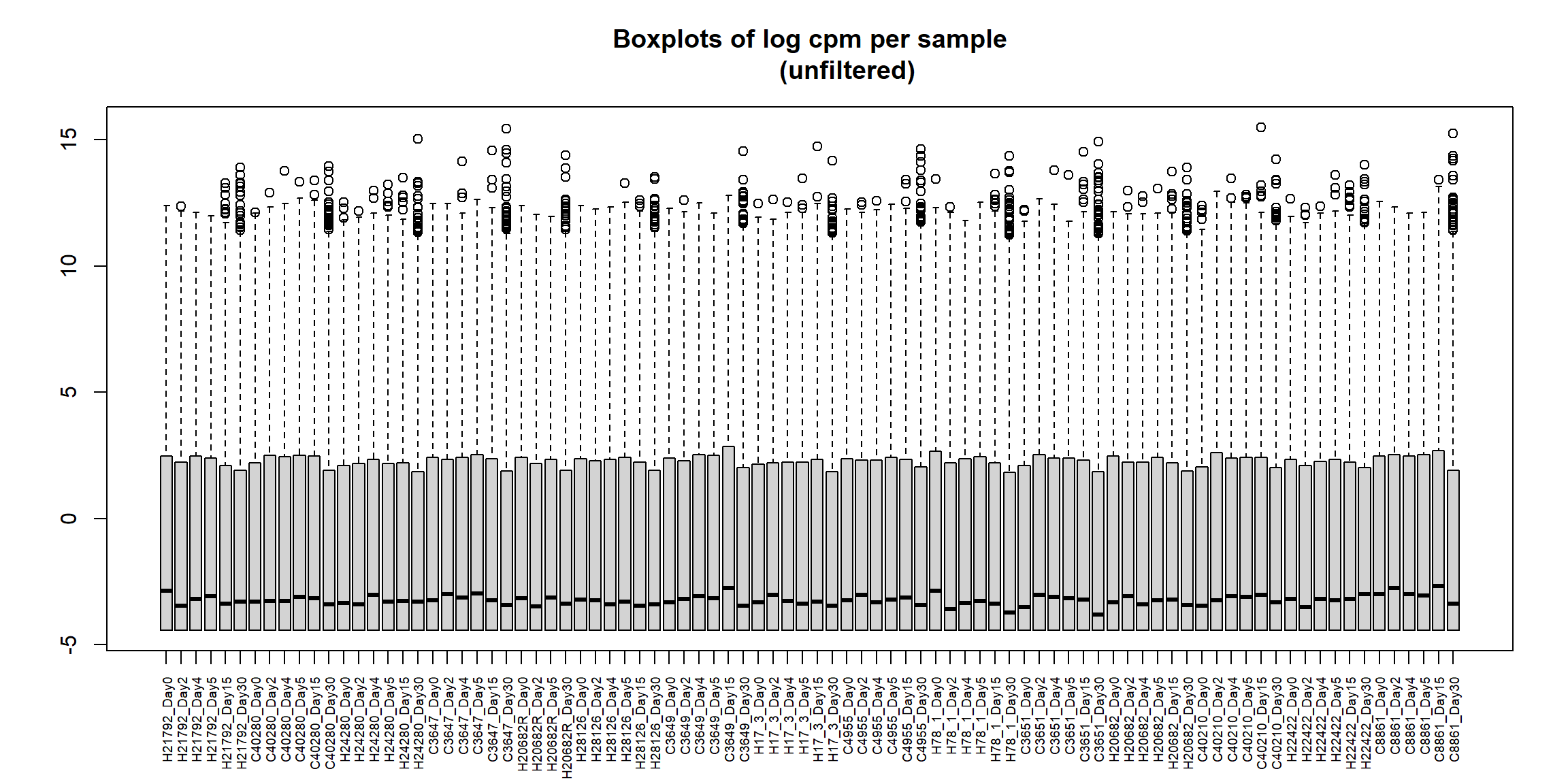
# saveRDS(RNA_log2cpm,"data/QC/concat/RNA_log2cpm.RDS")#####RowMu>0####
row_means <- rowMeans(RNA_log2cpm)
Filt_RMG0_RNA_fc <- RNA_fc[row_means >0,]
# saveRDS(Filt_RMG0_RNA_fc,"data/QC/concat/Filt_RMG0_RNA_fc.RDS")
Filt_RMG0_RNA_log2cpm <- cpm(Filt_RMG0_RNA_fc,log=TRUE)
# saveRDS(Filt_RMG0_RNA_log2cpm,"data/QC/concat/Filt_RMG0_RNA_log2cpm.RDS")
hist(Filt_RMG0_RNA_log2cpm, main = "Histogram of filtered counts using rowMeans > 0 method",
xlab =expression("Log"[2]*" counts-per-million"), col =5 )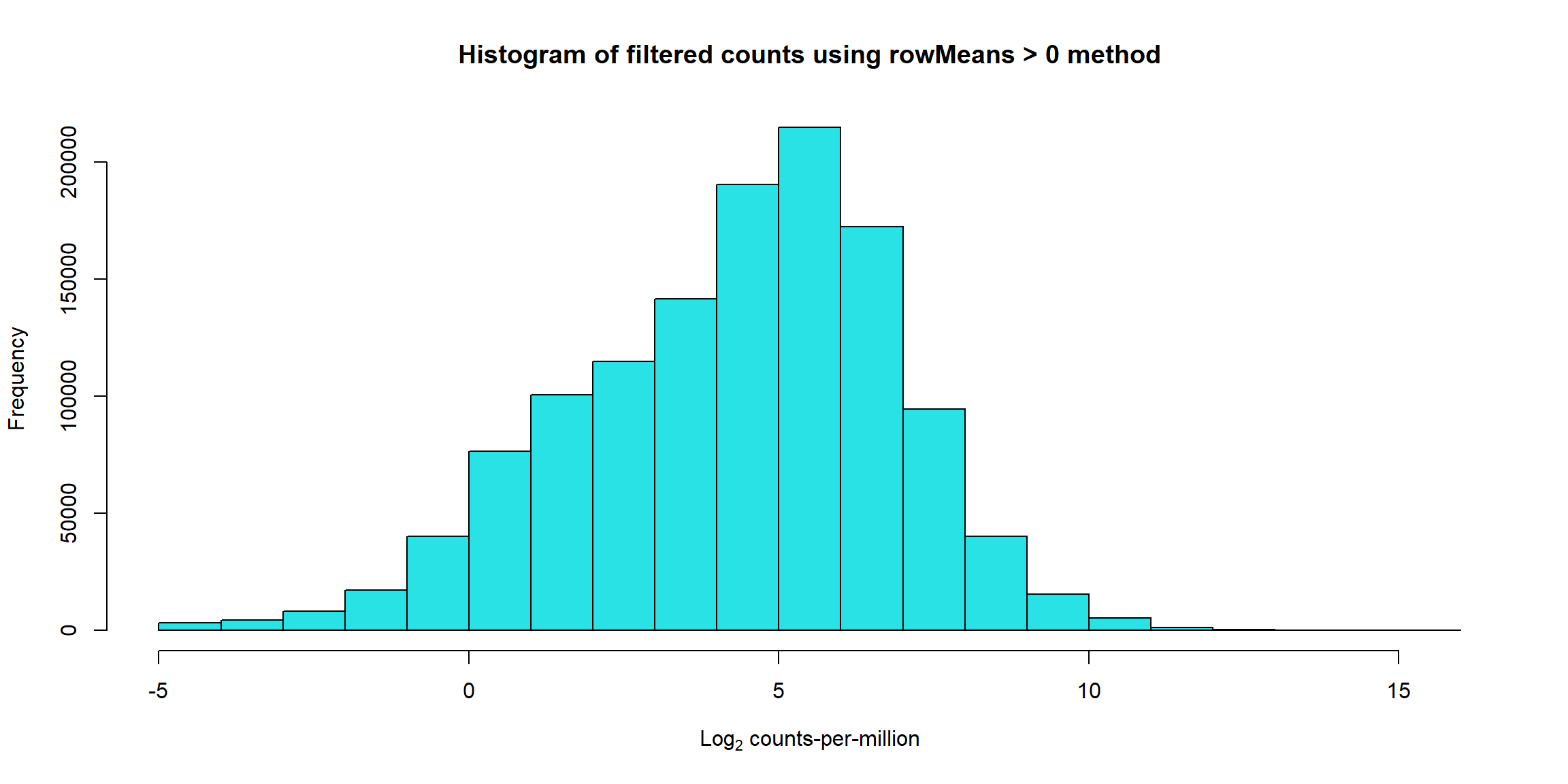
boxplot(Filt_RMG0_RNA_log2cpm, main = "Boxplots of log cpm per sample (RowMeans>0)",xaxt = "n", xlab= "")
axis(1,
at = 1:length(col_names), # positions (one per sample)
labels = col_names, # your labels vector
las = 2, # rotate text vertically (like srt=90)
cex.axis = 0.6) # shrink label size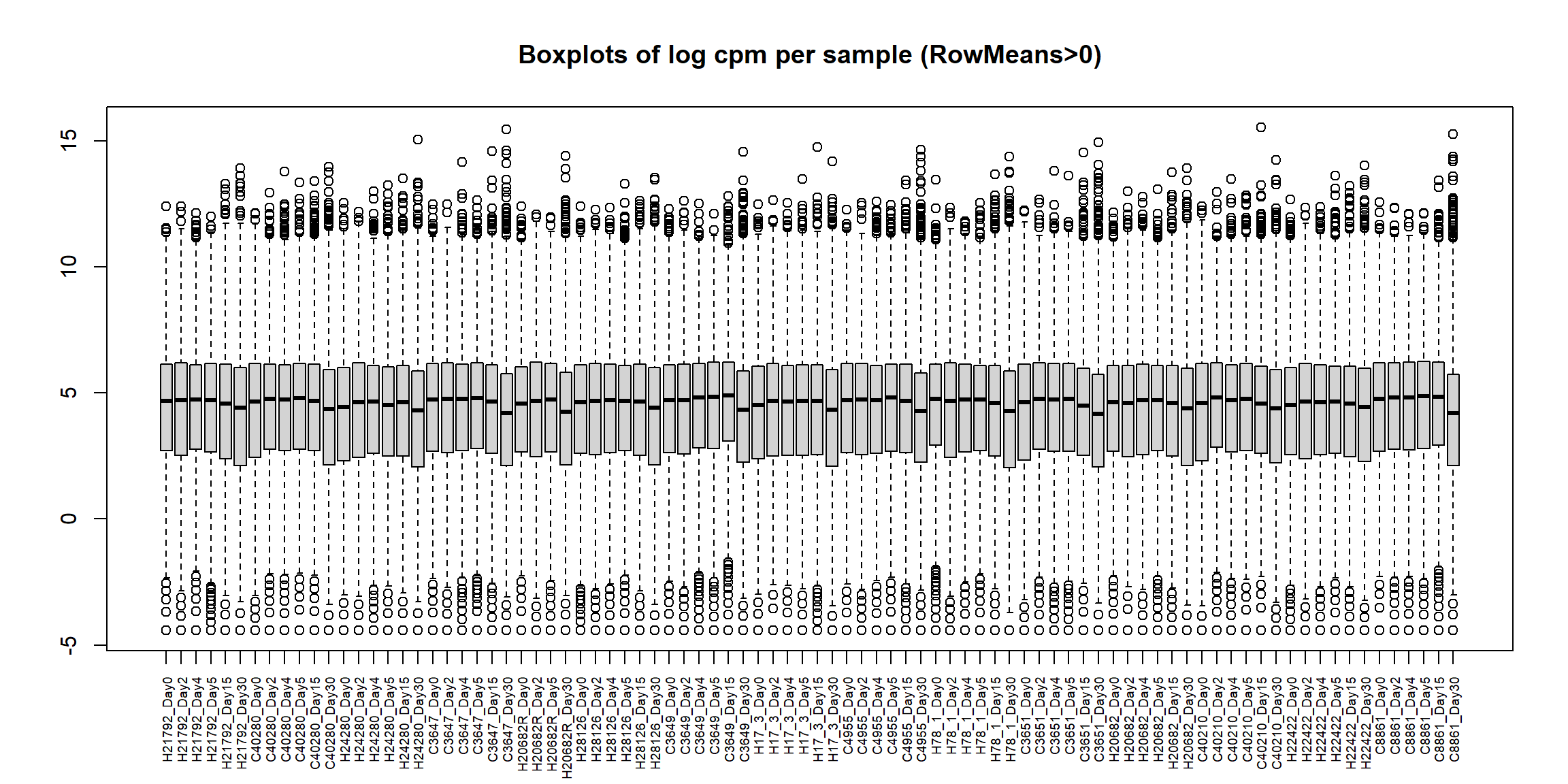
library(ggplot2)
library(dplyr)
# Define ordered day factor
day_levels <- c("Day0", "Day2", "Day4", "Day5", "Day15", "Day30")
total_reads_df <- total_reads_df %>%
mutate(
# Order Timepoint for bars
Timepoint = factor(Timepoint, levels = day_levels, ordered = TRUE),
# Individual text color
Individual_Color = ann_colors$individual_cor[Individual]
) %>%
# Order Sample for x-axis: first by Individual, then by Day
arrange(Individual, Timepoint) %>%
mutate(Sample = factor(Sample, levels = Sample)) # preserve order in ggplot
# Plot
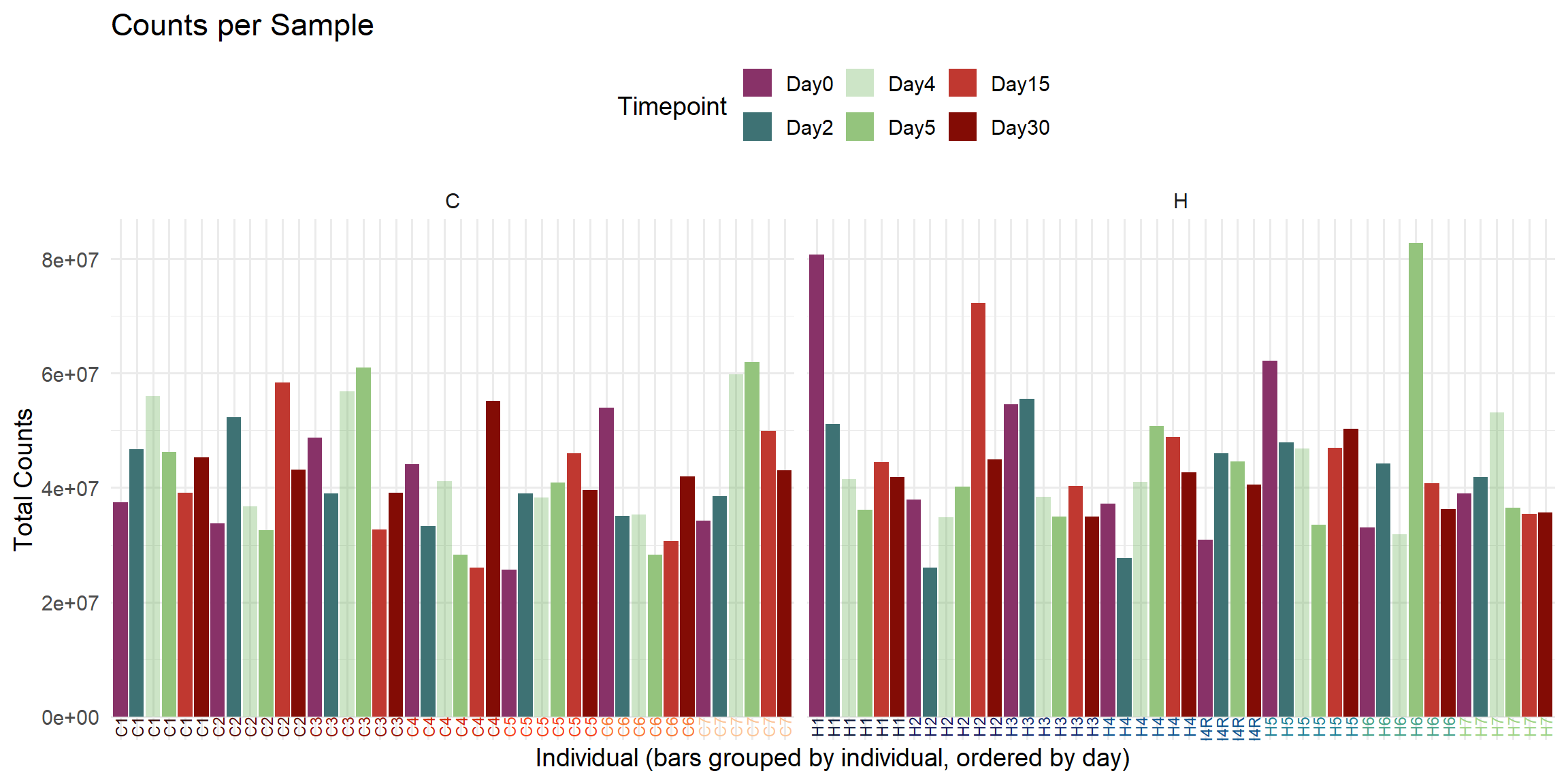
ggplot(total_reads_df, aes(x = Sample, y = Total_Counts, fill = Timepoint)) +
# Bars colored by Timepoint
geom_col() +
# Annotate Individual names under bars, colored by individual
geom_text(
aes(label = Individual, color = Individual_Color),
y = 0, angle = 90, hjust = 1, vjust = 0.5, size = 3
) +
# Manual Timepoint colors
scale_fill_manual(values = ann_colors$timepoint_cor) +
# Use exact color for individual labels
scale_color_identity() +
# Facet by Species if desired
facet_grid(~Species, scales = "free_x", space = "free_x") +
# Theme
theme_minimal(base_size = 14) +
theme(
axis.text.x = element_blank(), # hide default X labels
