Integrate lanes from all batches of EB pilot (Human data only, no doublets
Last updated: 2020-08-10
Checks: 7 0
Knit directory: Embryoid_Body_Pilot_Workflowr/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200804) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version f50ebd3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: output/.Rhistory
Untracked files:
Untracked: analysis/child/
Untracked: code/EB.getHumanMetadata.Rmd
Untracked: figure/
Untracked: output/mergedObjects/
Untracked: output/sampleQCrds/
Unstaged changes:
Modified: .Rprofile
Modified: .gitattributes
Modified: .gitignore
Modified: Embryoid_Body_Pilot_Workflowr.Rproj
Modified: README.md
Modified: _workflowr.yml
Modified: analysis/_site.yml
Modified: analysis/about.Rmd
Modified: analysis/index.Rmd
Modified: analysis/license.Rmd
Modified: code/README.md
Modified: data/README.md
Modified: output/README.md
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/IntegrateAnalysis.afterFilter.HarmonyBatchindividual.Rmd) and HTML (docs/IntegrateAnalysis.afterFilter.HarmonyBatchindividual.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | f50ebd3 | KLRhodes | 2020-08-10 | wflow_publish("analysis/Integrate*") |
| html | 421a225 | KLRhodes | 2020-08-10 | Build site. |
| Rmd | bc8ec6f | KLRhodes | 2020-08-10 | cleaning various versions of merging/intCurrent working directory |
library(Seurat)
library(harmony)
library(ggplot2)
library(DataCombine)
library(here)
library(RColorBrewer)
options(future.globals.maxSize= 15000*1024^2) #allow global exceeding 4GbRead in the files, add metadata, and create an object list
filelist<-list.files(here::here('output/sampleQCrds/'), full.names = T)
objectlist<- list()
for (i in 1:length(filelist)){
rds<- readRDS(filelist[i])
objectlist[i]<- rds
}Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecated
Warning in `[<-`(`*tmp*`, i, value = rds): implicit list embedding of S4 objects
is deprecatedcreate a merged seurat object
ids<-substr(basename(filelist),1,12)
merged<- merge(objectlist[[1]], c(objectlist[[2]], objectlist[[3]],objectlist[[4]],objectlist[[5]],objectlist[[6]],objectlist[[7]],objectlist[[8]],objectlist[[9]],objectlist[[10]],objectlist[[11]],objectlist[[12]],objectlist[[13]],objectlist[[14]],objectlist[[15]],objectlist[[16]]),add.cell.ids=ids, merge.data=T)#need to fix the individual names because they are slightly different from batch1
replacements<- data.frame(from= c("SNG-NA18511.variant2", "SNG-NA18858.variant2", "SNG-NA19160.variant2"), to=c("SNG-NA18511", "SNG-NA18858", "SNG-NA19160"))
