Gene set testing for Illumina HumanMethylation Arrays
Jovana Maksimovic
7/2/2019
Last updated: 2020-04-17
Checks: 7 0
Knit directory: methyl-geneset-testing/
This reproducible R Markdown analysis was created with workflowr (version 1.6.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200302) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0fec41a. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/.job/
Ignored: code/old/
Ignored: data/
Ignored: output/FDR-analysis/
Ignored: output/compare-methods/
Ignored: output/random-cpg-sims/
Unstaged changes:
Deleted: analysis/nullAnalysis.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/fdrAnalysis.Rmd) and HTML (docs/fdrAnalysis.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 0fec41a | Jovana Maksimovic | 2020-04-17 | Completed repeat of FDR analysis using TCGA data |
| Rmd | f42dc82 | JovMaksimovic | 2020-04-14 | Updated all FDR analysis code. |
| Rmd | 1ea3283 | JovMaksimovic | 2020-04-14 | Updated null analysis with KIRC TCGA data |
library(here)
library(minfi)
library(paletteer)
library(limma)
library(reshape2)
library(missMethyl)
library(ggplot2)
library(glue)
library(tibble)
library(dplyr)
library(curatedTCGAData)
library(MultiAssayExperiment)
library(TCGAutils)
library(DMRcate)Warning: replacing previous import
'GenomicRanges::.__C__GenomicRanges_OR_GenomicRangesList' by
'rtracklayer::.__C__GenomicRanges_OR_GenomicRangesList' when loading 'BSgenome'Warning: replacing previous import 'minfi::getMeth' by 'bsseq::getMeth' when
loading 'DMRcate'library(patchwork)
source(here("code/utility.R"))Load data
We are using publicly available kidney clear cell carcinoma (KIRC) 450k data from The Cancer Genome Atlas (TCGA). We are using only the normal samples to look at false discovery rate control by various methylation gene set testing methods.
Download the data using the curatedTCGAData Bioconductor package and then extract the normal samples. The data seems to have already been normalised(?) so we will only remove poor probes (>1 NA), SNP, multi-mapping and sex-chromosome probes.
kirc <- curatedTCGAData(diseaseCode = "KIRC", assays = "Methylation_methyl450",
