Genome quality assessment
Maeva Techer
2025-06-01
Last updated: 2025-06-01
Checks: 6 1
Knit directory:
locust-comparative-genomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221025) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4e391c3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: code/.DS_Store
Ignored: code/scripts/.DS_Store
Ignored: code/scripts/pal2nal.v14/.DS_Store
Ignored: data/.DS_Store
Ignored: data/DEG_results/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/americana/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cancellata/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cancellata/Thorax/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cubense/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/davidO/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/gregaria/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/nitens/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/piceifrons/.DS_Store
Ignored: data/DEG_results/RNAi/.DS_Store
Ignored: data/DEG_results/RNAi/All/.DS_Store
Ignored: data/DEG_results/RNAi/All_GFP/.DS_Store
Ignored: data/DEG_results/RNAi/All_control/.DS_Store
Ignored: data/DEG_results/RNAi/All_no_rRNA/.DS_Store
Ignored: data/DEG_results/RNAi/Head/.DS_Store
Ignored: data/DEG_results/RNAi/Head_control/.DS_Store
Ignored: data/DEG_results/RNAi/Head_no_rRNA/.DS_Store
Ignored: data/DEG_results/RNAi/Thorax/.DS_Store
Ignored: data/DEG_results/RNAi/Thorax_no_rRNA/.DS_Store
Ignored: data/DEG_results/gregaria/
Ignored: data/DEG_results/single_cell/.DS_Store
Ignored: data/WGCNA/.DS_Store
Ignored: data/WGCNA/input/.DS_Store
Ignored: data/WGCNA/input/Bulk_RNAseq/.DS_Store
Ignored: data/WGCNA/output/.DS_Store
Ignored: data/WGCNA/output/Bulk_RNAseq/.DS_Store
Ignored: data/behavioral_data/.DS_Store
Ignored: data/behavioral_data/Raw_data/.DS_Store
Ignored: data/list/.DS_Store
Ignored: data/list/Bulk_RNAseq/.DS_Store
Ignored: data/list/GO_Annotations/.DS_Store
Ignored: data/list/excluded_loci/.DS_Store
Ignored: data/orthofinder/.DS_Store
Ignored: data/orthofinder/Polyneoptera/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_iqtree/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_withDaust/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_withDaust/Orthogroups/.DS_Store
Ignored: data/orthofinder/Schistocerca/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/Orthogroups/.DS_Store
Ignored: data/overlap/.DS_Store
Ignored: data/overlap/Bulk_RNAseq/.DS_Store
Ignored: data/overlap/Bulk_RNAseq/cancellata/
Ignored: data/pathway_enrichment/.DS_Store
Ignored: data/pathway_enrichment/custom_sgregaria_orgdb/.DS_Store
Ignored: data/readcounts/.DS_Store
Ignored: data/readcounts/Bulk_RNAseq/.DS_Store
Ignored: data/readcounts/RNAi/.DS_Store
Untracked files:
Untracked: data/RefSeq/
Unstaged changes:
Modified: analysis/2_genome_quality.Rmd
Modified: analysis/2_orthologs-prediction.Rmd
Modified: analysis/2_synteny-graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/2_genome_quality.Rmd) and
HTML (docs/2_genome_quality.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 4e391c3 | Maeva TECHER | 2025-05-30 | add new analysis orthology, synteny |
| html | 4e391c3 | Maeva TECHER | 2025-05-30 | add new analysis orthology, synteny |
1. BUSCO assessment
To test whether core genes found in Insecta are present in the genomes that we will be analyzing, we run BUSCO to assess our Polyneoptera genome quality as recommended.