merged@meta.data<-FindReplace(merged@meta.data, "individual", replacements, from = "from", to= "to", exact=T, vector=F )Only exact matches will be replaced.#run PCA on full dataset pre-alignment
all.genes= rownames(merged)
merged<-FindVariableFeatures(merged,selection.method="vst", nfeatures = 5000)
#have previously used all genes (nfeatures=25000) and clustering by individual rather than batch (based on proportion of cells per cluster) was still observed downstream. Now using 5000 because it is the upper bound of what has been recommended in the literature.
merged<- ScaleData(merged, features = all.genes)Centering and scaling data matrixmerged<-RunPCA(merged, npcs = 100, verbose=F)DimPlot(merged, reduction = "pca", group.by = "Batch")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
Now, running harmony to integrate. Here, using Batch, SampleID(10x Lane), and individual to integrate. Since Batch and Lane are confounded, this may over correct for Batch.
merged<- RunHarmony(merged, c("Batch", "individual"), plot_convergence = T, assay.use = "SCT")Harmony 1/10Harmony 2/10Harmony 3/10Harmony converged after 3 iterationsWarning: Invalid name supplied, making object name syntactically valid. New
object name is Seurat..ProjectDim.SCT.harmony; see ?make.names for more details
on syntax validity
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
Visualize Harmony embeddings
DimPlot(merged, reduction="harmony", group.by= c("individual", "Batch"), combine=F)[[1]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[2]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
Now Running UMAP and identifying clusters, etc
merged<- RunUMAP(merged, reduction = "harmony", dims = 1:100, verbose = F)Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per sessionmerged<- FindNeighbors(merged, reduction="harmony", dims = 1:100, verbose = F)
merged<- FindClusters(merged, resolution=1, verbose = F)
merged<- FindClusters(merged, resolution=0.8, verbose = F)
merged<- FindClusters(merged, resolution=0.5, verbose = F)
merged<- FindClusters(merged, resolution=0.1, verbose = F)SAVING merged/aligned/reclustered object
path<- here::here("output/mergedObjects/")
saveRDS(merged, file=paste0(path,'Harmony.Batchindividual.rds'))#reassign idents
Idents(merged)<- 'SCT_snn_res.1'VizDimLoadings(merged, dims = 1:2, reduction = "harmony")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
VizDimLoadings(merged, dims = 3:4, reduction = "harmony")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
VizDimLoadings(merged, dims = 5:6, reduction = "harmony")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