dry.run = FALSE)snapshotDate(): 2019-10-22see ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentationloading from cacherequire("rhdf5")see ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentationloading from cachesee ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentationloading from cachesee ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentationloading from cachesee ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentationloading from cacheharmonizing input:
removing 6576 sampleMap rows not in names(experiments)
removing 218 colData rownames not in sampleMap 'primary'kirc <- splitAssays(kirc, c("11")) # extract only the normal samples
exp <- experiments(kirc)[[1]]
meta <- colData(kirc)
betas <- as.matrix(assay(exp))
colnames(betas) <- substr(colnames(betas), 1, 12)
m <- match(colnames(betas), meta$patientID)
meta <- meta[m, ]
head(meta[, 1:5])DataFrame with 6 rows and 5 columns
patientID years_to_birth vital_status days_to_death
<character> <integer> <integer> <integer>
TCGA-A3-3357 TCGA-A3-3357 62 0 NA
TCGA-A3-3367 TCGA-A3-3367 72 0 NA
TCGA-A3-3370 TCGA-A3-3370 48 0 NA
TCGA-A3-3373 TCGA-A3-3373 54 0 NA
TCGA-A3-3376 TCGA-A3-3376 51 1 1696
TCGA-A3-3385 TCGA-A3-3385 46 0 NA
days_to_last_followup
<integer>
TCGA-A3-3357 2688