axis.ticks.x = element_blank(),
legend.position = "top"
) +
labs(
title = "Counts per Sample",
x = "Individual (bars grouped by individual, ordered by day)",
y = "Total Counts",
fill = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
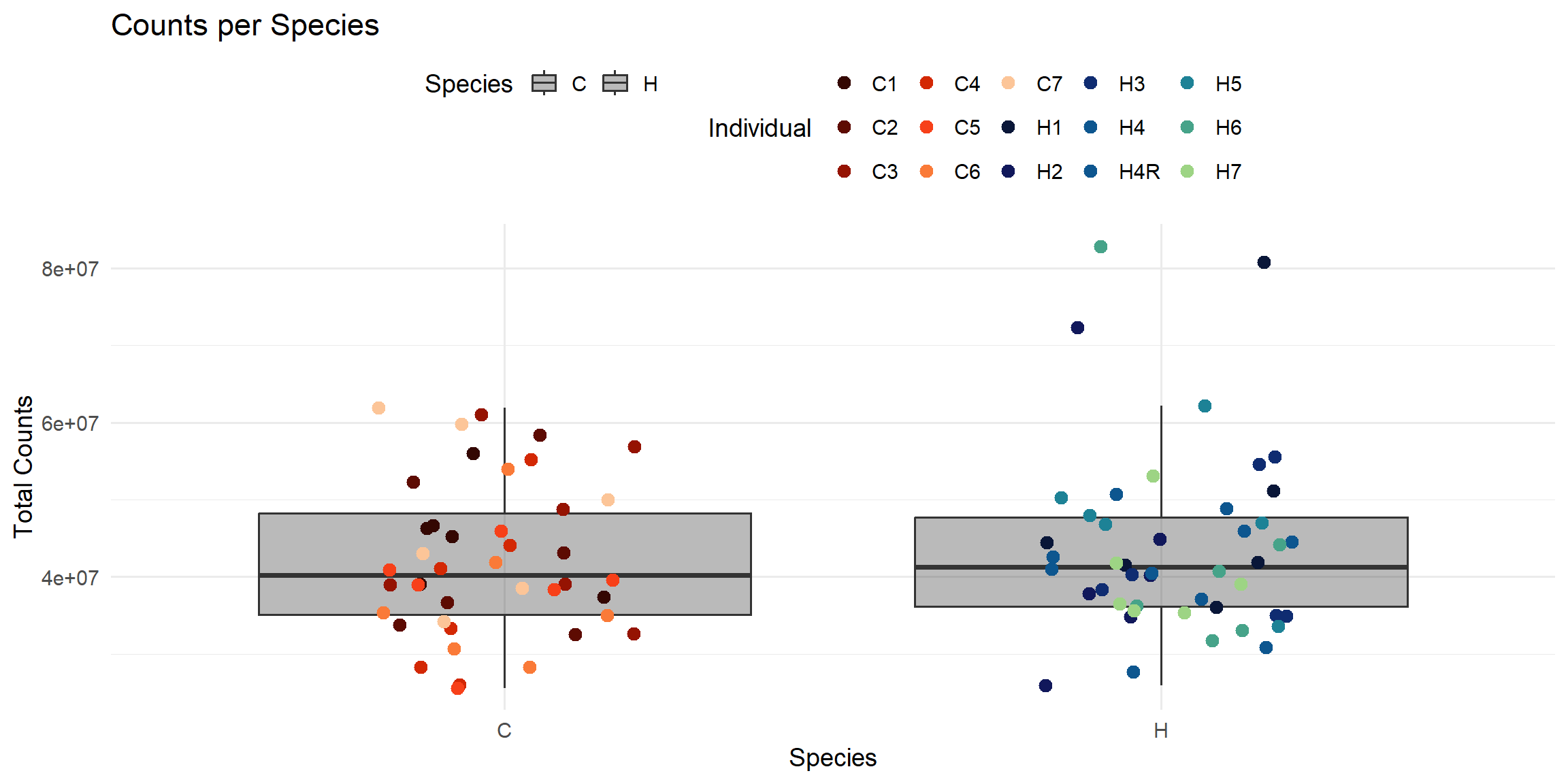
ggplot(total_reads_df, aes(x = Species, y = Total_Counts)) +
# Boxplot for each species
geom_boxplot(aes(fill = Species), alpha = 0.3, outlier.shape = NA) +
# Overlay individual points colored by individual
geom_jitter(aes(color = Individual), width = 0.2, size = 3) +
# Use individual colors
scale_color_manual(values = ann_colors$individual_cor) +
# Optional: species fill colors (light grey / black)
scale_fill_manual(values = c("H" = "#17171717", "C" = "#17171717")) +
theme_minimal(base_size = 14) +
theme(
legend.position = "top"
) +
labs(
title = "Counts per Species",
x = "Species",
y = "Total Counts",
color = "Individual",
fill = "Species"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
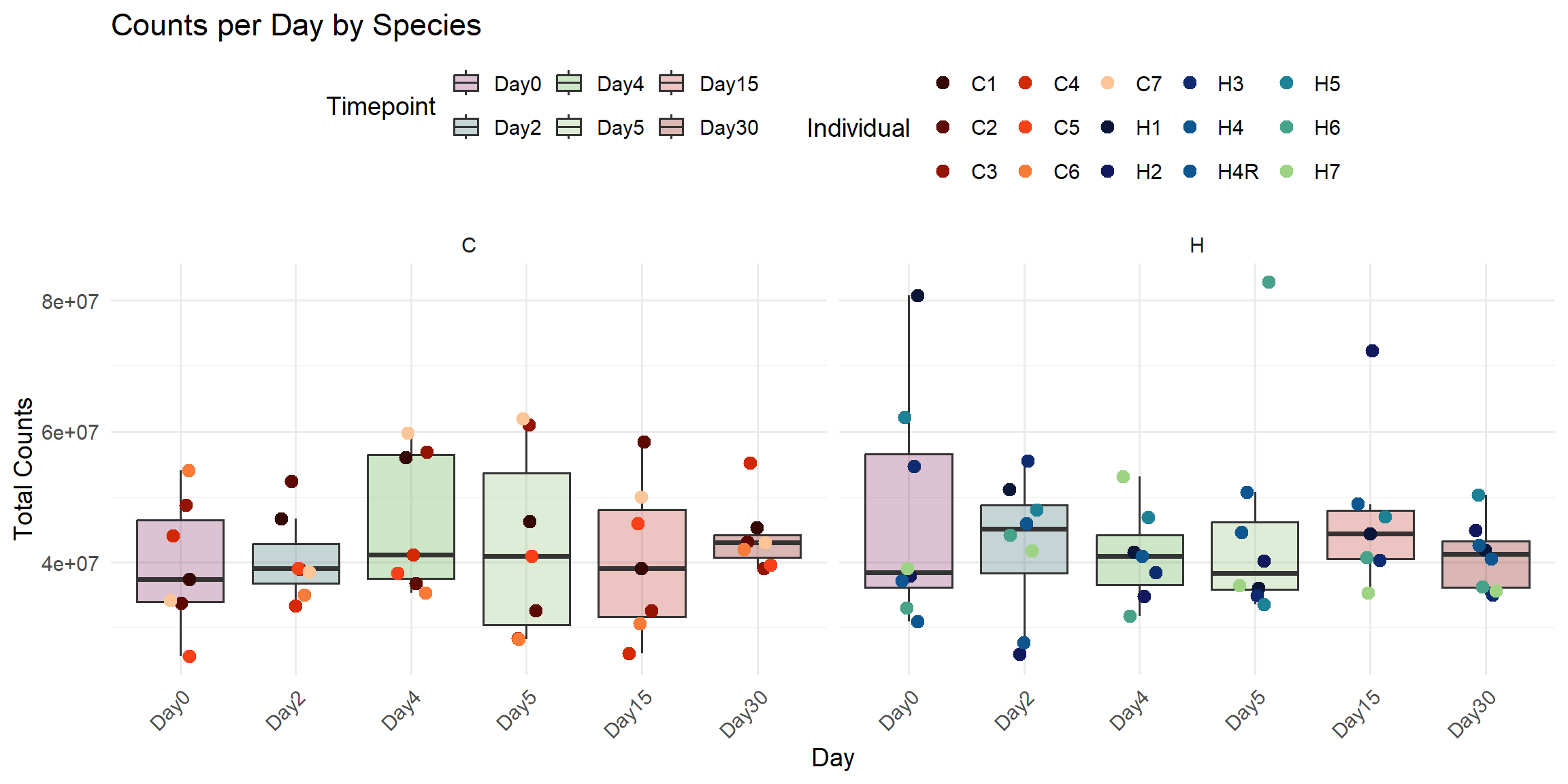
ggplot(total_reads_df, aes(x = Timepoint, y = Total_Counts)) +
# Boxplot per day
geom_boxplot(aes(fill = Timepoint), alpha = 0.3, outlier.shape = NA) +
# Overlay individual points colored by individual
geom_jitter(aes(color = Individual), width = 0.15, size = 3) +
# Use individual colors
scale_color_manual(values = ann_colors$individual_cor) +
# Use timepoint colors for boxplots
scale_fill_manual(values = ann_colors$timepoint_cor) +
# Separate x-axis by species
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 45, hjust = 1)
) +
labs(
title = "Counts per Day by Species",
x = "Day",
y = "Total Counts",
color = "Individual",
fill = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
# Plot
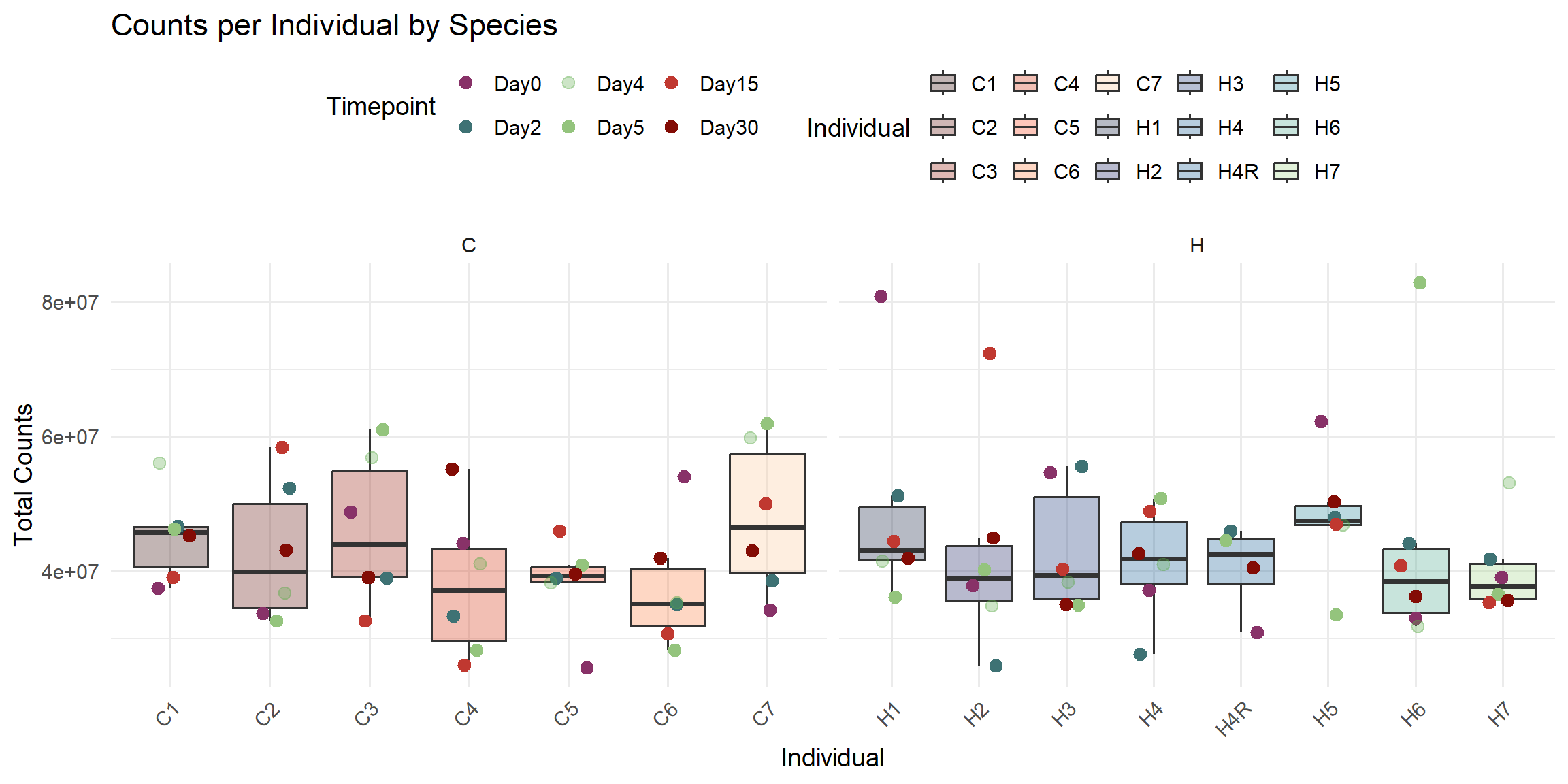
ggplot(total_reads_df, aes(x = Individual, y = Total_Counts)) +
# Boxplots colored by individual
geom_boxplot(aes(fill = Individual), alpha = 0.3, outlier.shape = NA) +
# Overlay points colored by Timepoint
geom_jitter(aes(color = Timepoint), width = 0.2, size = 3) +