xlim <- c(min(merged@reductions$harmony@cell.embeddings[,'harmony_1']),
max(merged@reductions$harmony@cell.embeddings[,'harmony_1']))
ylim <- c(min(merged@reductions$harmony@cell.embeddings[,'harmony_2']),
max(merged@reductions$harmony@cell.embeddings[,'harmony_2']))
individuals <- table(merged$individual)
individuals <- individuals[individuals>50]
individuals <- names(individuals)
for (i in individuals)
{
print(DimPlot(merged, reduction = "harmony", group.by = c("Batch"), pt.size = 0.01,
cells = WhichCells(merged, expression = individual == i)) +
xlim(xlim) + ylim(ylim) + ggtitle(i))
}
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |

| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |

| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap")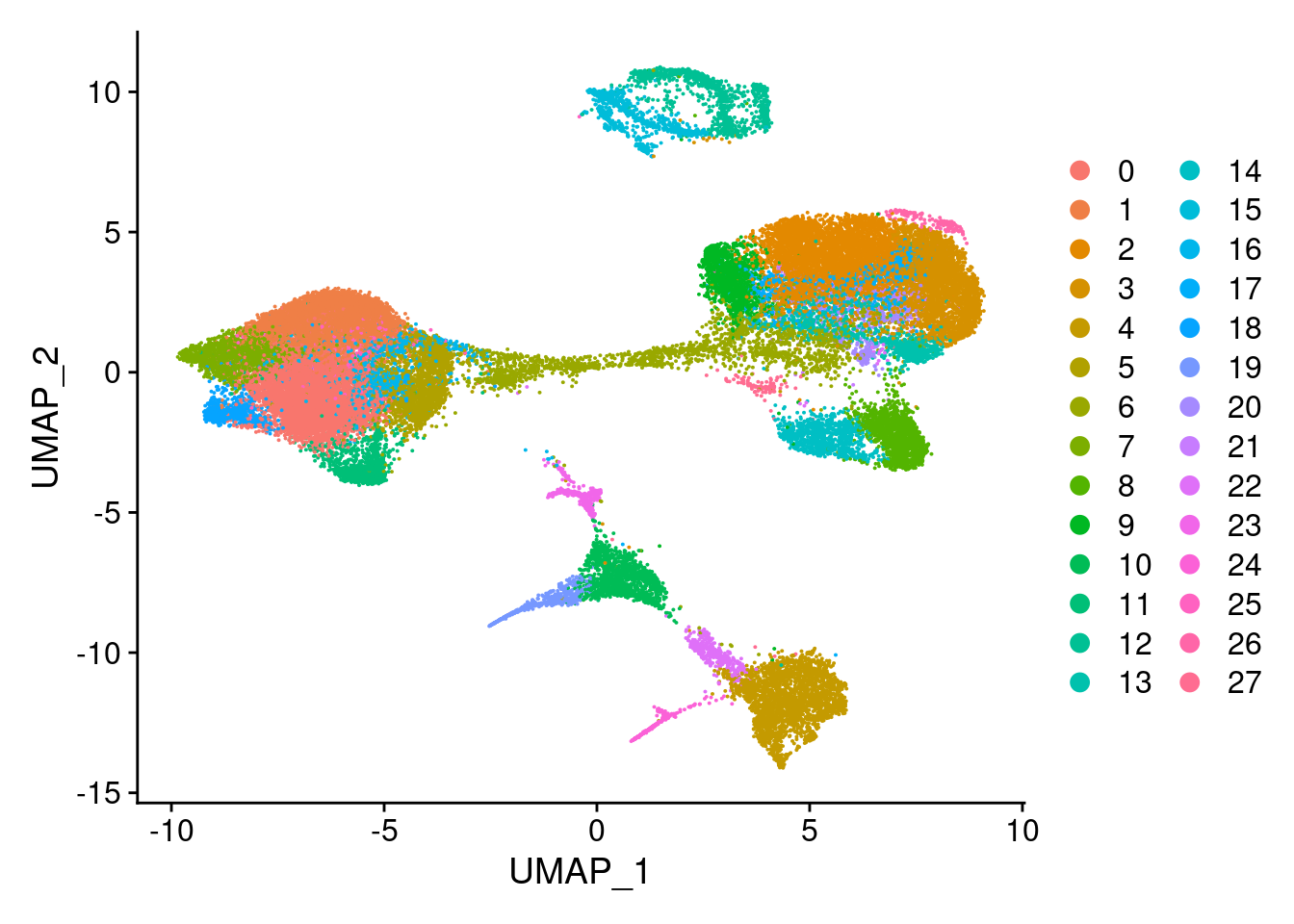
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "Batch")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "individual")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
xlim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_1']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_1']))
ylim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_2']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_2']))
for (i in individuals)
{
print(DimPlot(merged, reduction = "umap",
cells = WhichCells(merged, expression = individual == i)) +
xlim(xlim) + ylim(ylim) + ggtitle(i))
}
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |

| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
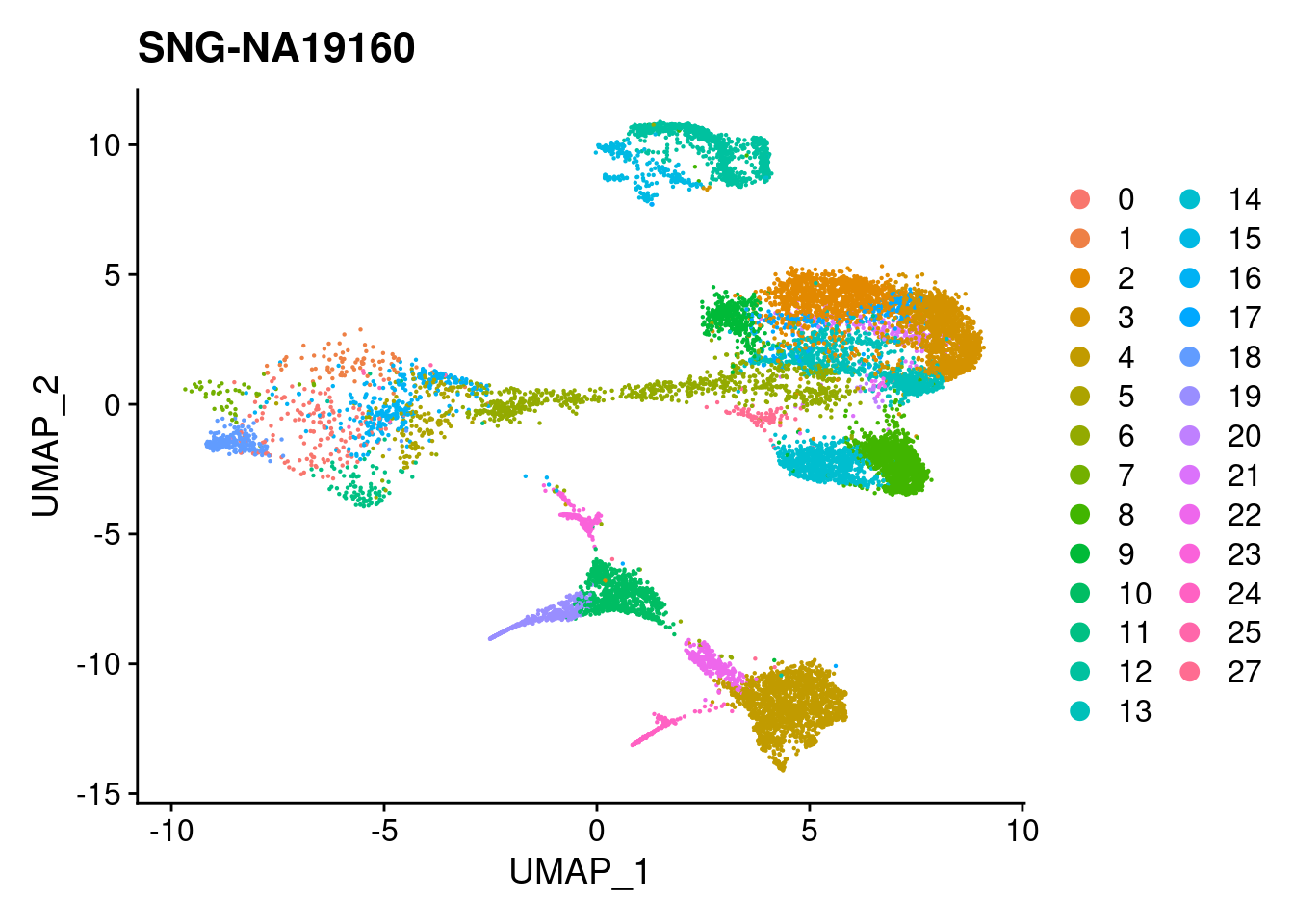
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
plots2<- DimPlot(merged, group.by = "individual", split.by = "Batch")
plots2
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, group.by = "Batch", split.by = c("individual"))
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, group.by = "SCT_snn_res.1", split.by = c("Batch"), label=T)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "harmony", group.by = "SCT_snn_res.1", split.by = "Batch", combine = F)[[1]]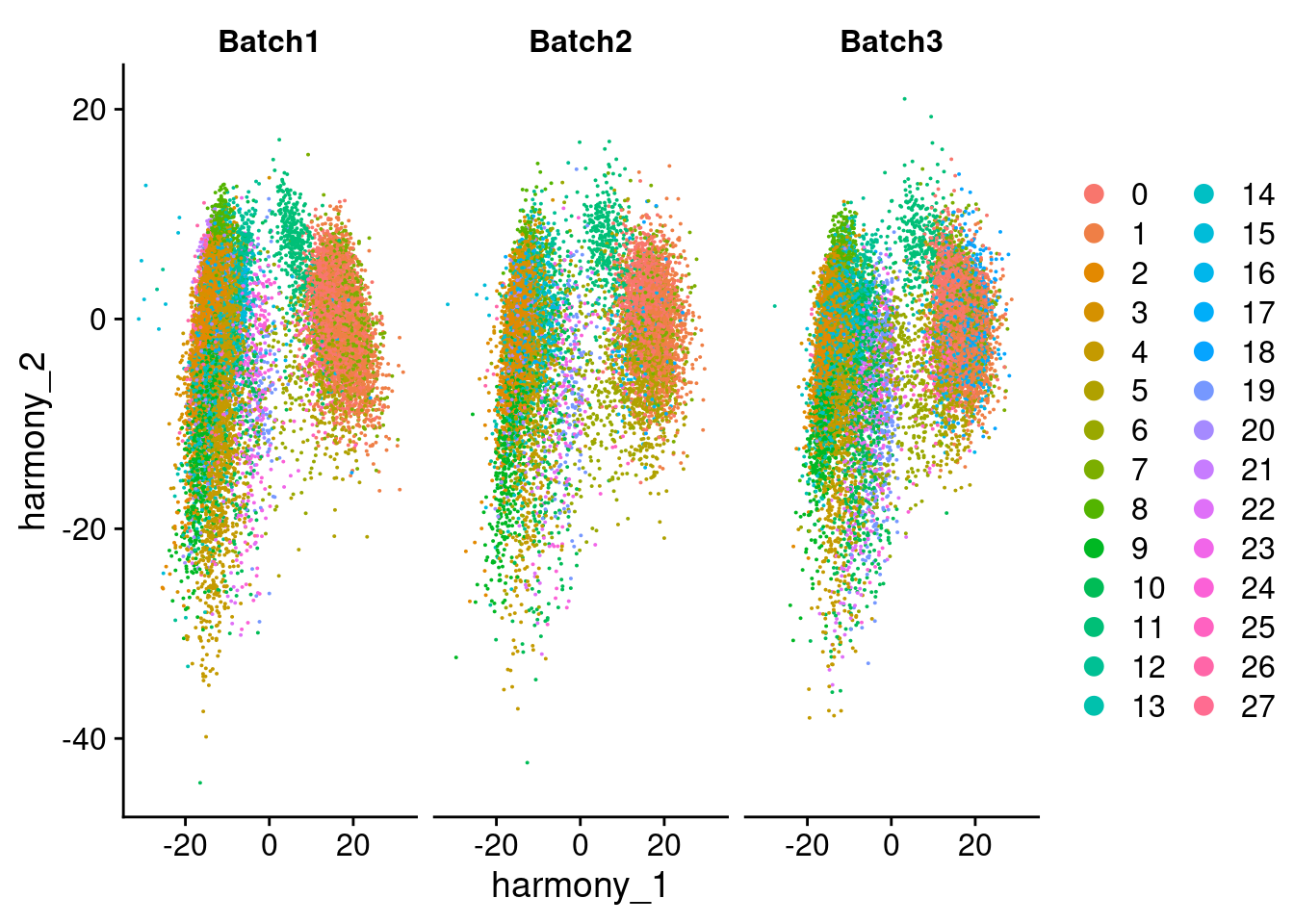
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
VlnPlot(merged, features = c("POU5F1", "PAX6", "TNNT2", "SOX17", "HAND1", "LUM"), ncol=2)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#pluripotent markers
FeaturePlot(merged, features = c("POU5F1", "SOX2", "NANOG"), pt.size = 0.2, ncol=3)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#Endoderm markers (first 3 definitive endo, 4-6 liver markers, )