TCGA-A3-3367 2270
TCGA-A3-3370 2274
TCGA-A3-3373 1621
TCGA-A3-3376 NA
TCGA-A3-3385 1993betasNoNA <- betas[rowSums(is.na(betas)) == 0, ]
mds <- plotMDS(betasNoNA, gene.selection = "common", plot = FALSE)
dat <- tibble(x = mds$x, y = mds$y, gender = meta$gender)
ggplot(dat, aes(x = x, y = y, colour = gender)) +
geom_point() +
labs(x = "Principal Component 1", y = "Principal Component 2",
colour = "Gender")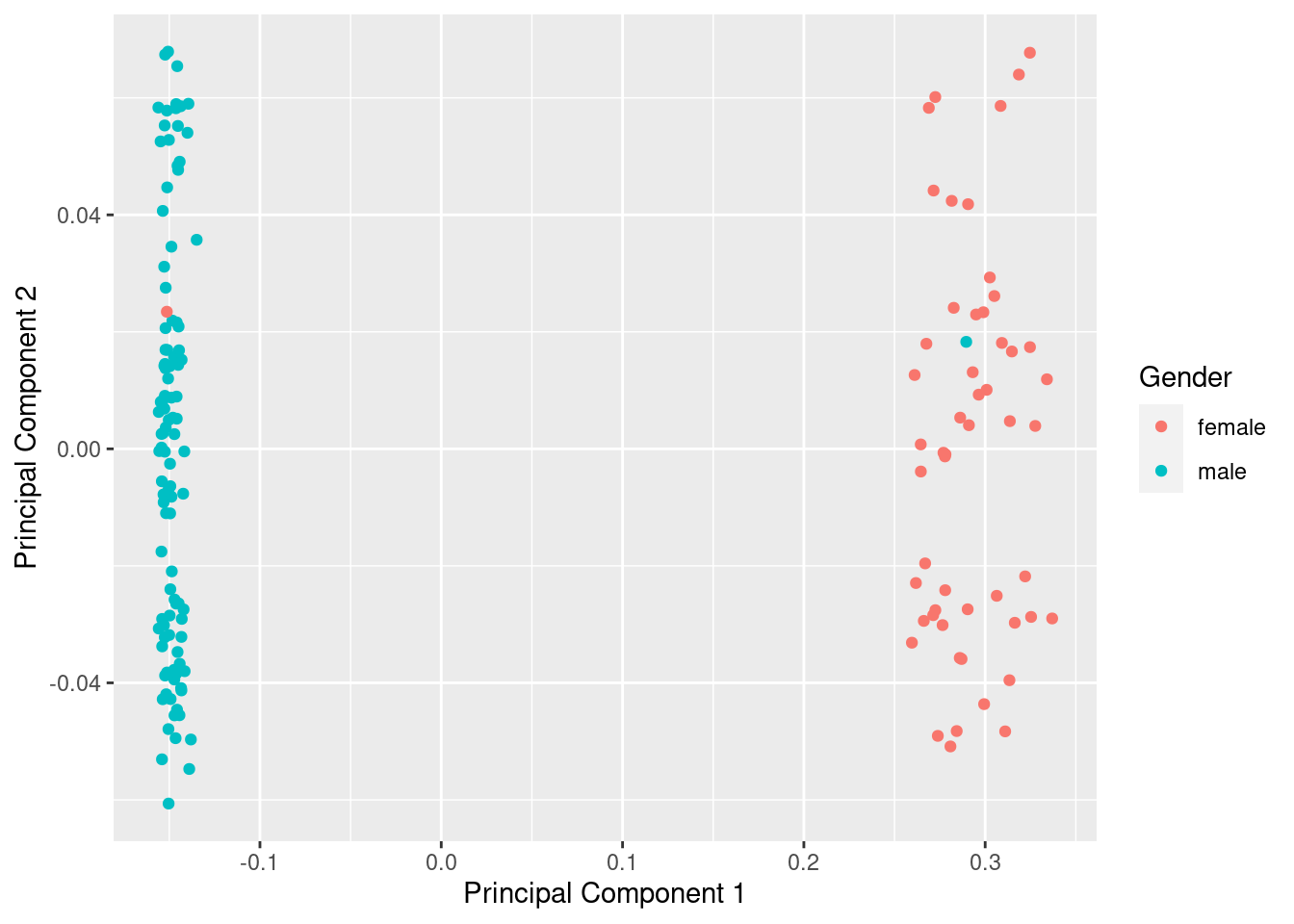
betasFlt <- rmSNPandCH(betasNoNA, rmXY = TRUE, rmcrosshyb = TRUE)
mds <- plotMDS(betasFlt, gene.selection = "common", plot = FALSE)
dat <- tibble(x = mds$x, y = mds$y, gender = meta$gender)
ggplot(dat, aes(x = x, y = y, colour = gender)) +
geom_point() +
labs(x = "Principal Component 1", y = "Principal Component 2",
colour = "Gender")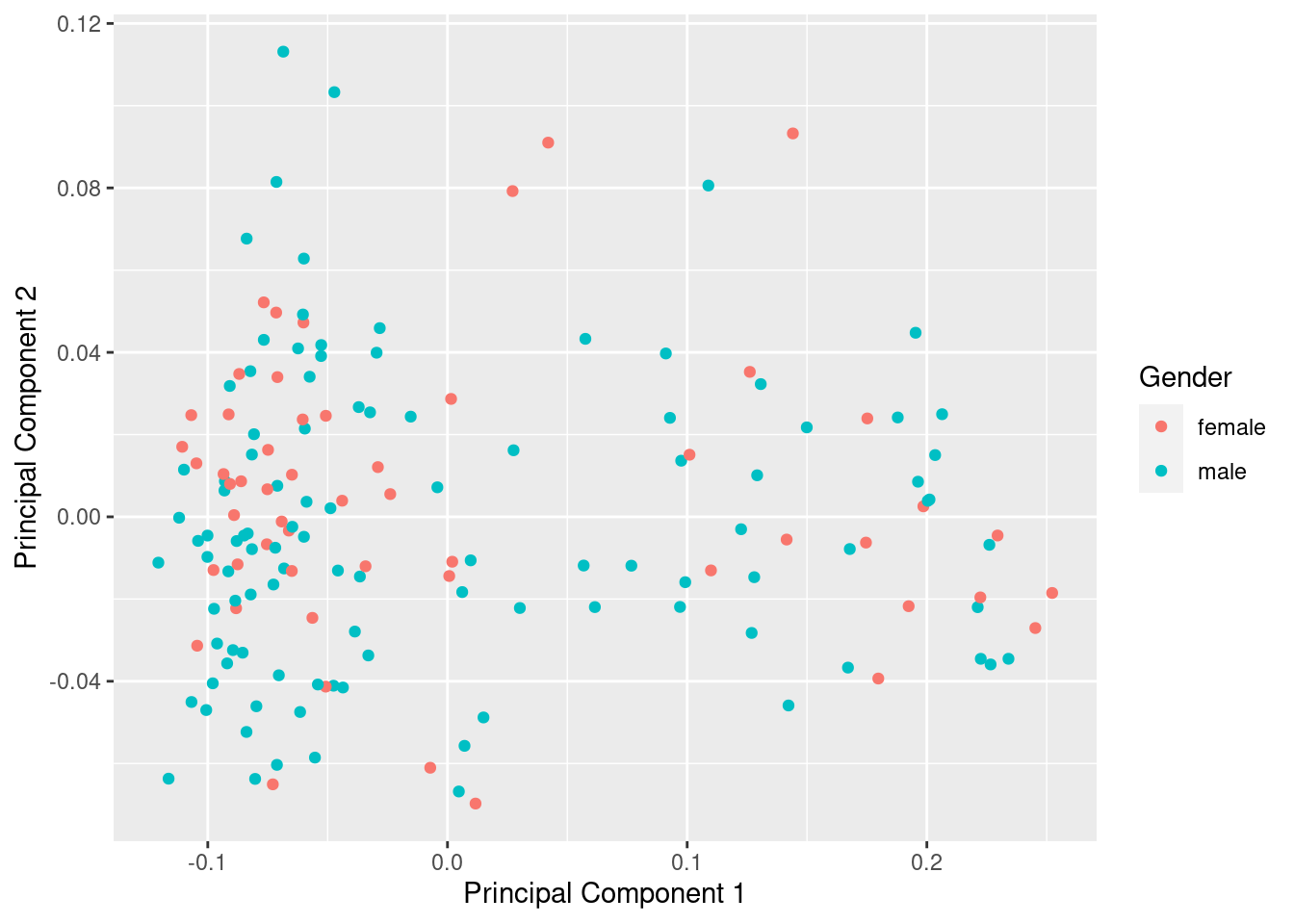
dat$age <- meta$years_to_birth
dat$race <- sub(" or", "\nor", meta$race)
dat$ethnicity <- sub(" or", "\nor", meta$ethnicity)
a <- ggplot(dat, aes(x = x, y = y, colour = age)) +
geom_point() +
labs(x = "Principal Component 1", y = "Principal Component 2",
colour = "Age") +
viridis::scale_color_viridis(direction = -1)