# Use manual colors
scale_fill_manual(values = ann_colors$individual_cor) +
scale_color_manual(values = ann_colors$timepoint_cor) +
# Facet by species
facet_wrap(~Species, scales = "free_x", nrow = 1) +
# Theme
theme_minimal(base_size = 14) +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 45, hjust = 1)
) +
labs(
title = "Counts per Individual by Species",
x = "Individual",
y = "Total Counts",
fill = "Individual",
color = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
library(tidyverse)
total_reads_df <- RNA_fc %>%
as.data.frame() %>%
mutate(Gene = rownames(.)) %>%
dplyr::select(-Gene) %>%
summarise(across(everything(), \(x) sum(x, na.rm = TRUE))) %>%
pivot_longer(
cols = everything(),
names_to = "Sample",
values_to = "Total_Reads"
) %>%
left_join(RNA_Metadata, by = c("Sample" = "SampleName"))library(ggplot2)
library(dplyr)
# Define ordered day factor
day_levels <- c("Day0", "Day2", "Day4", "Day5", "Day15", "Day30")
total_reads_df <- total_reads_df %>%
mutate(
# Order Timepoint for bars
Timepoint = factor(Timepoint, levels = day_levels, ordered = TRUE),
# Individual text color
Individual_Color = ann_colors$individual_cor[Individual]
) %>%
# Order Sample for x-axis: first by Individual, then by Day
arrange(Individual, Timepoint) %>%
mutate(Sample = factor(Sample, levels = Sample)) # preserve order in ggplot
# Plot
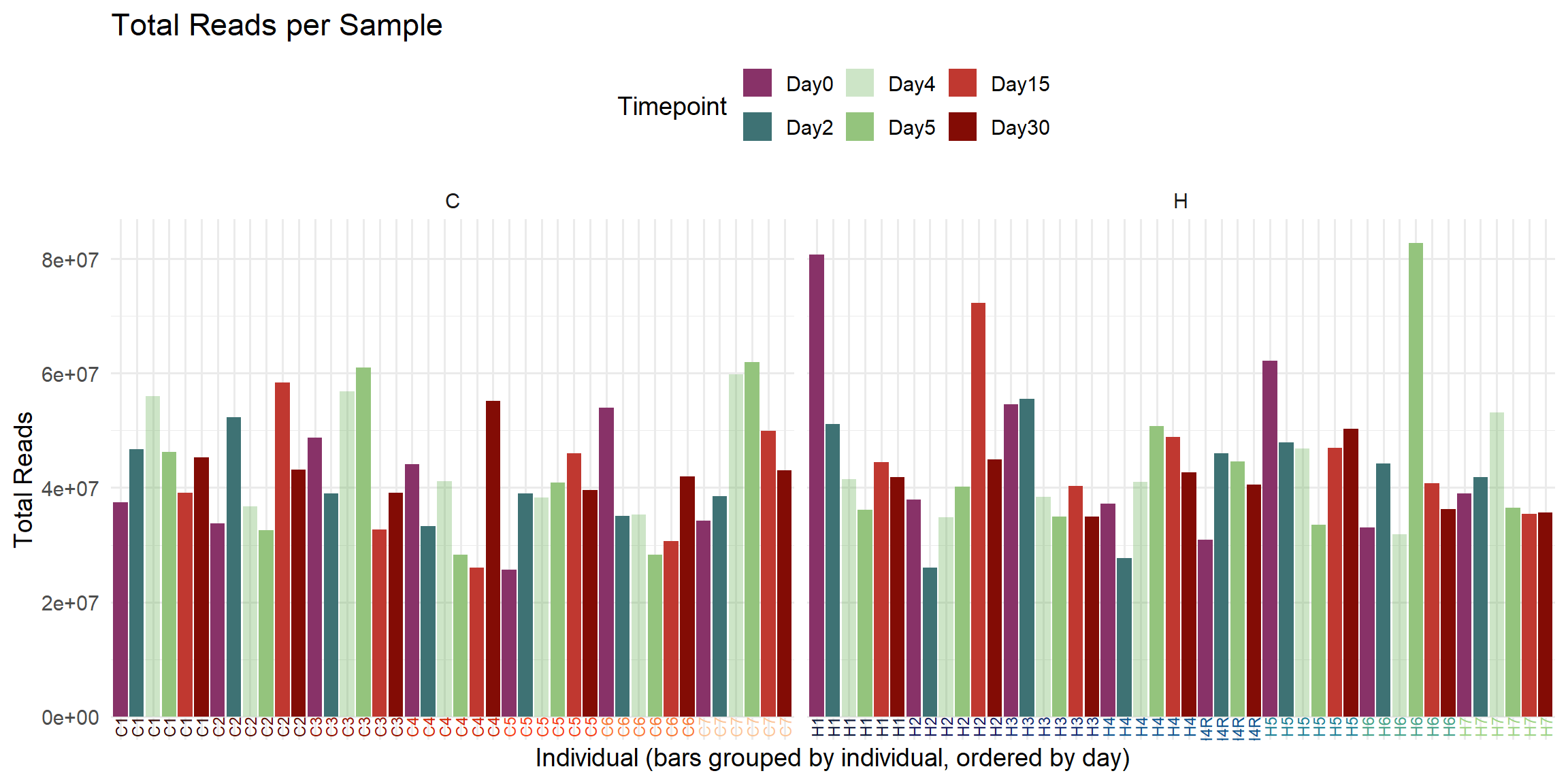
ggplot(total_reads_df, aes(x = Sample, y = Total_Reads.x, fill = Timepoint)) +
# Bars colored by Timepoint
geom_col() +
# Annotate Individual names under bars, colored by individual
geom_text(
aes(label = Individual, color = Individual_Color),
y = 0, angle = 90, hjust = 1, vjust = 0.5, size = 3
) +
# Manual Timepoint colors
scale_fill_manual(values = ann_colors$timepoint_cor) +
# Use exact color for individual labels
scale_color_identity() +
# Facet by Species if desired
facet_grid(~Species, scales = "free_x", space = "free_x") +
# Theme
theme_minimal(base_size = 14) +
theme(
axis.text.x = element_blank(), # hide default X labels
axis.ticks.x = element_blank(),
legend.position = "top"
) +
labs(
title = "Total Reads per Sample",