FeaturePlot(merged, features = c("SOX17","CLDN6","FOXA2", "TTR", "AFP", "FGB"), pt.size = 0.2, combine = F)[[1]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[2]]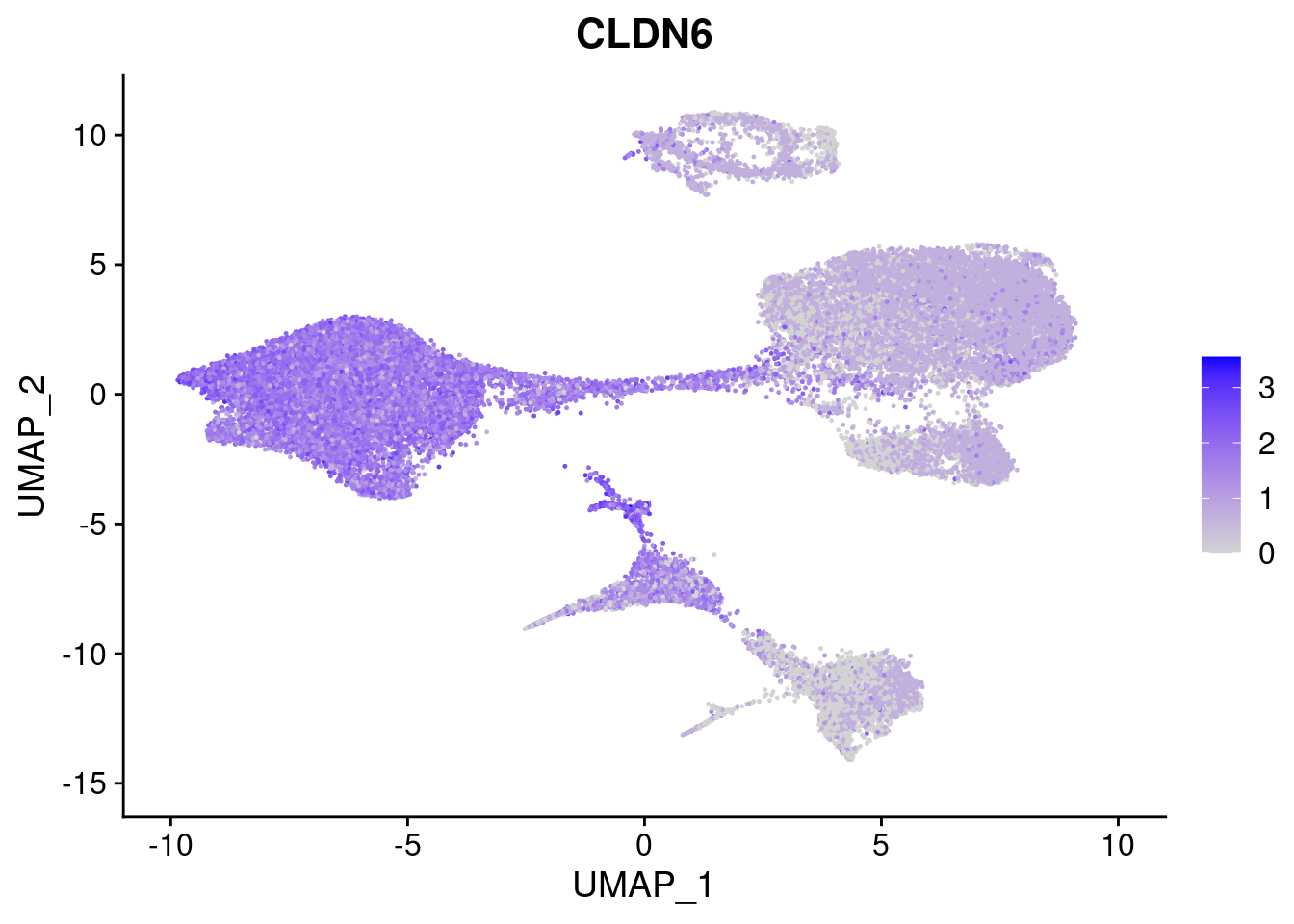
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[3]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[4]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[5]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[6]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#Mesoderm Markers (first 3 early meso markers, 4-6 heart markers, 7-9 endothelial markers (which comes from mesoderm), then some other general muscle markers)
FeaturePlot(merged, features = c("HAND1", "BMP4", "TNNT2","KDR", "GNG11", "ECSCR", "COL3A1", "ACTC1"), pt.size = 0.2, combine=F)[[1]]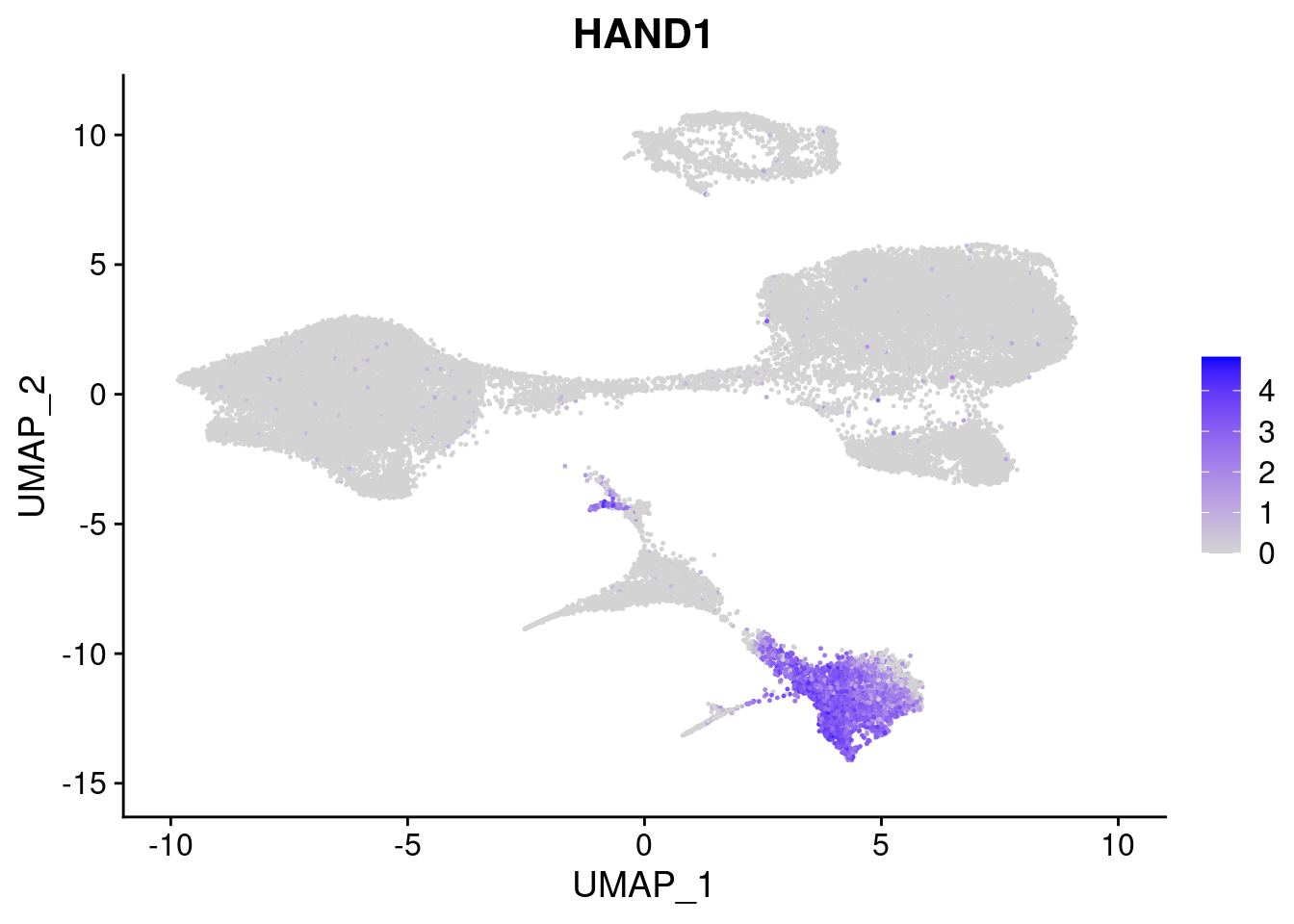
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[2]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[3]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[4]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[5]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[6]]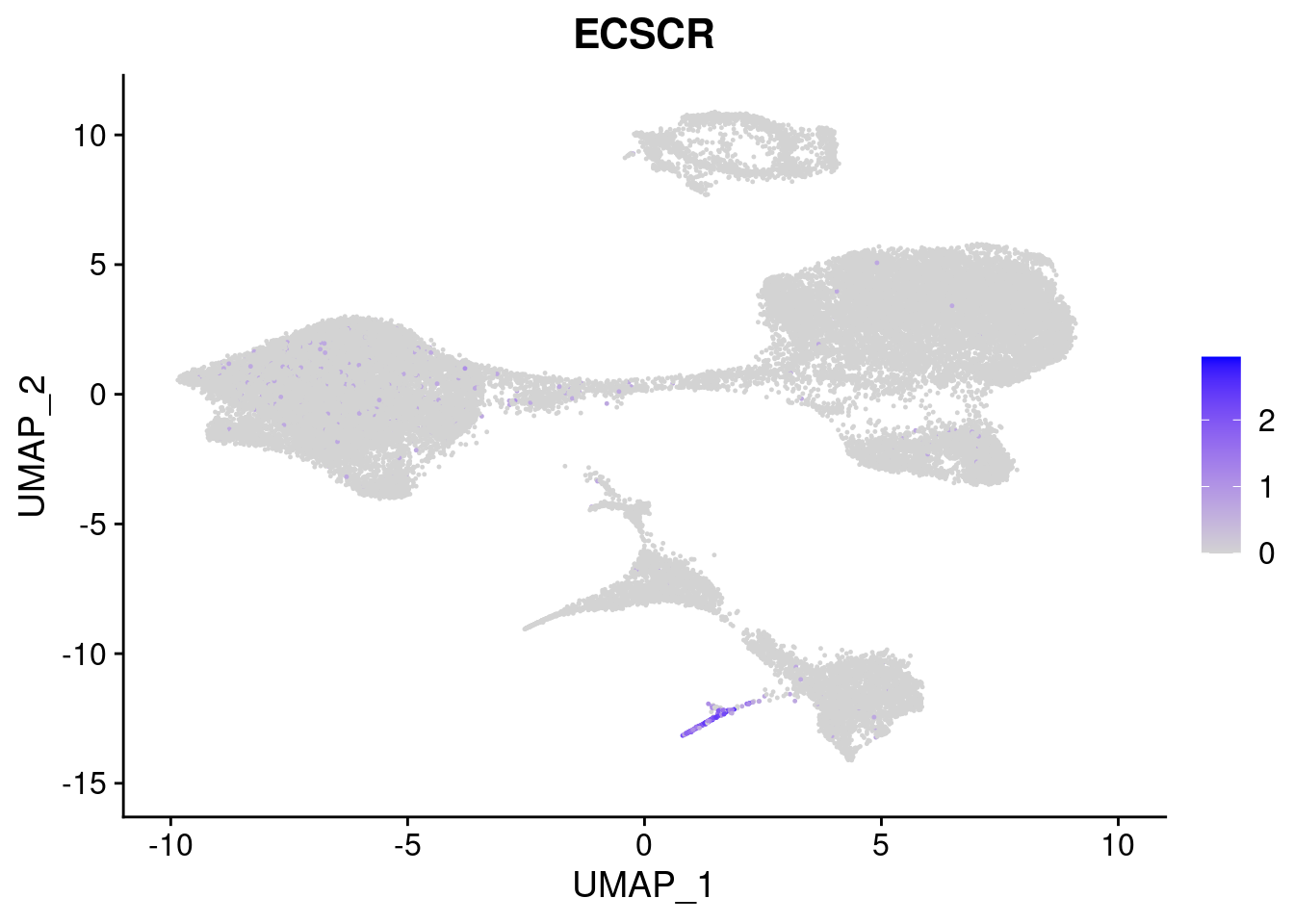
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[7]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[8]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#Ectoderm Markers (3-1 early ectoderm markers, 4-6schwann cell (myelinating, non myelinating, or precursor), 7-8 oligodendrocytes, 9-10 radial glia)
FeaturePlot(merged, features = c("PAX6", "GBX2", "NES", "MPZ", "SOX10","GAP43", "OLIG1", "OLIG2", "VIM", "HES5"), pt.size = 0.2, ncol=3, combine=F)[[1]]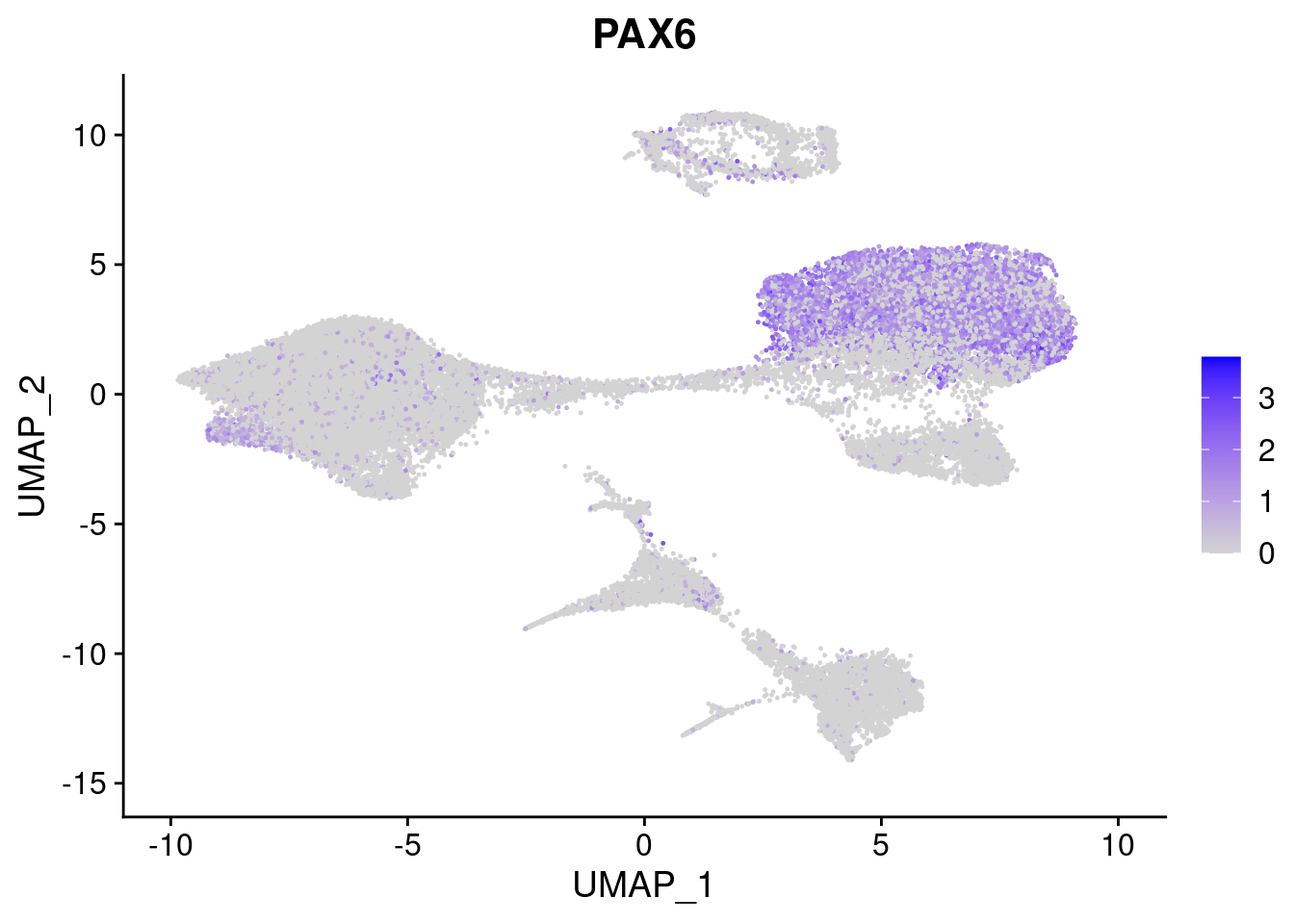
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[2]]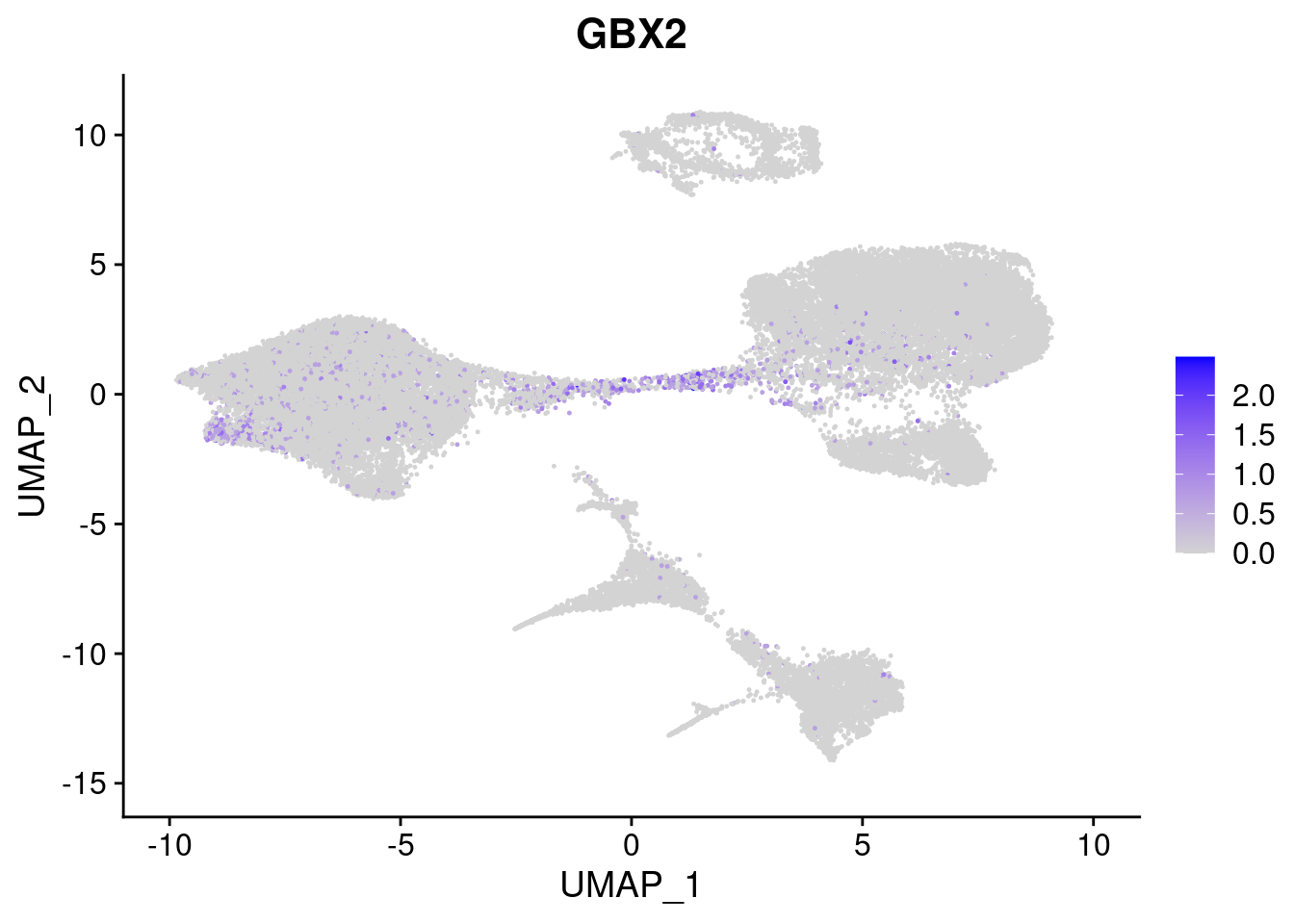
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[3]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[4]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[5]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[6]]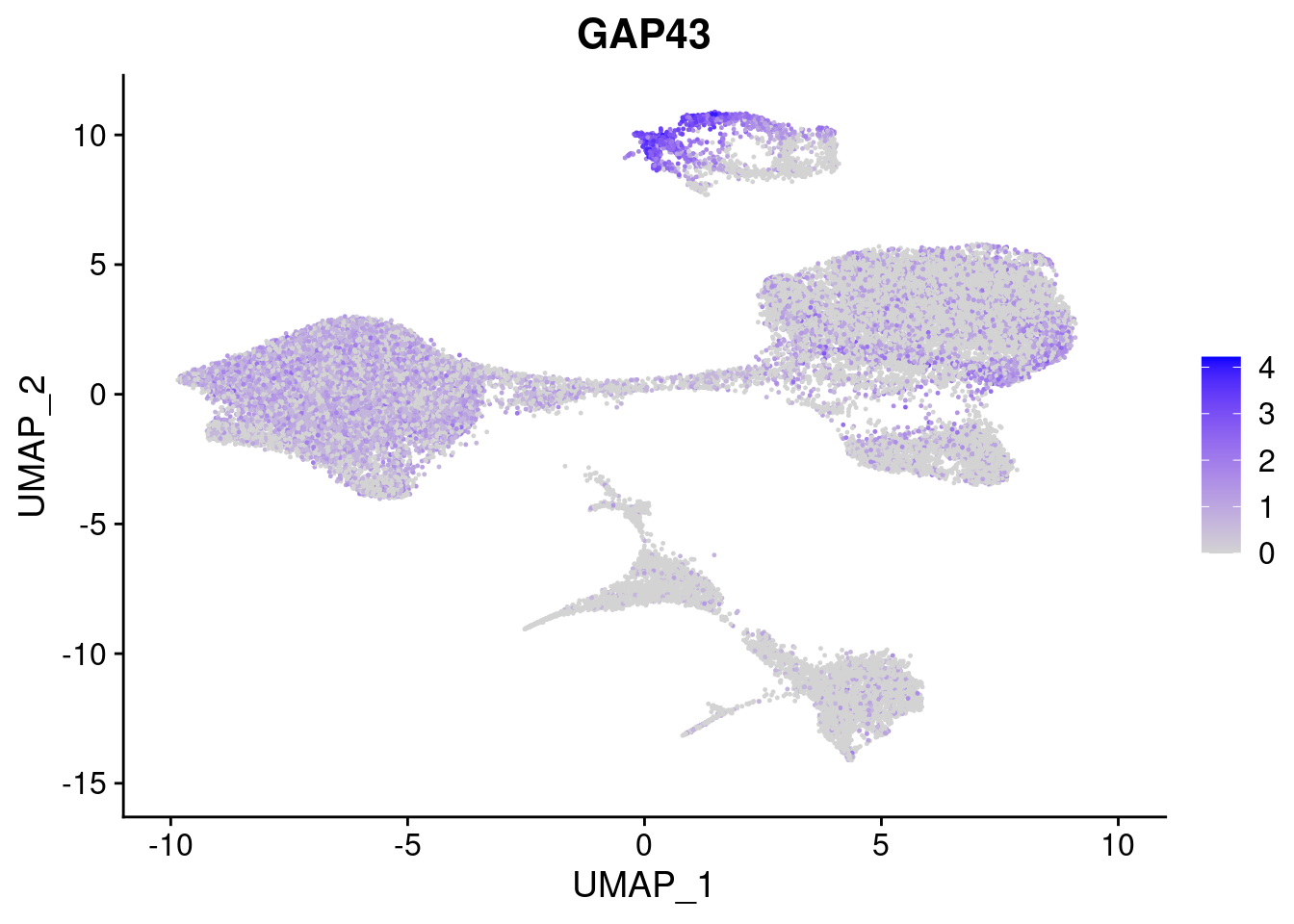
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[7]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[8]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[9]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
[[10]]
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#More ectoderm, specifically neurons
#immature neurons: NEUROD1
#Mature Neurons: MAP2, SYP
#dopaminergic: TH, FOXA2,
FeaturePlot(merged, features = c("MAP2", "SYP","NEUROD1", "TH" ), pt.size = 0.2, ncol=3)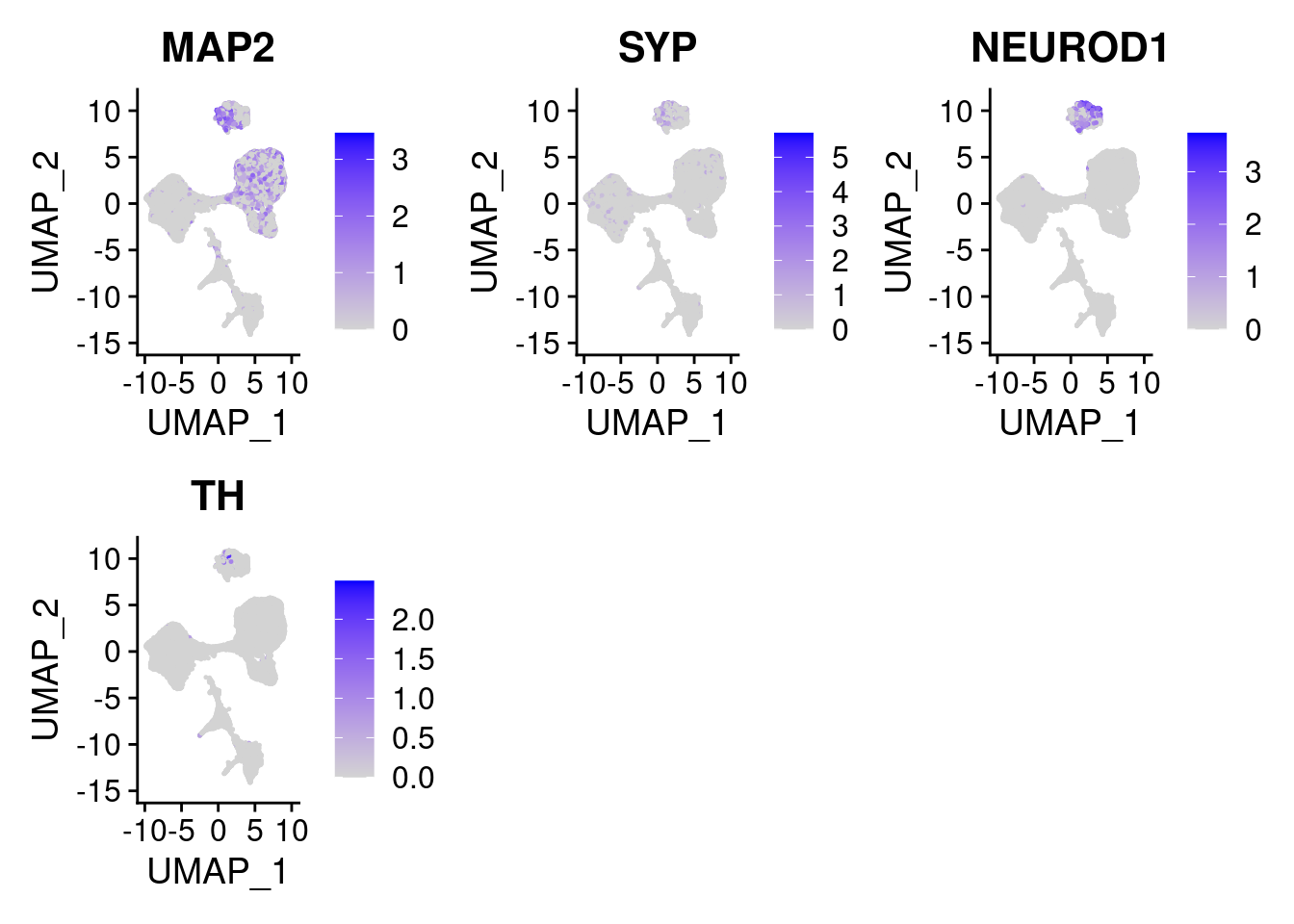
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
Identify cluster markers
#how many cells per cluster?