We ran in protein mode using the protein files
XXX_proteins.faa downloaded from NCBI and stored in the
folder 1_RawData using the following custom script for
GRACE busco_assess.sh:
#!/bin/bash
#SBATCH --job-name=busco_all_proteins
#SBATCH --time=3-00:00:00
#SBATCH --cpus-per-task=48
#SBATCH --mem=250G
#SBATCH --ntasks=1
#SBATCH --output=busco_allproteins.%j.out
#SBATCH --error=busco_allproteins.%j.err
# Load required modules
module purge
ml GCC/12.2.0 OpenMPI/4.1.4 BUSCO/5.7.1 WebProxy
# Set environment variable for NumExpr to avoid "safe limit of 8"
export NUMEXPR_MAX_THREADS=$SLURM_CPUS_PER_TASK
# Set lineage and mode
LINEAGE=insecta_odb10
MODE=protein
# Set directories
INPUT_DIR=$PWD
OUTPUT_DIR=$PWD/busco_results
mkdir -p $OUTPUT_DIR
# Loop through all *_proteins.faa files
for FILE in ${INPUT_DIR}/*_proteins.faa; do
BASENAME=$(basename "$FILE" _proteins.faa)
echo "🔎 Starting BUSCO for: $BASENAME"
busco -i "$FILE" \
-o "${BASENAME}_busco" \
-l "$LINEAGE" \
-m "$MODE" \
-c $SLURM_CPUS_PER_TASK \
--out_path "$OUTPUT_DIR" \
--force
echo "✅ BUSCO run complete for $FILE"
done
echo "🎉 All BUSCO protein assessments completed!"Just launch the script with:
#chmod 777 busco_assess.sh
sbatch busco_assess.shFor 13 genomes, and using 48 CPUs, the run finished in ~5 hours and
only used ~9 Gb of memory. We extracted the results from each species
folder
busco_results/{SPECIES}_busco/short_summary.specific.insecta_odb10.{SPECIES}_busco.txt.
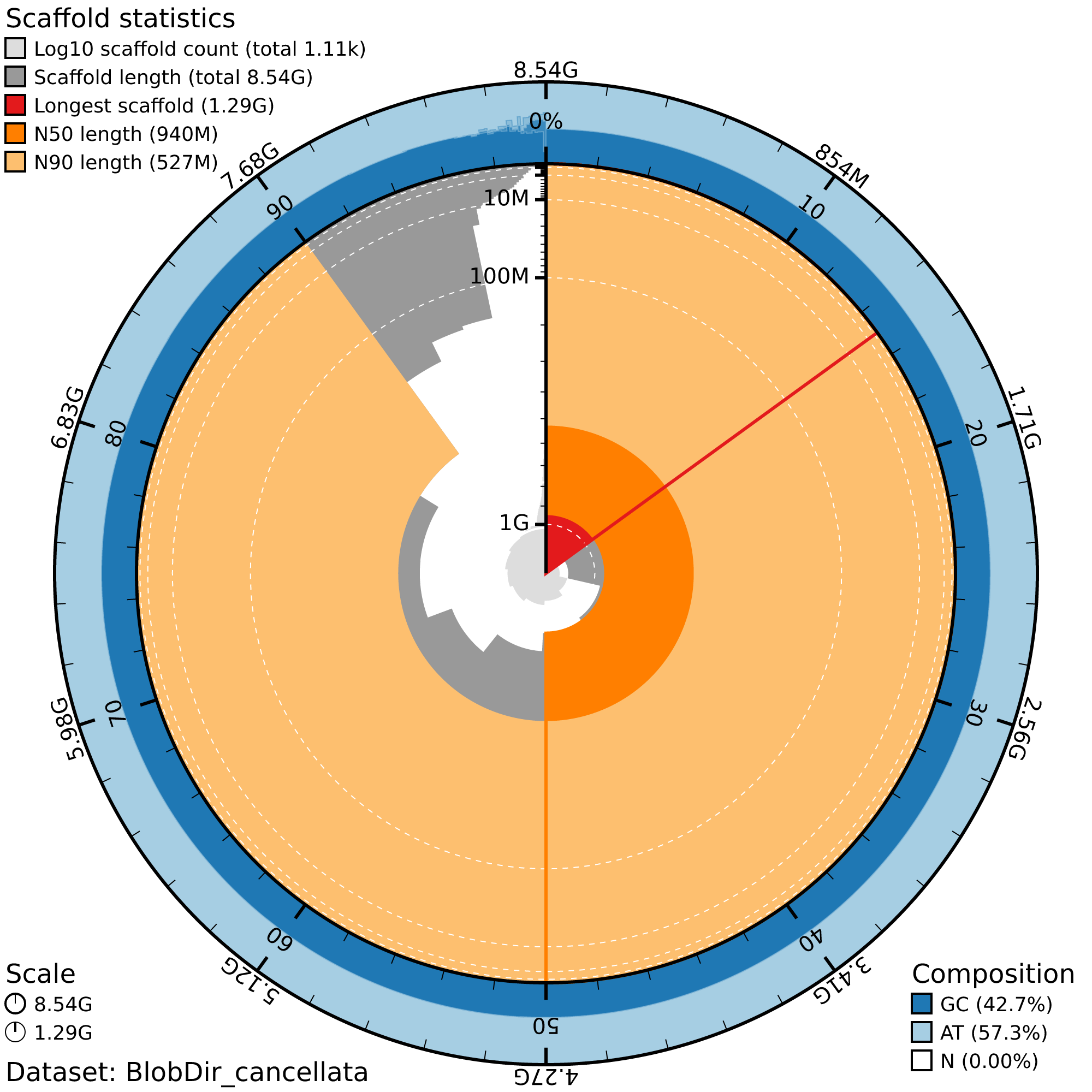
2. Genome quality with Blobtools
We make snail plot with blobtools using the following commands.
Unfortunately, because blobtools does not support indexing
with .csi and our contigs are over 512 Mbp, we only plotted the GC, and