x = "Individual (bars grouped by individual, ordered by day)",
y = "Total Reads",
fill = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
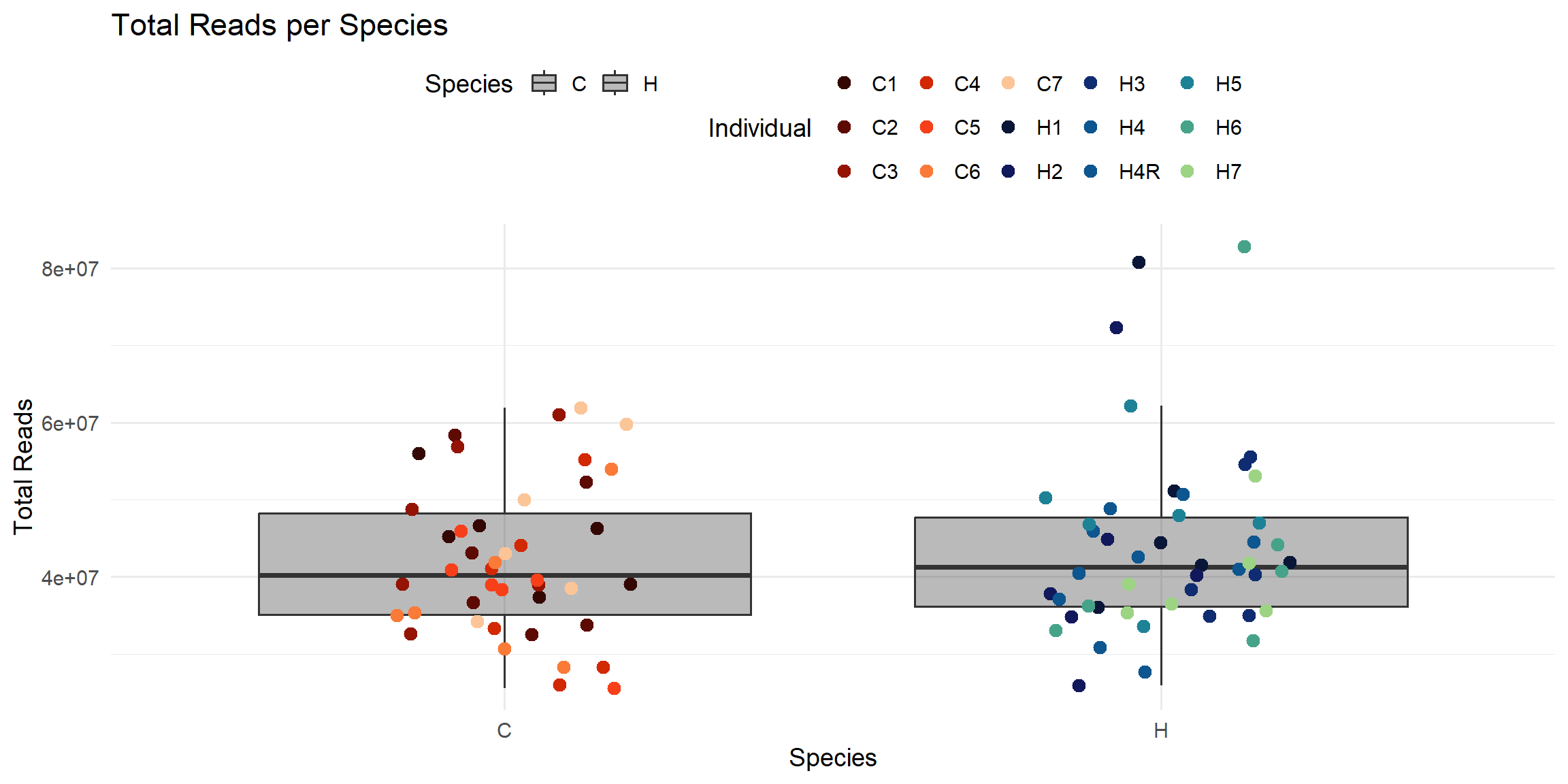
ggplot(total_reads_df, aes(x = Species, y = Total_Reads.x)) +
# Boxplot for each species
geom_boxplot(aes(fill = Species), alpha = 0.3, outlier.shape = NA) +
# Overlay individual points colored by individual
geom_jitter(aes(color = Individual), width = 0.2, size = 3) +
# Use individual colors
scale_color_manual(values = ann_colors$individual_cor) +
# Optional: species fill colors (light grey / black)
scale_fill_manual(values = c("H" = "#17171717", "C" = "#17171717")) +
theme_minimal(base_size = 14) +
theme(
legend.position = "top"
) +
labs(
title = "Total Reads per Species",
x = "Species",
y = "Total Reads",
color = "Individual",
fill = "Species"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
ggplot(total_reads_df, aes(x = Timepoint, y = Total_Reads.x)) +
# Boxplot per day
geom_boxplot(aes(fill = Timepoint), alpha = 0.3, outlier.shape = NA) +
# Overlay individual points colored by individual
geom_jitter(aes(color = Individual), width = 0.15, size = 3) +
# Use individual colors
scale_color_manual(values = ann_colors$individual_cor) +
# Use timepoint colors for boxplots
scale_fill_manual(values = ann_colors$timepoint_cor) +
# Separate x-axis by species
facet_wrap(~Species, scales = "free_x") +
theme_minimal(base_size = 14) +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 45, hjust = 1)
) +
labs(
title = "Total Reads per Day by Species",
x = "Day",
y = "Total Reads",
color = "Individual",
fill = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
# Plot
ggplot(total_reads_df, aes(x = Individual, y = Total_Reads.x)) +
# Boxplots colored by individual
geom_boxplot(aes(fill = Individual), alpha = 0.3, outlier.shape = NA) +
# Overlay points colored by Timepoint
geom_jitter(aes(color = Timepoint), width = 0.2, size = 3) +
# Use manual colors
scale_fill_manual(values = ann_colors$individual_cor) +
scale_color_manual(values = ann_colors$timepoint_cor) +