b <- ggplot(dat, aes(x = x, y = y, colour = race)) +
geom_point() +
labs(x = "Principal Component 1", y = "Principal Component 2",
colour = "Race") +
theme(legend.text = element_text(size = 7))
c <- ggplot(dat, aes(x = x, y = y, colour = ethnicity)) +
geom_point() +
labs(x = "Principal Component 1", y = "Principal Component 2",
colour = "Ethnicity") +
theme(legend.text = element_text(size = 7))
(b + c) / a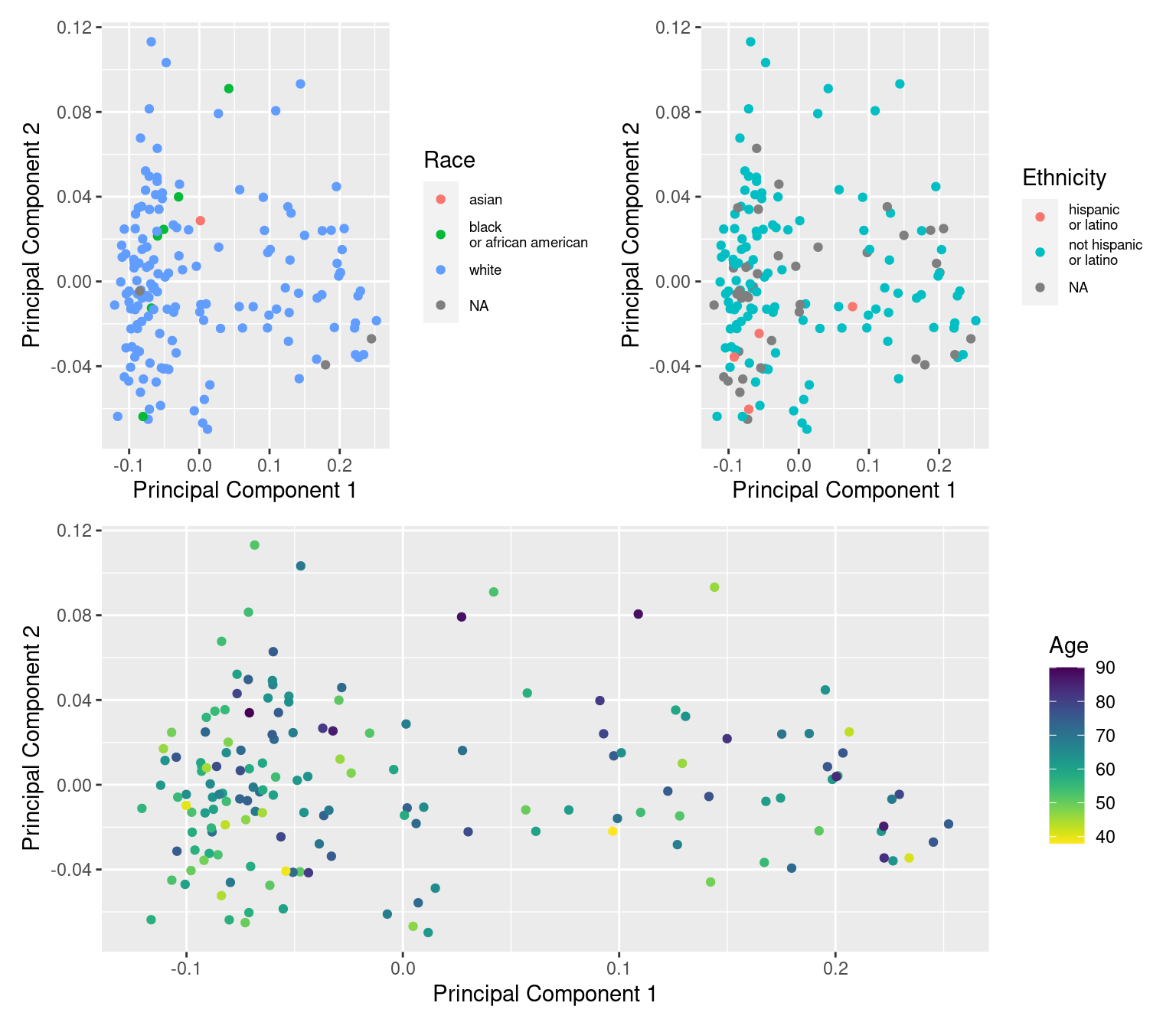
dat <- as_tibble(melt(betasFlt))
colnames(dat) <- c("cpg", "ID", "beta")
p <- ggplot(dat, aes(x = beta)) +
geom_line(aes(color = ID), stat="density", size=0.5, alpha=0.5,
show.legend = FALSE)
p + labs(x = "Beta value", y = "Density")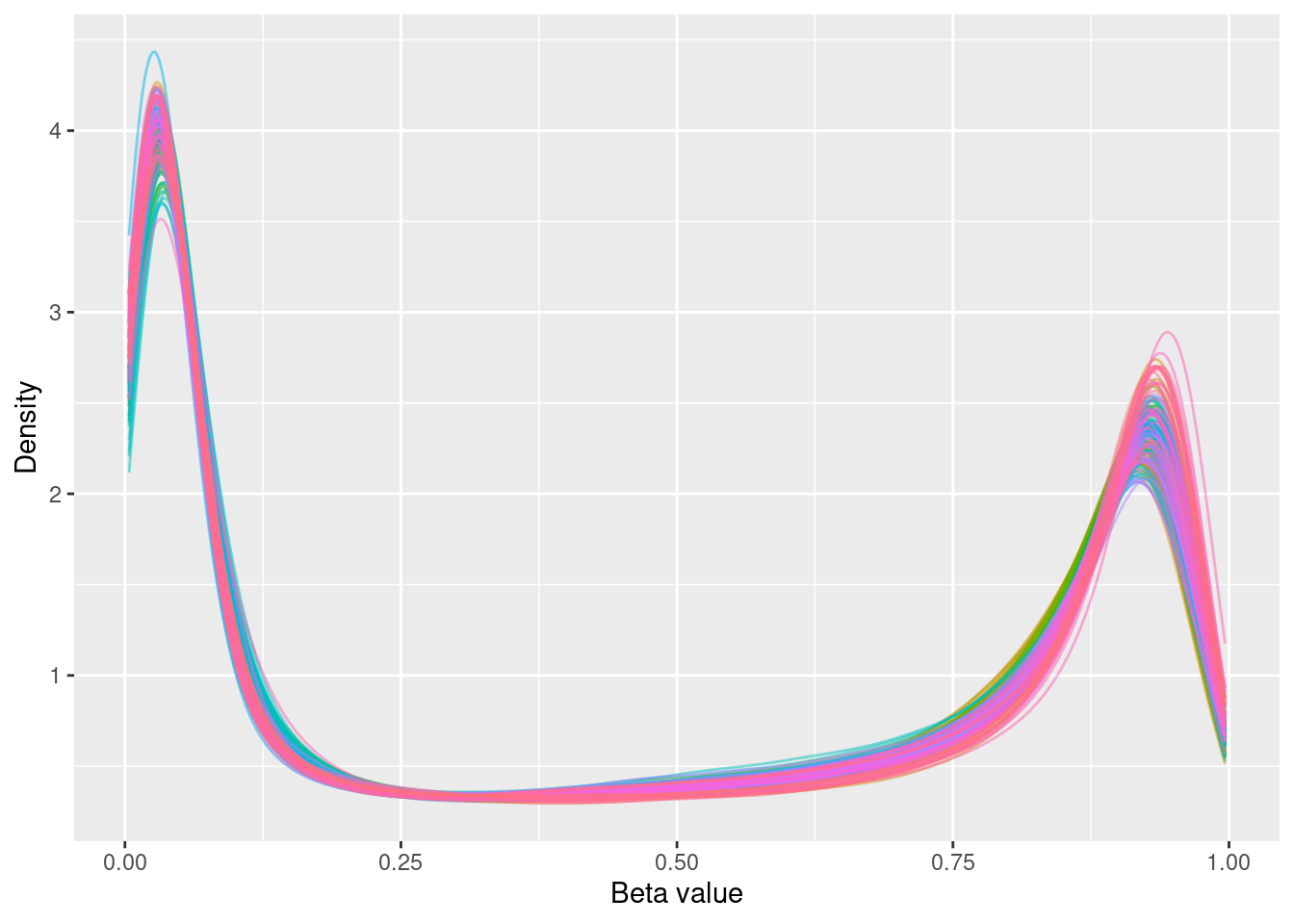
FDR analysis
Run 100 null simulations comparing 5, 10, 20, 40, 80 randomly selected samples per “group”. There should be no significant differential methylation between the groups as the data contains no signal. Consequently, we expect about 5% false gene set discoveries across the 100 simulations from methods that are “holding their size”. We will compare gometh (with top 1000 and 5000 ranked CpGs selected), MethylGSA:GLM, MethylGSM:ORA, MethylGSA:GSEA and ChAMP:ebGSEA for the complete list of BROAD gene sets provided in the ChAMP package.