t1<-table(merged@meta.data$SCT_snn_res.1, merged@meta.data$Batch)
t1
Batch1 Batch2 Batch3
0 2377 1416 2350
1 2128 1191 1821
2 2014 1306 1157
3 1708 721 878
4 1361 435 871
5 406 855 741
6 412 512 1075
7 687 258 647
8 778 221 526
9 550 490 442
10 249 273 859
11 445 410 309
12 615 207 329
13 416 141 540
14 443 190 404
15 332 203 320
16 36 92 592
17 256 245 207
18 11 55 633
19 105 120 369
20 289 255 43
21 568 3 0
22 82 75 252
23 238 64 94
24 103 60 134
25 83 38 99
26 101 19 29
27 80 10 29#percent of cells in each cluster per batch
t1colsum<- colSums(t1)
percT1<-t1/t1colsum
percT1
Batch1 Batch2 Batch3
0 0.1408759557 0.1435377598 0.1492063492
1 0.2157121135 0.0756190476 0.1079239021
2 0.1278730159 0.0774017661 0.1172833249
3 0.1012268121 0.0730866700 0.0557460317
4 0.1379624937 0.0276190476 0.0516209329
5 0.0257777778 0.0506726723 0.0751140395
6 0.0244177088 0.0519006589 0.0682539683
7 0.0696401419 0.0163809524 0.0383452854
8 0.0493968254 0.0130978486 0.0533198175
9 0.0325964559 0.0496705525 0.0280634921
10 0.0252407501 0.0173333333 0.0509097375
11 0.0282539683 0.0242991762 0.0313228586
12 0.0364487643 0.0209832742 0.0208888889
13 0.0421692854 0.0089523810 0.0320037930
14 0.0281269841 0.0112605938 0.0409528637
15 0.0196764061 0.0205778003 0.0203174603
16 0.0036492651 0.0058412698 0.0350856398
17 0.0162539683 0.0145202394 0.0209832742
18 0.0006519291 0.0055752661 0.0401904762
19 0.0106436898 0.0076190476 0.0218692586
20 0.0183492063 0.0151129023 0.0043588444
21 0.0336632490 0.0003041054 0.0000000000
22 0.0083122149 0.0047619048 0.0149351034
23 0.0151111111 0.0037930421 0.0095286366
24 0.0061044272 0.0060821085 0.0085079365
25 0.0084135834 0.0024126984 0.0058673621
26 0.0064126984 0.0011260594 0.0029396858
27 0.0047413027 0.0010136847 0.0018412698heatmap(t(percT1))
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#how many cells per cluster from each individual?
t2<-table(merged@meta.data$SCT_snn_res.1, merged@meta.data$individual)
t2
SNG-NA18511 SNG-NA18858 SNG-NA19160
0 363 5633 147
1 199 4859 82
2 3005 237 1235
3 1575 154 1578
4 831 31 1805
5 113 1741 148
6 871 289 839
7 84 1466 42
8 155 34 1336
9 971 103 408
10 524 38 819
11 106 965 93
12 139 19 993
13 89 11 997
14 135 7 895
15 581 37 237
16 411 16 293
17 416 56 236
18 450 5 244
19 155 15 424
20 133 435 19
21 328 33 210
22 108 6 295
23 83 135 178
24 90 3 204
25 18 193 9
26 0 149 0
27 16 0 103t2colsums<-colSums(t2)
percT2<- t2/t2colsums
percT2
SNG-NA18511 SNG-NA18858 SNG-NA19160
0 0.0303791112 0.3379124175 0.0105991780
1 0.0119376125 0.3503497008 0.0068624990
2 0.2166702718 0.0198342958 0.0740851830
3 0.1318101933 0.0092381524 0.1137789314
4 0.0498500300 0.0022352008 0.1510586660
5 0.0081476675 0.1457025693 0.0088782244
6 0.0728931291 0.0173365327 0.0604946283
7 0.0050389922 0.1057033672 0.0035149385
8 0.0111760040 0.0028454264 0.0801439712
9 0.0812620303 0.0061787642 0.0294181268
10 0.0314337133 0.0027399236 0.0685413005
11 0.0076429447 0.0807598962 0.0055788842
12 0.0116327726 0.0011397720 0.0715985291
13 0.0053389322 0.0007931358 0.0834379446
14 0.0097339390 0.0005858231 0.0536892621
15 0.0486233158 0.0022195561 0.0170884707
16 0.0246550690 0.0011536520 0.0245208804
17 0.0299949528 0.0046865847 0.0141571686
18 0.0376600552 0.0002999400 0.0175931935
19 0.0092981404 0.0010815488 0.0354841409
20 0.0095897325 0.0364047201 0.0011397720
21 0.0274499958 0.0019796041 0.0151416829
22 0.0064787043 0.0004326195 0.0246882584
23 0.0059845699 0.0112980166 0.0106778644
24 0.0075320110 0.0001799640 0.0147090634
25 0.0010797840 0.0139159276 0.0007532011
26 0.0000000000 0.0124696627 0.0000000000
27 0.0013390242 0.0000000000 0.0074266349heatmap(t(percT2))
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
cormat<-round(cor(percT2),2)
library(reshape2)
melted_cormat<-melt(cormat)
ugly<-ggplot(data= melted_cormat, aes(x=Var1, y=Var2, fill=value)) +
geom_tile() +
ggtitle("Pairwise Pearson Correlation of the percent of cells from \neach cell line assigned to each Seurat Cluster")
get_lower_tri<- function(cormat){
cormat[upper.tri(cormat)]<-NA
return(cormat)
}
lower_tri<- get_lower_tri(cormat)
melted_tri<- melt(lower_tri)
pretty<-ggplot(data= melted_tri, aes(x=Var1, y=Var2, fill=value)) +
geom_tile(color="white") +
scale_fill_gradient2(low="blue", high="red", mid="white", midpoint = 0, limit= c(-1,1), space= "Lab", name="Pearson\nCorrelation") +
theme_minimal() +
ggtitle("Pairwise Pearson Correlation of the percent of cells from \neach cell line assigned to each Seurat Cluster")
pretty
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#exploring similarity in the number of cells per individual between batches
merged.Batch1<- (subset(merged, Batch == "Batch1"))
b1t<- table(merged.Batch1$SCT_snn_res.1, merged.Batch1$individual)
remove("merged.Batch1")
b1tcolsums<- colSums(b1t)
percb1t<- b1t/b1tcolsums
merged.Batch2<- (subset(merged, Batch == "Batch2"))
b2t<- table(merged.Batch2$SCT_snn_res.1, merged.Batch2$individual)
remove("merged.Batch2")
b2tcolsums<- colSums(b2t)
percb2t<- b2t/b2tcolsums
merged.Batch3<- (subset(merged, Batch == "Batch3"))
b3t<- table(merged.Batch3$SCT_snn_res.1, merged.Batch3$individual)
remove("merged.Batch3")
b3tcolsums<- colSums(b3t)