# Facet by species
facet_wrap(~Species, scales = "free_x", nrow = 1) +
# Theme
theme_minimal(base_size = 14) +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 45, hjust = 1)
) +
labs(
title = "Total Reads per Individual by Species",
x = "Individual",
y = "Total Reads",
fill = "Individual",
color = "Timepoint"
)
| Version | Author | Date |
|---|---|---|
| 0958201 | John D. Hurley | 2026-03-23 |
######Cor_HeatMap####
Cor_Filt_RMG0_RNA_log2cpm <- cor(Filt_RMG0_RNA_log2cpm, method = "spearman")
individual <- RNA_Metadata$Individual
species <- RNA_Metadata$Species
timepoint <- RNA_Metadata$Timepoint
timepoint <- factor(timepoint,levels = c("Day0","Day2","Day4","Day5","Day15","Day30"))
Cor_metadata <- data.frame(
sample_cor = colnames(Filt_RMG0_RNA_log2cpm),
species_cor = species,
timepoint_cor = timepoint,
individual_cor = individual
)
ann_colors <- list(
timepoint_cor = c(
"Day0" = "#883268", # Purple
"Day2" = "#3E7274", # blue
"Day4" = "#5AAA464D", # light green
"Day5" = "#94C47D", # Green
"Day15" = "#C03830", # red
"Day30" = "#830C05" # dark red
),
species_cor = c(
"H" = "#171717", # black
"C" = "#17171717" # light grey
),
individual_cor = c(
H1 = "#091638", #Blue-Green Darkest
H2 = "#11185B",
H3 = "#0F2C71",
H4 = "#0D568F",
H4R = "#0D568F",
H5 = "#1D8296",
H6 = "#46A389",
H7 = "#9DD484", #Blue-Green Lightest
C1 = "#340702", #Brown-Orange darkest
C2 = "#5D0B02",
C3 = "#951302",
C4 = "#D32804",
C5 = "#F74019",
C6 = "#FA7A38",
C7 = "#FCC598"
)
)
rownames(Cor_metadata) <- Cor_metadata$sample_cor
# saveRDS(Cor_Filt_RMG0_RNA_log2cpm, "data/QC/concat/Cor_Filt_RMG0_RNA_log2cpm.RDS")
# saveRDS(Cor_metadata, "data/QC/concat/Cor_RNA_metadata.RDS")
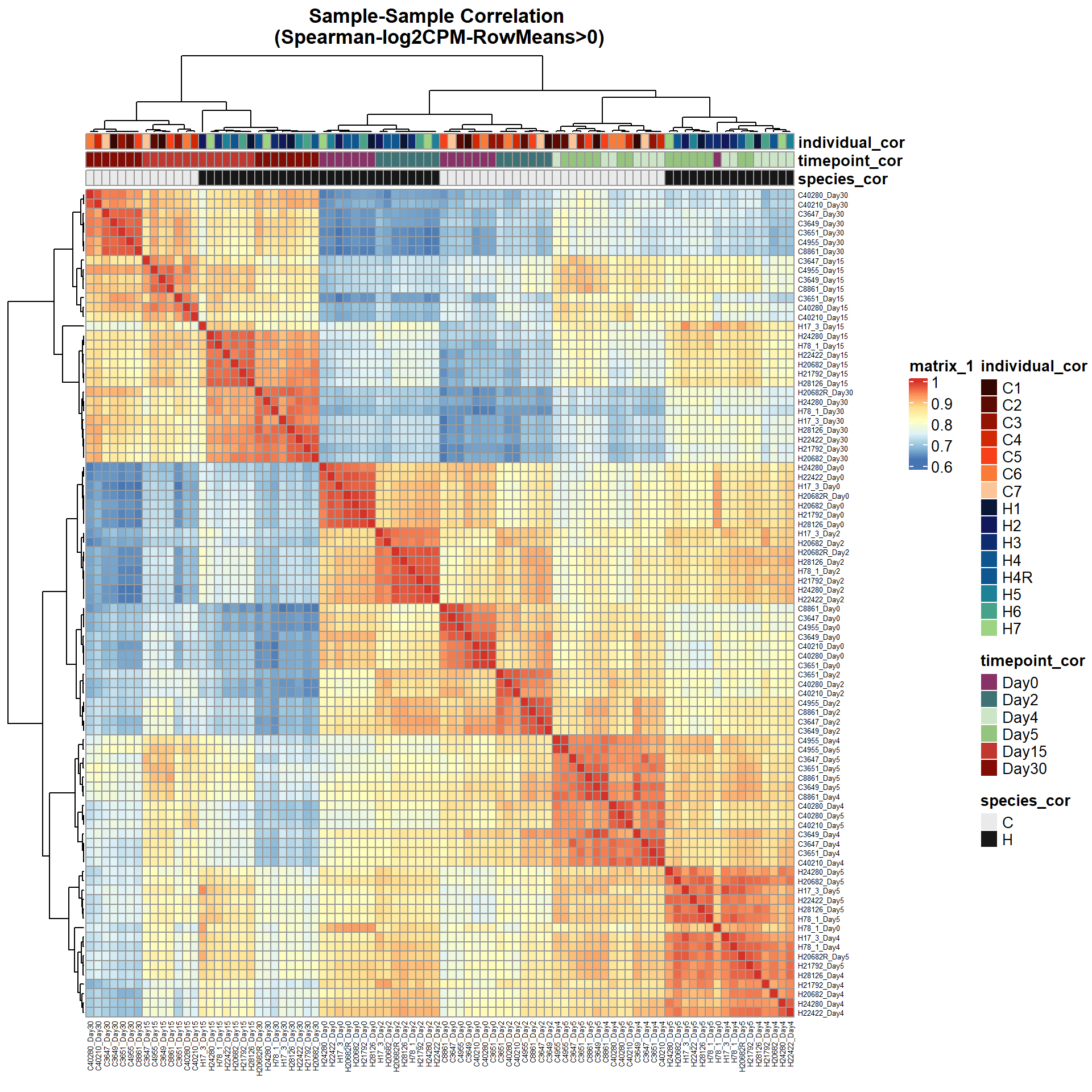
# saveRDS(ann_colors,"data/QC/concat/ann_colors.RDS")print(
pheatmap(Cor_Filt_RMG0_RNA_log2cpm,
fontsize_row = 5,
fontsize_col = 5,
annotation_col = Cor_metadata[, c("species_cor", "timepoint_cor","individual_cor")],
annotation_colors = ann_colors,
clustering_distance_rows = "correlation",
clustering_distance_cols = "correlation",
main = "Sample-Sample Correlation \n(Spearman-log2CPM-RowMeans>0)")
)
####Subset####