length of our scaffolds and not the coverage.
First we
srun --ntasks 1 --cpus-per-task 24 --mem 50G --time 05:00:00 --pty bash
ml WebProxy
# Check that the singularity container is properly installed
singularity exec /sw/hprc/sw/bio/containers/blobtoolkit/blobtoolkit_4.4.5.sif blobtools --help
# Create a BlobDir for each species
singularity exec /sw/hprc/sw/bio/containers/blobtoolkit/blobtoolkit_4.4.5.sif blobtools create --fasta /scratch/group/songlab/maeva/psmc-schistocerca/reference/cancellata.fasta BlobDir_cancellata
# Add mapping coverage using bwa mapping we
# Does not plot on the snail plot
#singularity exec /sw/hprc/sw/bio/containers/blobtoolkit/blobtoolkit_4.4.5.sif blobtools add --cov ../results/bam/americana.sorted.bam BlobDir_americana
# Make a snail plot
singularity exec /sw/hprc/sw/bio/containers/blobtoolkit/blobtoolkit_4.4.5.sif blobtools view --view snail --plot BlobDir_cancellataExample of snailplot obtained for S. cancellata
sessionInfo()R version 4.4.2 (2024-10-31)
Platform: aarch64-apple-darwin20
Running under: macOS Sequoia 15.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Tokyo
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] vctrs_0.6.5 cli_3.6.5 knitr_1.49 rlang_1.1.6
[5] xfun_0.51 stringi_1.8.4 promises_1.3.2 jsonlite_1.9.1
[9] workflowr_1.7.1 glue_1.8.0 rprojroot_2.0.4 git2r_0.35.0
[13] htmltools_0.5.8.1 httpuv_1.6.15 sass_0.4.9 rmarkdown_2.29
[17] evaluate_1.0.3 jquerylib_0.1.4 tibble_3.2.1 fastmap_1.2.0
[21] yaml_2.3.10 lifecycle_1.0.4 whisker_0.4.1 stringr_1.5.1
[25] compiler_4.4.2 fs_1.6.5 Rcpp_1.0.14 pkgconfig_2.0.3
[29] rstudioapi_0.17.1 later_1.4.1 digest_0.6.37 R6_2.6.1
[33] pillar_1.10.2 magrittr_2.0.3 bslib_0.9.0 tools_4.4.2
[37] cachem_1.1.0