percb3t<- b3t/b3tcolsumscols1<- c("Batch1_18511","Batch1_18858","Batch1_19160", "Batch2_18511", "Batch2_18858","Batch2_19160",
"Batch3_18511","Batch3_18858", "Batch3_19160")
cols2<- c("Batch1_18511", "Batch2_18511", "Batch3_18511","Batch1_18858", "Batch2_18858", "Batch3_18858","Batch1_19160", "Batch2_19160", "Batch3_19160")
fullpercs<- as.data.frame(cbind(percb1t[,1:3], percb2t,percb3t))
colnames(fullpercs)<-cols1
fullpercs<- cbind(fullpercs$Batch1_18511, fullpercs$Batch2_18511, fullpercs$Batch3_18511,
fullpercs$Batch1_18858, fullpercs$Batch2_18858, fullpercs$Batch3_18858,
fullpercs$Batch1_19160, fullpercs$Batch2_19160, fullpercs$Batch3_19160)
colnames(fullpercs)<-cols2
fullpercs_cor<- round(cor(fullpercs),2)
fullpercs_melt<- melt(fullpercs_cor)
ggplot(data= fullpercs_melt, aes(x=Var1, y=Var2, fill=value)) +
geom_tile(color="white") +
scale_fill_gradient2(low="blue", high="red", mid="white", midpoint = 0, limit= c(-1,1), space= "Lab", name="Pearson\nCorrelation") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 30, hjust=1))+
ggtitle("Pairwise Pearson Correlation of the percent of cells from \neach cell line assigned to each Seurat Cluster (res 1)")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#now clustering individual_Batch samples with hierarchical clustering/they will get reordered based on similarity
beauty<- colorRampPalette(brewer.pal(9,"Purples"))(200)
rownames(fullpercs)<- c(0:(nrow(fullpercs)-1))
heatmap(as.matrix(fullpercs), scale="none", col=beauty, cexCol = .7, cexRow=.6)
text(1:ncol(fullpercs),labels=names(fullpercs),srt=30)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#generate a heatmap of the proportion of cells from each individual_batch in each seurat cluster. dendrograms based on similarity of the vectors. should be colored by the value(proportion), but some of the cluster/sample values to seem to match with the colorReclustering with less resolution, check if everything is robust
#reassign idents
Idents(merged)<- 'SCT_snn_res.0.5'DimPlot(merged, reduction = "umap")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "Batch")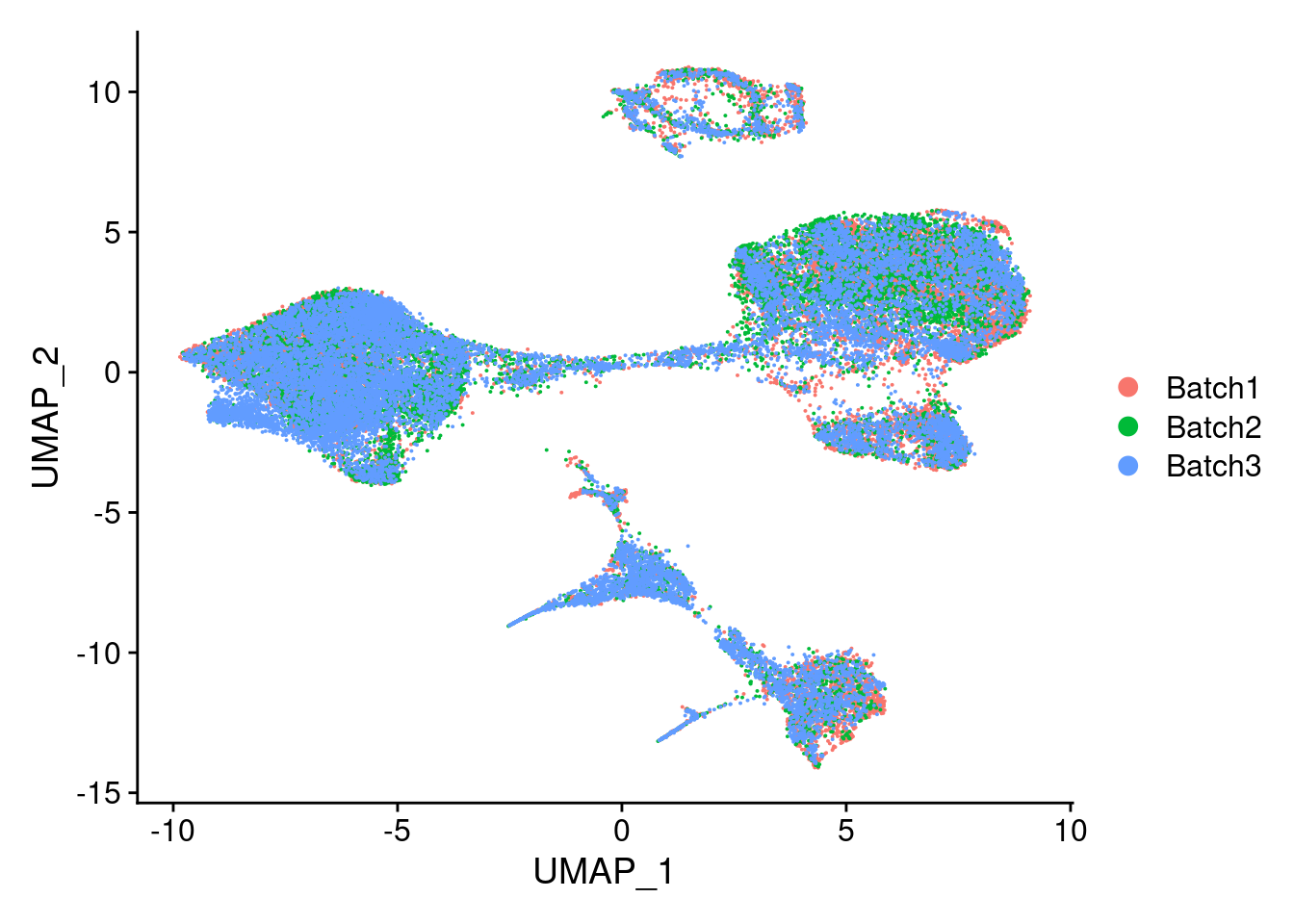
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "individual")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
xlim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_1']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_1']))
ylim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_2']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_2']))
for (i in individuals)
{
print(DimPlot(merged, reduction = "umap",
cells = WhichCells(merged, expression = individual == i)) +
xlim(xlim) + ylim(ylim) + ggtitle(i))
}
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |

| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
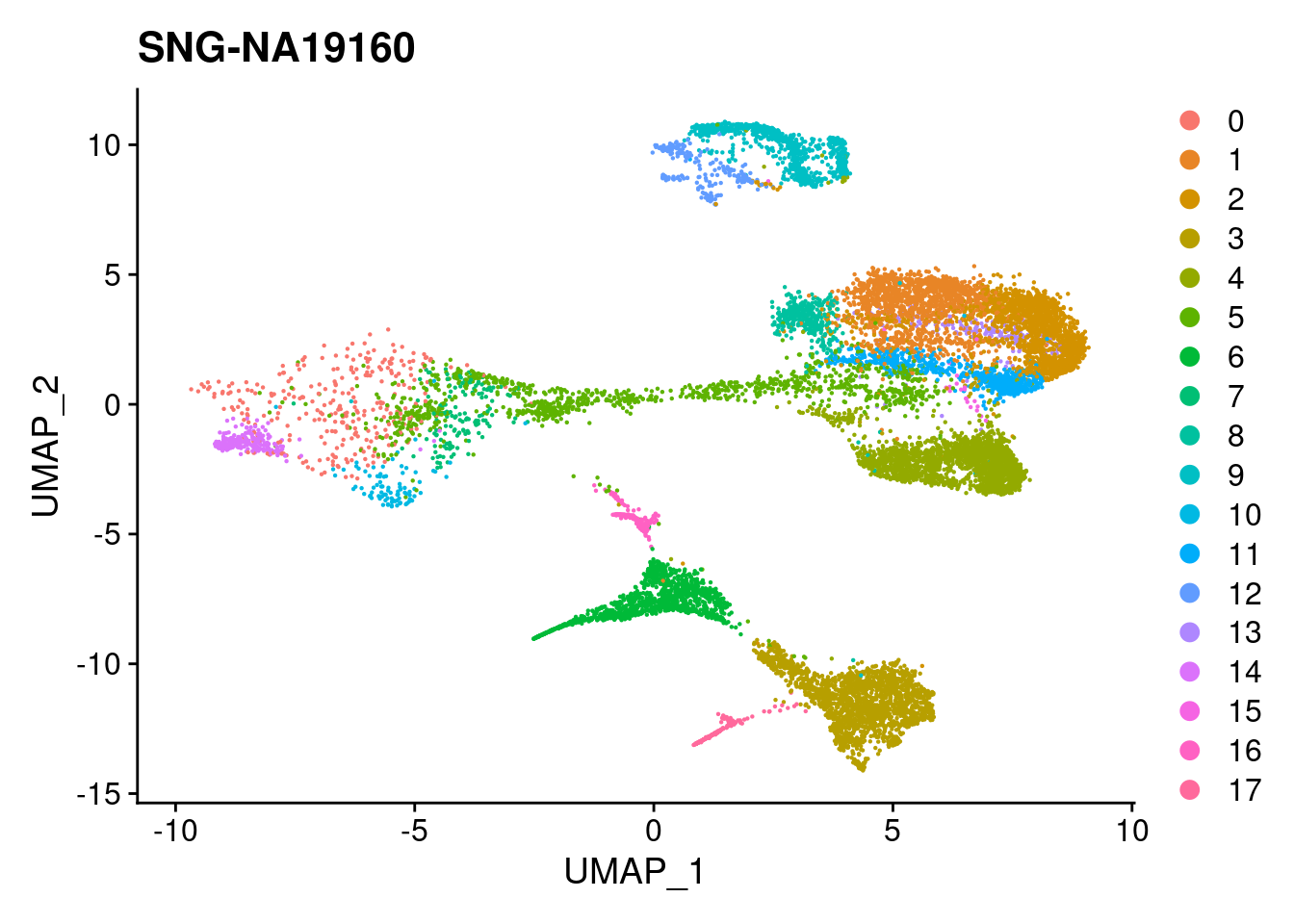
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#exploring similarity in the number of cells per individual between batches
merged.Batch1<- (subset(merged, Batch == "Batch1"))
b1t<- table(merged.Batch1$SCT_snn_res.0.5, merged.Batch1$individual)
remove("merged.Batch1")
b1tcolsums<- colSums(b1t)
percb1t<- b1t/b1tcolsums
merged.Batch2<- (subset(merged, Batch == "Batch2"))
b2t<- table(merged.Batch2$SCT_snn_res.0.5, merged.Batch2$individual)
remove("merged.Batch2")
b2tcolsums<- colSums(b2t)
percb2t<- b2t/b2tcolsums
merged.Batch3<- (subset(merged, Batch == "Batch3"))
b3t<- table(merged.Batch3$SCT_snn_res.0.5, merged.Batch3$individual)
remove("merged.Batch3")
b3tcolsums<- colSums(b3t)
percb3t<- b3t/b3tcolsumscols1<- c("Batch1_18511","Batch1_18858","Batch1_19160", "Batch2_18511", "Batch2_18858","Batch2_19160",
"Batch3_18511","Batch3_18858", "Batch3_19160")
cols2<- c("Batch1_18511", "Batch2_18511", "Batch3_18511","Batch1_18858", "Batch2_18858", "Batch3_18858","Batch1_19160", "Batch2_19160", "Batch3_19160")
fullpercs<- as.data.frame(cbind(percb1t[,1:3], percb2t,percb3t))
colnames(fullpercs)<-cols1
fullpercs<- cbind(fullpercs$Batch1_18511, fullpercs$Batch2_18511, fullpercs$Batch3_18511,
fullpercs$Batch1_18858, fullpercs$Batch2_18858, fullpercs$Batch3_18858,
fullpercs$Batch1_19160, fullpercs$Batch2_19160, fullpercs$Batch3_19160)
colnames(fullpercs)<-cols2
fullpercs_cor<- round(cor(fullpercs),2)
fullpercs_melt<- melt(fullpercs_cor)
ggplot(data= fullpercs_melt, aes(x=Var1, y=Var2, fill=value)) +
geom_tile(color="white") +
scale_fill_gradient2(low="blue", high="red", mid="white", midpoint = 0, limit= c(-1,1), space= "Lab", name="Pearson\nCorrelation") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 30, hjust=1))+
ggtitle("Pairwise Pearson Correlation of the percent of cells from \neach cell line assigned to each Seurat Cluster\ncluster res. 0.5")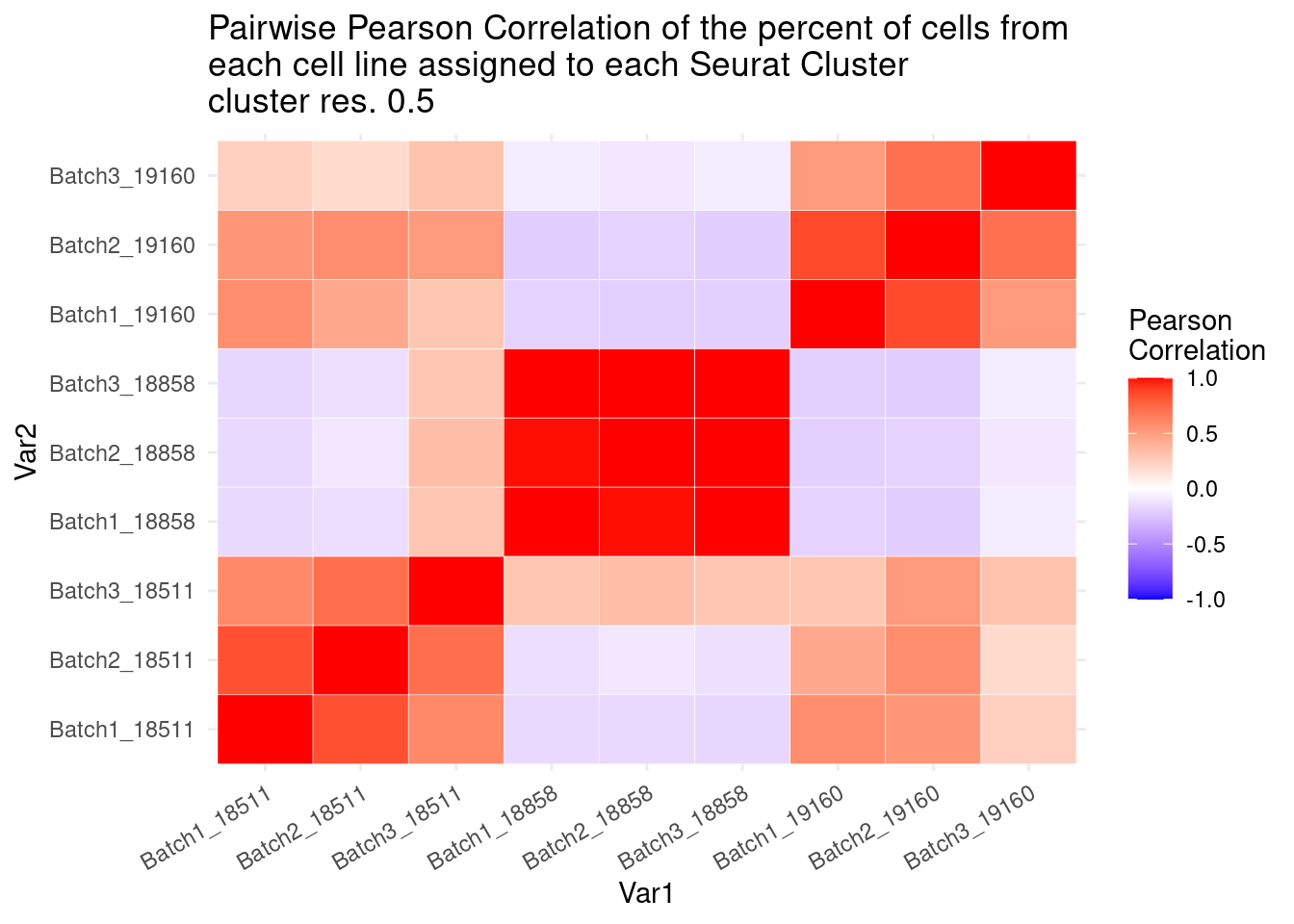
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#now clustering individual_Batch samples with hierarchical clustering/they will get reordered based on similarity
beauty<- colorRampPalette(brewer.pal(9,"Purples"))(200)
rownames(fullpercs)<- c(0:(nrow(fullpercs)-1))
heatmap(as.matrix(fullpercs), scale="none", col=beauty, cexCol = .7, cexRow=.6)