RNA_Metadata_No4 <- RNA_Metadata %>%
filter(Timepoint != "Day4")
RNA_fc_NoD4 <- RNA_fc %>%
dplyr::select(-ends_with("_Day4"))
RNA_log2cpm_NoD4 <- cpm(RNA_fc_NoD4,log=TRUE)
dim(RNA_log2cpm_NoD4)[1] 44125 74dim(RNA_fc)[1] 44125 88row_means_NoD4 <- rowMeans(RNA_log2cpm_NoD4)
Filt_RMG0_RNA_fc_NoD4 <- RNA_fc_NoD4[row_means_NoD4 >0,]
dim(Filt_RMG0_RNA_fc_NoD4)[1] 14084 74Filt_RMG0_RNA_log2cpm_NoD4 <- cpm(Filt_RMG0_RNA_fc_NoD4,log=TRUE)
# saveRDS(RNA_Metadata_No4,"data/QC/concat/RNA_Metatdata_No4.RDS")
# saveRDS(Filt_RMG0_RNA_fc_NoD4,"data/QC/concat/Filt_RMG0_RNA_fc_NoD4.RDS")
# saveRDS(Filt_RMG0_RNA_log2cpm_NoD4, "data/QC/concat/Filt_RMG0_RNA_log2cpm_NoD4.RDS")######Cor_HeatMap####
Cor_Filt_RMG0_RNA_log2cpm_NoD4 <- cor(Filt_RMG0_RNA_log2cpm_NoD4, method = "spearman")
Cor_metadata_No4 <- Cor_metadata %>%
dplyr::filter(timepoint_cor !="Day4")
ann_colors_No4 <- ann_colors
ann_colors_No4$timepoint_cor <- ann_colors$timepoint_cor[
names(ann_colors$timepoint_cor) != "Day4"
]
# saveRDS(Cor_metadata_No4, "data/QC/concat/Cor_metadata_No4.RDS")
# saveRDS(Cor_Filt_RMG0_RNA_log2cpm_NoD4, "data/QC/concat/Cor_Filt_RMG0_RNA_log2cpm_NoD4.RDS")
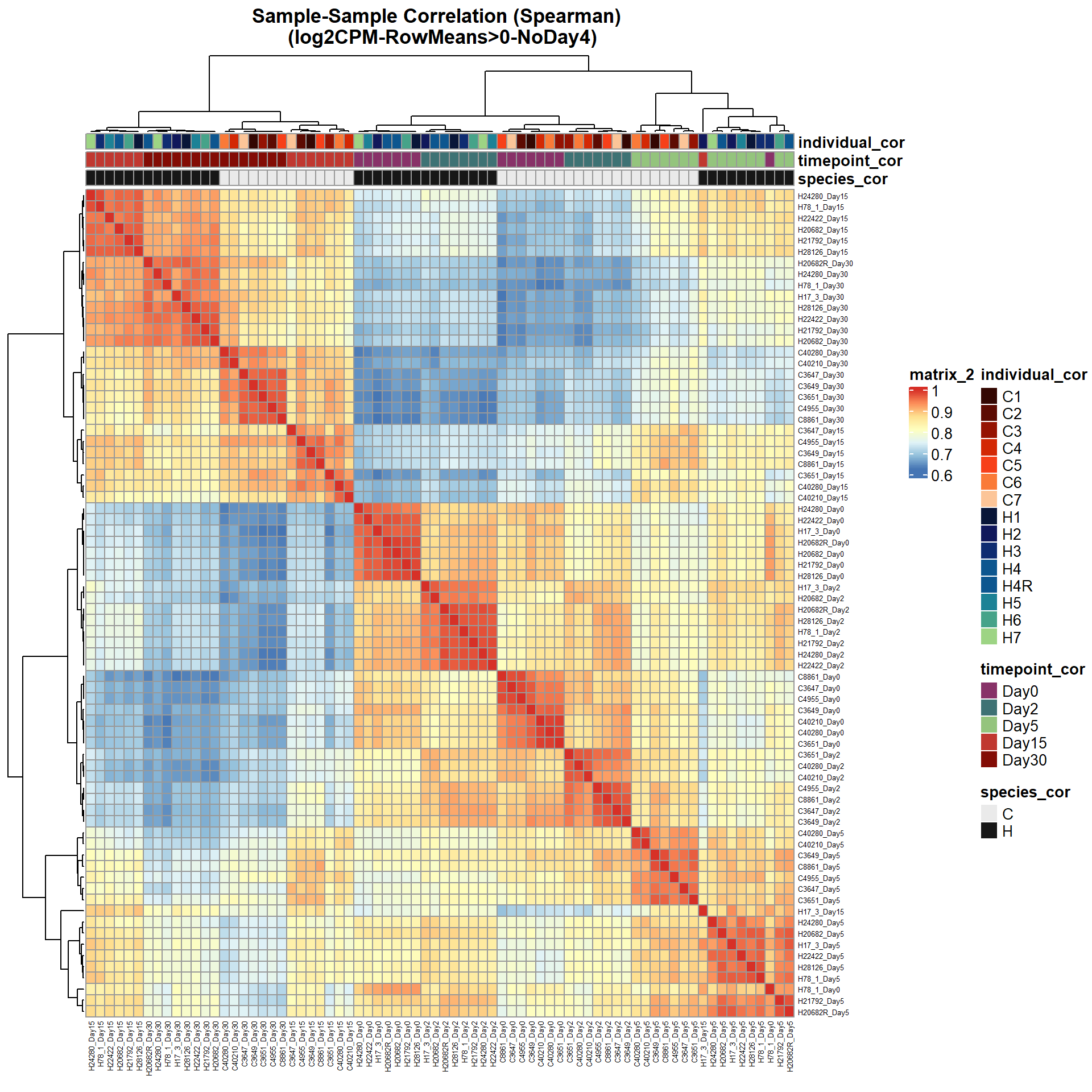
# saveRDS(ann_colors_No4,"data/QC/concat/ann_colors_no4.RDS")print(
pheatmap(Cor_Filt_RMG0_RNA_log2cpm_NoD4,
fontsize_row = 5,
fontsize_col = 5,
annotation_col = Cor_metadata_No4[, c("species_cor", "timepoint_cor","individual_cor")],
annotation_colors = ann_colors_No4,
clustering_distance_rows = "correlation",
clustering_distance_cols = "correlation",
main = "Sample-Sample Correlation (Spearman) \n (log2CPM-RowMeans>0-NoDay4)")
)
markers <- marker_genes$Ensembl.ID
log2cpm_long <- Filt_RMG0_RNA_log2cpm %>%
as.data.frame() %>%
rownames_to_column("Ensembl.ID") %>%
filter(Ensembl.ID %in% markers) %>%
pivot_longer(
cols = -Ensembl.ID,
names_to = "Sample",
values_to = "log2CPM"
)
log2cpm_long <- log2cpm_long %>%
left_join(RNA_Metadata, by = c("Sample" = "SampleName"))
log2cpm_long <- log2cpm_long %>%
left_join(marker_genes, by = "Ensembl.ID")
log2cpm_long$Timepoint <- factor(
log2cpm_long$Timepoint,
levels = c("Day0", "Day2", "Day4", "Day5", "Day15", "Day30")
)
log2cpm_long_NoR <- log2cpm_long %>%
filter(Individual != "H4R")
library(dplyr)
mean_df <- log2cpm_long_NoR %>%
group_by(Stage, Timepoint, Species) %>%
summarise(
mean_log2CPM = mean(log2CPM, na.rm = TRUE),
.groups = "drop"
)
log2cpm_long_NoR$Stage <- factor(
log2cpm_long_NoR$Stage,
levels = c(
"Pluripotency",
"Mesoderm",
"CardiacMesoderm",
"CardiacProgenitors",
"EarlyCardiomyocytes",
"MatureCardiomyocytes"
)
)library(ggplot2)
ggplot(log2cpm_long_NoR, aes(
x = Timepoint,
y = log2CPM
)) +
geom_boxplot(
aes(fill = Stage),
alpha = 0.3,
outlier.shape = NA
) +
geom_line(
data = mean_df,
aes(
x = Timepoint,
y = mean_log2CPM,
group = interaction(Stage, Species),
linetype = Species
),
color = "black",
linewidth = 1.2
) +
geom_point(
data = mean_df,
aes(
x = Timepoint,
y = mean_log2CPM,
shape = Species
),
color = "black",
size = 2
) +
scale_linetype_manual(values = c(
C = "dotted",
H = "solid"
)) +
scale_shape_manual(values = c(
C = 16,
H = 17
)) +
facet_wrap(~ factor(Stage, levels = levels(log2cpm_long_NoR$Stage))) +
theme_classic() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))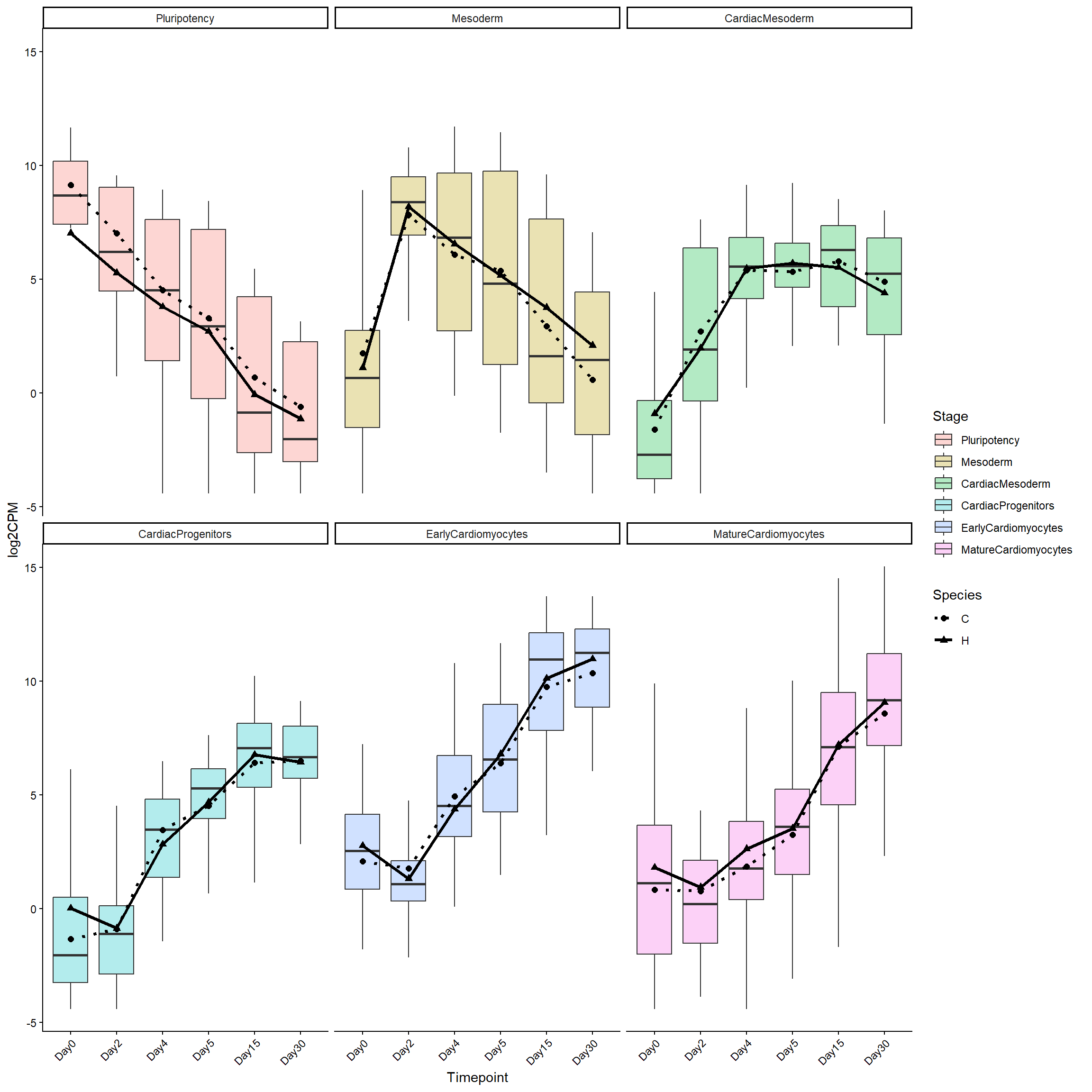
# git -> commit all changes
# git -> push
wflow_publish("analysis/RNA_CorrelationHeatMap_Ensemble.Rmd")
sessionInfo()R version 4.5.1 (2025-06-13 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 11 x64 (build 26100)
Matrix products: default
LAPACK version 3.12.1
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] scales_1.4.0 ComplexHeatmap_2.24.1
[3] ggfortify_0.4.19 readxl_1.4.5
[5] RUVSeq_1.42.0 EDASeq_2.42.0
[7] ShortRead_1.66.0 GenomicAlignments_1.44.0
[9] SummarizedExperiment_1.38.1 MatrixGenerics_1.20.0
[11] matrixStats_1.5.0 Rsamtools_2.24.0
[13] GenomicRanges_1.60.0 Biostrings_2.76.0
[15] GenomeInfoDb_1.44.3 XVector_0.48.0
[17] BiocParallel_1.42.1 lubridate_1.9.5
[19] forcats_1.0.1 stringr_1.6.0
[21] purrr_1.2.1 tidyr_1.3.2
[23] tidyverse_2.0.0 Cormotif_1.54.0
[25] affy_1.86.0 pheatmap_1.0.13
[27] org.Hs.eg.db_3.21.0 AnnotationDbi_1.70.0
[29] IRanges_2.42.0 S4Vectors_0.46.0