text(1:ncol(fullpercs),labels=names(fullpercs),srt=30)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#generate a heatmap of the raw proportion of cells from each individual_batch in each seurat cluster. dendrograms based on similarity of the vectors. should be colored by the value(proportion), but some of the cluster/sample values to seem to match with the color#reassign idents
Idents(merged)<- 'SCT_snn_res.0.1'DimPlot(merged, reduction = "umap")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "Batch")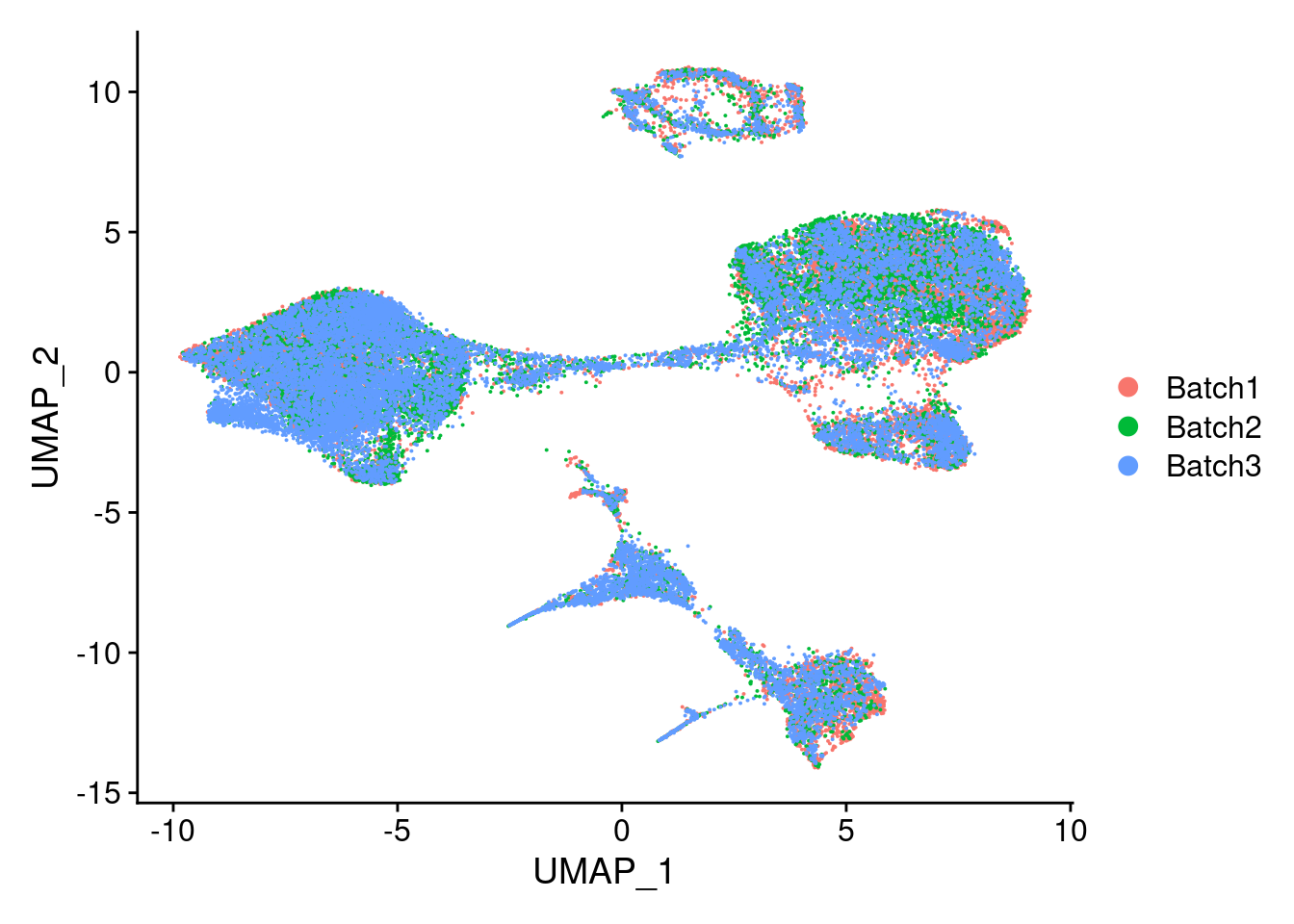
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "individual")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
xlim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_1']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_1']))
ylim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_2']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_2']))
for (i in individuals)
{
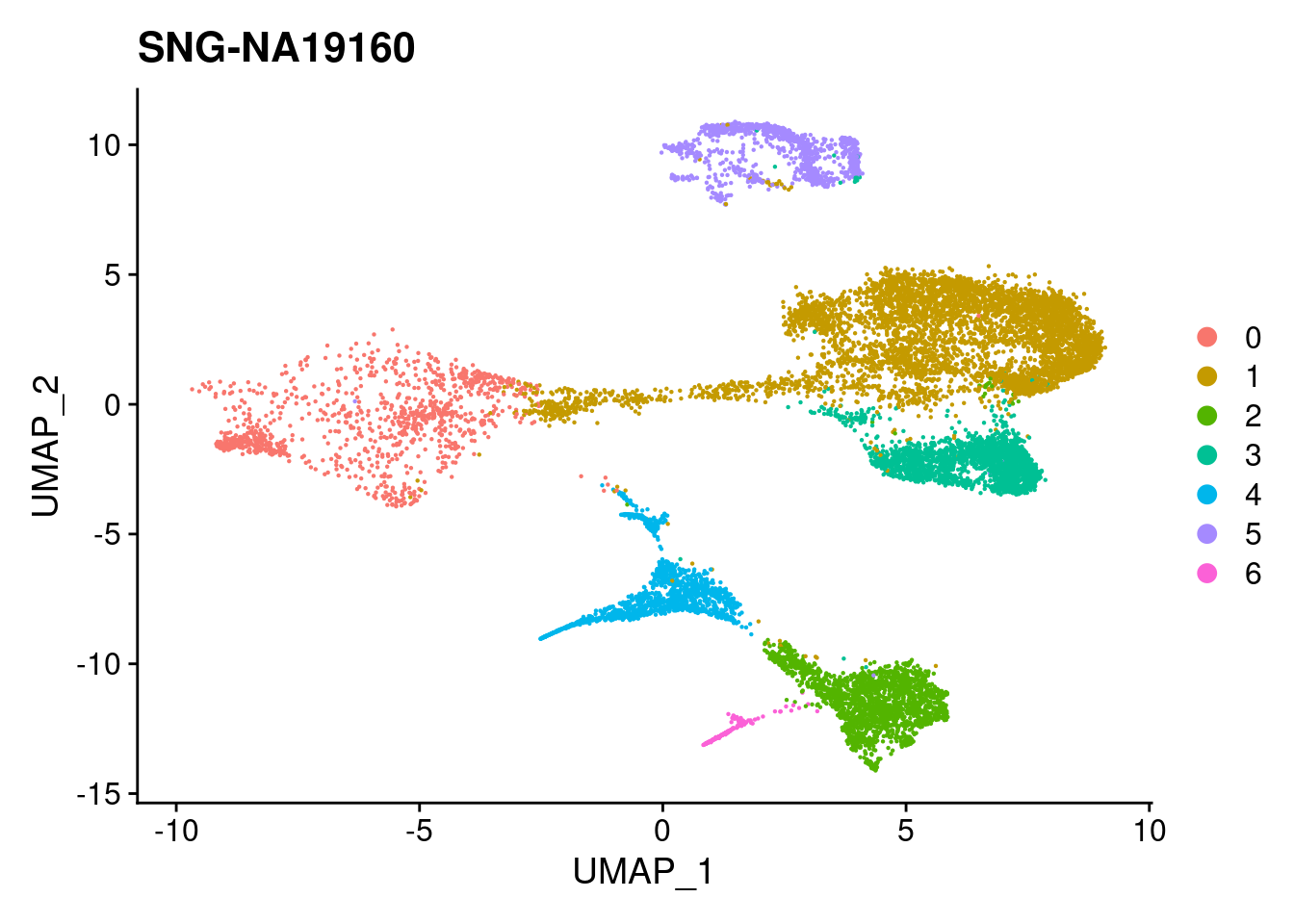
print(DimPlot(merged, reduction = "umap",
cells = WhichCells(merged, expression = individual == i)) +
xlim(xlim) + ylim(ylim) + ggtitle(i))
}
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
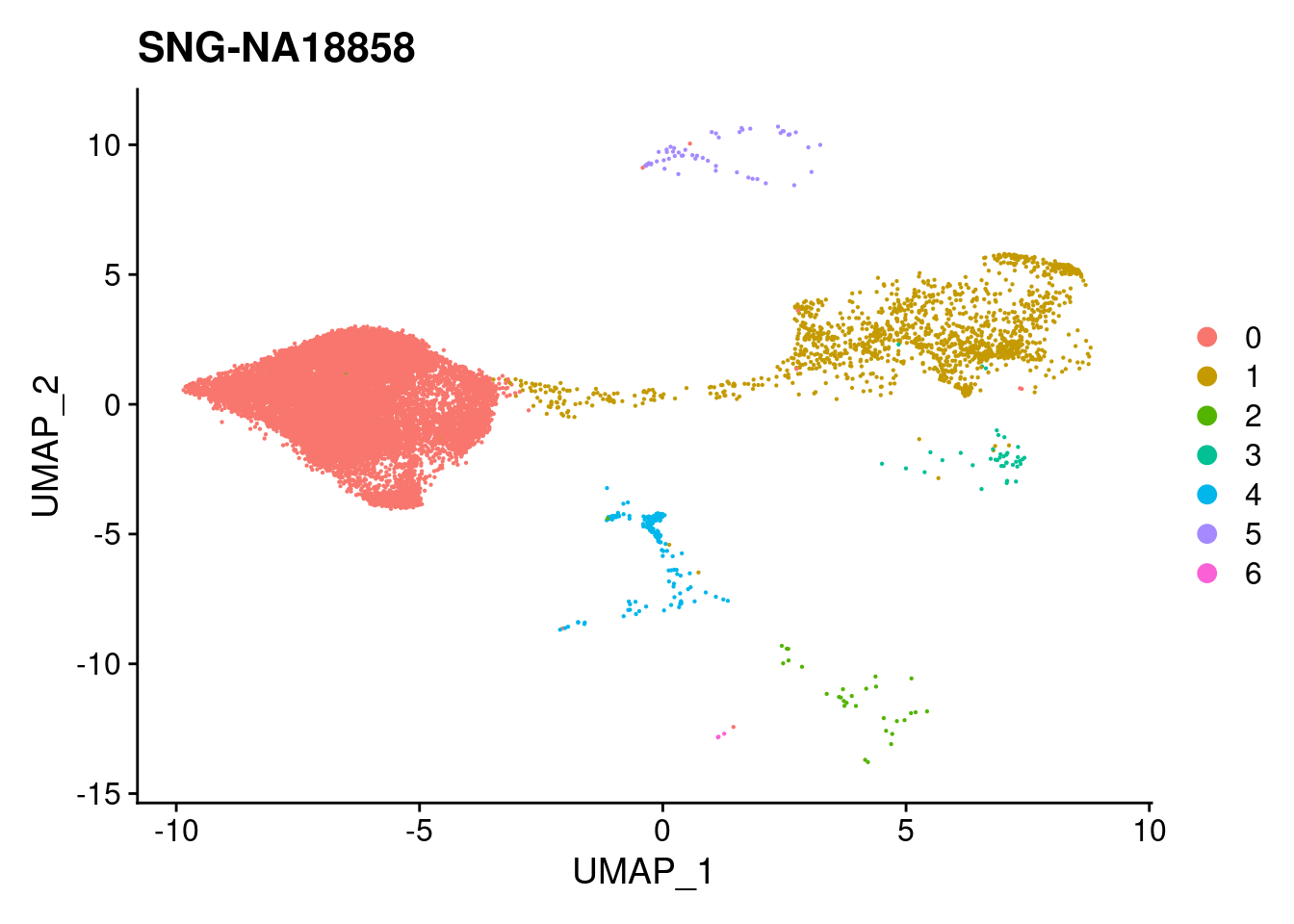
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#exploring similarity in the number of cells per individual between batches
merged.Batch1<- (subset(merged, Batch == "Batch1"))
b1t<- table(merged.Batch1$SCT_snn_res.0.1, merged.Batch1$individual)
remove("merged.Batch1")
b1tcolsums<- colSums(b1t)
percb1t<- b1t/b1tcolsums
merged.Batch2<- (subset(merged, Batch == "Batch2"))
b2t<- table(merged.Batch2$SCT_snn_res.0.1, merged.Batch2$individual)
remove("merged.Batch2")
b2tcolsums<- colSums(b2t)
percb2t<- b2t/b2tcolsums
merged.Batch3<- (subset(merged, Batch == "Batch3"))
b3t<- table(merged.Batch3$SCT_snn_res.0.1, merged.Batch3$individual)
remove("merged.Batch3")
b3tcolsums<- colSums(b3t)
percb3t<- b3t/b3tcolsumscols1<- c("Batch1_18511","Batch1_18858","Batch1_19160", "Batch2_18511", "Batch2_18858","Batch2_19160",
"Batch3_18511","Batch3_18858", "Batch3_19160")
cols2<- c("Batch1_18511", "Batch2_18511", "Batch3_18511","Batch1_18858", "Batch2_18858", "Batch3_18858","Batch1_19160", "Batch2_19160", "Batch3_19160")
fullpercs<- as.data.frame(cbind(percb1t[,1:3], percb2t,percb3t))
colnames(fullpercs)<-cols1
fullpercs<- cbind(fullpercs$Batch1_18511, fullpercs$Batch2_18511, fullpercs$Batch3_18511,
fullpercs$Batch1_18858, fullpercs$Batch2_18858, fullpercs$Batch3_18858,
fullpercs$Batch1_19160, fullpercs$Batch2_19160, fullpercs$Batch3_19160)
colnames(fullpercs)<-cols2
fullpercs_cor<- round(cor(fullpercs),2)
fullpercs_melt<- melt(fullpercs_cor)
ggplot(data= fullpercs_melt, aes(x=Var1, y=Var2, fill=value)) +
geom_tile(color="white") +
scale_fill_gradient2(low="blue", high="red", mid="white", midpoint = 0, limit= c(-1,1), space= "Lab", name="Pearson\nCorrelation") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 30, hjust=1))+
ggtitle("Pairwise Pearson Correlation of the percent of cells from \neach cell line assigned to each Seurat Cluster\ncluster res. 0.1")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#now clustering individual_Batch samples with hierarchical clustering/they will get reordered based on similarity
beauty<- colorRampPalette(brewer.pal(9,"Purples"))(200)
#fullnonorm<- as.data.frame(cbind(b1t[,1:3], b2t,b3t))
#colnames(fullnonorm)<-cols1
#heatmap((as.matrix(fullnonorm)), scale="column", col= beauty)
rownames(fullpercs)<- c(0:(nrow(fullpercs)-1))
heatmap(as.matrix(fullpercs), scale="none", col=beauty, cexCol = .7, cexRow=.6)
text(1:ncol(fullpercs),labels=names(fullpercs),srt=30)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#generate a heatmap of the raw proportion of cells from each individual_batch in each seurat cluster. dendrograms based on similarity of the vectors. should be colored by the value(proportion), but some of the cluster/sample values to seem to match with the color#reassign idents
Idents(merged)<- 'SCT_snn_res.0.8'DimPlot(merged, reduction = "umap")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "Batch")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
DimPlot(merged, reduction = "umap", group.by = "individual")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
xlim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_1']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_1']))
ylim <- c(min(merged@reductions$umap@cell.embeddings[,'UMAP_2']),
max(merged@reductions$umap@cell.embeddings[,'UMAP_2']))
for (i in individuals)
{
print(DimPlot(merged, reduction = "umap",
cells = WhichCells(merged, expression = individual == i)) +
xlim(xlim) + ylim(ylim) + ggtitle(i))
}
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |

| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |

| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#exploring similarity in the number of cells per individual between batches
merged.Batch1<- (subset(merged, Batch == "Batch1"))
b1t<- table(merged.Batch1$SCT_snn_res.0.8, merged.Batch1$individual)
remove("merged.Batch1")
b1tcolsums<- colSums(b1t)
percb1t<- b1t/b1tcolsums
merged.Batch2<- (subset(merged, Batch == "Batch2"))
b2t<- table(merged.Batch2$SCT_snn_res.0.8, merged.Batch2$individual)
remove("merged.Batch2")
b2tcolsums<- colSums(b2t)
percb2t<- b2t/b2tcolsums
merged.Batch3<- (subset(merged, Batch == "Batch3"))