[31] Biobase_2.68.0 BiocGenerics_0.54.1
[33] generics_0.1.4 readr_2.1.6
[35] ggrepel_0.9.6 dplyr_1.1.4
[37] tibble_3.3.1 ggplot2_4.0.2
[39] edgeR_4.6.3 limma_3.64.3
[41] workflowr_1.7.2
loaded via a namespace (and not attached):
[1] later_1.4.5 BiocIO_1.18.0 bitops_1.0-9
[4] filelock_1.0.3 R.oo_1.27.1 cellranger_1.1.0
[7] preprocessCore_1.70.0 XML_3.99-0.20 lifecycle_1.0.5
[10] httr2_1.2.2 pwalign_1.4.0 doParallel_1.0.17
[13] rprojroot_2.1.1 processx_3.8.6 lattice_0.22-7
[16] MASS_7.3-65 magrittr_2.0.4 sass_0.4.10
[19] rmarkdown_2.30 jquerylib_0.1.4 yaml_2.3.12
[22] httpuv_1.6.16 otel_0.2.0 DBI_1.3.0
[25] RColorBrewer_1.1-3 abind_1.4-8 R.utils_2.13.0
[28] RCurl_1.98-1.17 rappdirs_0.3.4 git2r_0.36.2
[31] circlize_0.4.17 GenomeInfoDbData_1.2.14 codetools_0.2-20
[34] DelayedArray_0.34.1 xml2_1.5.2 tidyselect_1.2.1
[37] shape_1.4.6.1 UCSC.utils_1.4.0 farver_2.1.2
[40] BiocFileCache_2.16.2 jsonlite_2.0.0 GetoptLong_1.1.0
[43] iterators_1.0.14 foreach_1.5.2 tools_4.5.1
[46] progress_1.2.3 Rcpp_1.1.1 glue_1.8.0
[49] gridExtra_2.3 SparseArray_1.8.1 xfun_0.56
[52] withr_3.0.2 BiocManager_1.30.27 fastmap_1.2.0
[55] latticeExtra_0.6-31 callr_3.7.6 digest_0.6.39
[58] timechange_0.4.0 R6_2.6.1 colorspace_2.1-2
[61] Cairo_1.7-0 jpeg_0.1-11 biomaRt_2.64.0
[64] RSQLite_2.4.5 R.methodsS3_1.8.2 rtracklayer_1.68.0
[67] prettyunits_1.2.0 httr_1.4.8 S4Arrays_1.8.1
[70] whisker_0.4.1 pkgconfig_2.0.3 gtable_0.3.6
[73] blob_1.3.0 S7_0.2.1 hwriter_1.3.2.1
[76] htmltools_0.5.9 clue_0.3-66 png_0.1-8
[79] knitr_1.51 rstudioapi_0.18.0 tzdb_0.5.0
[82] rjson_0.2.23 curl_7.0.0 cachem_1.1.0
[85] GlobalOptions_0.1.3 parallel_4.5.1 restfulr_0.0.16
[88] pillar_1.11.1 vctrs_0.7.1 promises_1.5.0
[91] dbplyr_2.5.2 cluster_2.1.8.1 evaluate_1.0.5
[94] GenomicFeatures_1.60.0 cli_3.6.5 locfit_1.5-9.12
[97] compiler_4.5.1 rlang_1.1.7 crayon_1.5.3
[100] labeling_0.4.3 interp_1.1-6 aroma.light_3.38.0
[103] ps_1.9.1 getPass_0.2-4 fs_1.6.6
[106] stringi_1.8.7 deldir_2.0-4 Matrix_1.7-4
[109] hms_1.1.4 bit64_4.6.0-1 KEGGREST_1.48.1
[112] statmod_1.5.1 memoise_2.0.1 affyio_1.78.0
[115] bslib_0.10.0 bit_4.6.0