b3t<- table(merged.Batch3$SCT_snn_res.0.8, merged.Batch3$individual)
remove("merged.Batch3")
b3tcolsums<- colSums(b3t)
percb3t<- b3t/b3tcolsumscols1<- c("Batch1_18511","Batch1_18858","Batch1_19160", "Batch2_18511", "Batch2_18858","Batch2_19160",
"Batch3_18511","Batch3_18858", "Batch3_19160")
cols2<- c("Batch1_18511", "Batch2_18511", "Batch3_18511","Batch1_18858", "Batch2_18858", "Batch3_18858","Batch1_19160", "Batch2_19160", "Batch3_19160")
fullpercs<- as.data.frame(cbind(percb1t[,1:3], percb2t,percb3t))
colnames(fullpercs)<-cols1
fullpercs<- cbind(fullpercs$Batch1_18511, fullpercs$Batch2_18511, fullpercs$Batch3_18511,
fullpercs$Batch1_18858, fullpercs$Batch2_18858, fullpercs$Batch3_18858,
fullpercs$Batch1_19160, fullpercs$Batch2_19160, fullpercs$Batch3_19160)
colnames(fullpercs)<-cols2
fullpercs_cor<- round(cor(fullpercs),2)
fullpercs_melt<- melt(fullpercs_cor)
ggplot(data= fullpercs_melt, aes(x=Var1, y=Var2, fill=value)) +
geom_tile(color="white") +
scale_fill_gradient2(low="blue", high="red", mid="white", midpoint = 0, limit= c(-1,1), space= "Lab", name="Pearson\nCorrelation") +
theme_minimal() +
ggtitle("Pairwise Pearson Correlation of the percent of cells from \neach cell line assigned to each Seurat Cluster\ncluster res. 0.8")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#now clustering individual_Batch samples with hierarchical clustering/they will get reordered based on similarity
beauty<- colorRampPalette(brewer.pal(9,"Purples"))(200)
rownames(fullpercs)<- c(0:(nrow(fullpercs)-1))
heatmap(as.matrix(fullpercs), scale="none", col=beauty, cexCol = .7, cexRow=.6)
text(1:ncol(fullpercs),labels=names(fullpercs),srt=30)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
#generate a heatmap of the raw proportion of cells from each individual_batch in each seurat cluster. dendrograms based on similarity of the vectors. should be colored by the value(proportion), but some of the cluster/sample values to seem to match with the colorVlnPlot(merged, features= "percent.mt", group.by = "SCT_snn_res.1", pt.size = 0)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
merged[["percent.rps"]]<- PercentageFeatureSet(merged, pattern = "^RPS")
merged[["percent.rpl"]]<- PercentageFeatureSet(merged, pattern = "^RPL")
merged[["percent.rp"]]<- merged[["percent.rps"]]+merged[["percent.rpl"]]
VlnPlot(merged, features= "percent.rp", group.by = "SCT_snn_res.1", pt.size=0)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
FeaturePlot(merged, features = "nFeature_RNA")
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
head(merged)An object of class Seurat
2 features across 42488 samples within 2 assays
Active assay: SCT (1 features, 1 variable features)
1 other assay present: RNA
3 dimensional reductions calculated: pca, harmony, umapVlnPlot(merged, features= "nFeature_RNA", group.by = "SCT_snn_res.1", pt.size=0)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
FeaturePlot(merged, features = c("POU5F1", "SOX17", "HAND1", "PAX6"), pt.size = 0.2, ncol=2, combine=T)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
FeaturePlot(merged, features = c("FGB", "ECSCR", "NEUROD1", "SOX10"), pt.size = 0.2, ncol=2)
| Version | Author | Date |
|---|---|---|
| 421a225 | KLRhodes | 2020-08-10 |
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reshape2_1.4.4 RColorBrewer_1.1-2 here_0.1-11 DataCombine_0.2.21
[5] ggplot2_3.3.2 harmony_1.0 Rcpp_1.0.5 Seurat_3.2.0
[9] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rtsne_0.15 colorspace_1.4-1 deldir_0.1-28
[4] ellipsis_0.3.1 ggridges_0.5.2 rprojroot_1.3-2
[7] fs_1.4.2 spatstat.data_1.4-3 farver_2.0.3
[10] leiden_0.3.3 listenv_0.8.0 npsurv_0.4-0
[13] ggrepel_0.8.2 RSpectra_0.16-0 codetools_0.2-16
[16] splines_3.6.1 lsei_1.2-0 knitr_1.29
[19] polyclip_1.10-0 jsonlite_1.7.0 ica_1.0-2
[22] cluster_2.1.0 png_0.1-7 uwot_0.1.8
[25] shiny_1.5.0 sctransform_0.2.1 compiler_3.6.1
[28] httr_1.4.2 backports_1.1.8 Matrix_1.2-18
[31] fastmap_1.0.1 lazyeval_0.2.2 later_1.1.0.1
[34] htmltools_0.5.0 tools_3.6.1 rsvd_1.0.3
[37] igraph_1.2.5 gtable_0.3.0 glue_1.4.1
[40] RANN_2.6.1 dplyr_1.0.0 rappdirs_0.3.1
[43] spatstat_1.64-1 vctrs_0.3.2 gdata_2.18.0
[46] ape_5.3 nlme_3.1-140 lmtest_0.9-37
[49] xfun_0.16 stringr_1.4.0 globals_0.12.5
[52] mime_0.9 miniUI_0.1.1.1 lifecycle_0.2.0
[55] irlba_2.3.3 gtools_3.8.2 goftest_1.2-2
[58] future_1.18.0 MASS_7.3-51.4 zoo_1.8-8
[61] scales_1.1.1 promises_1.1.1 spatstat.utils_1.17-0
[64] parallel_3.6.1 yaml_2.2.1 reticulate_1.16
[67] pbapply_1.4-2 gridExtra_2.3 rpart_4.1-15
[70] stringi_1.4.6 caTools_1.18.0 rlang_0.4.7
[73] pkgconfig_2.0.3 bitops_1.0-6 evaluate_0.14
[76] lattice_0.20-38 ROCR_1.0-7 purrr_0.3.4
[79] tensor_1.5 labeling_0.3 patchwork_1.0.1
[82] htmlwidgets_1.5.1 cowplot_1.0.0 tidyselect_1.1.0
[85] RcppAnnoy_0.0.16 plyr_1.8.6 magrittr_1.5
[88] R6_2.4.1 gplots_3.0.4 generics_0.0.2
[91] withr_2.2.0 pillar_1.4.6 whisker_0.4
[94] mgcv_1.8-28 fitdistrplus_1.0-14 survival_3.2-3
[97] abind_1.4-5 tibble_3.0.3 future.apply_1.6.0
[100] crayon_1.3.4 KernSmooth_2.23-15 plotly_4.9.2.1
[103] rmarkdown_2.3 grid_3.6.1 data.table_1.13.0
[106] git2r_0.26.1 digest_0.6.25 xtable_1.8-4
[109] tidyr_1.1.0 httpuv_1.5.4 munsell_0.5.0
[112] viridisLite_0.3.0