outFile <- here("data/TCGA.KIRC.rds")
if(!file.exists(outFile)){
saveRDS(betasFlt, file = outFile)
}Load the results of the FDR simulations.
inFile <- here("output/FDR-analysis/FDR-analysis.rds")
if(file.exists(inFile)) dat <- as_tibble(readRDS(inFile))The plots below shows that MethylGSA-ORA does not control the false discovery rate very well as the median proportion of p-value 0.05 for the 100 simulations is greater than 0.5. MethylGSA-GSEA does better, although its median false discovery rate is still relatively high at around 0.25. MethylGSA-GLM has good false discovery rate control with median FDR at around 0.05. ChAMP-ebGSEA is only slightly anti-conservative using both the wicox test (WT) and known population median test (KPMT) with a median FDR at around 0.06-0.08. MissMethyl-gometh is very consistent although somewhat conservative with a median FDR at around 0.02-0.03.
dat %>% group_by(simNo, sampleNo, method) %>%
summarise(psig = sum(pvalue < 0.05)/length(pvalue)) -> psigData
p <- ggplot(psigData, aes(x = sampleNo, y = psig, fill = method)) +
geom_violin(scale = "width", width = 0.8)
p + stat_summary(geom = "point", size = 1, color = "white",
position = position_dodge(0.8),
show.legend = FALSE, fun = median) +
geom_hline(yintercept=0.05, linetype="dashed", color = "red") +
labs(y="Prop. gene sets with p-value < 0.05", x="No. samples",
fill="Method")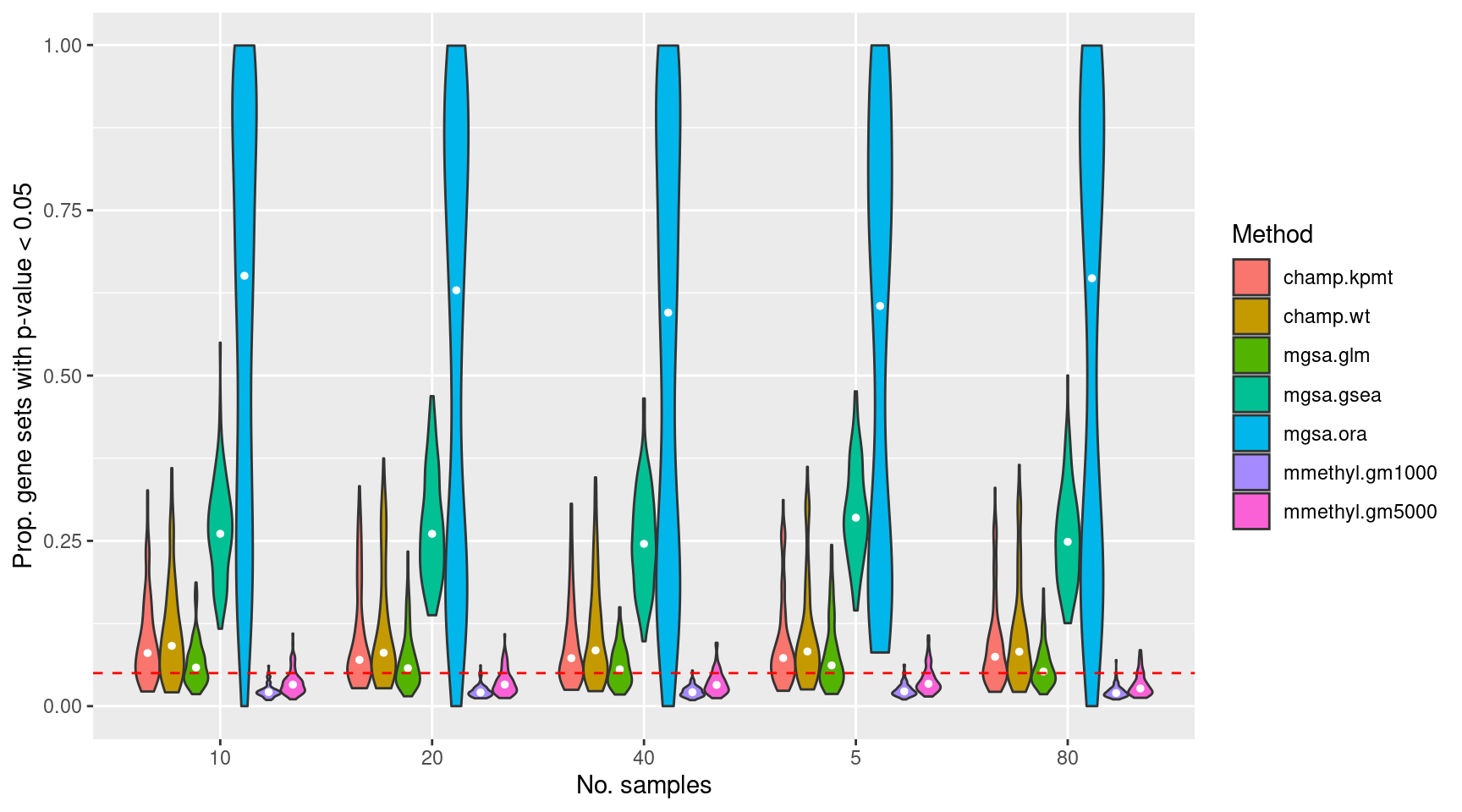
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /config/RStudio/R/3.6.1/lib64/R/lib/libRblas.so
LAPACK: /config/RStudio/R/3.6.1/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] DMRcatedata_2.2.1 ExperimentHub_1.12.0
[3] AnnotationHub_2.18.0 BiocFileCache_1.10.2
[5] dbplyr_1.4.2 rhdf5_2.28.0
[7] patchwork_1.0.0 DMRcate_2.0.7
[9] TCGAutils_1.6.2 curatedTCGAData_1.8.1
[11] MultiAssayExperiment_1.12.6 dplyr_0.8.3
[13] tibble_2.1.3 glue_1.4.0
[15] ggplot2_3.3.0 missMethyl_1.20.4
[17] reshape2_1.4.3 limma_3.42.2
[19] paletteer_1.1.0 minfi_1.32.0
[21] bumphunter_1.26.0 locfit_1.5-9.1
[23] iterators_1.0.12 foreach_1.5.0
[25] Biostrings_2.54.0 XVector_0.24.0
[27] SummarizedExperiment_1.16.1 DelayedArray_0.12.3
[29] BiocParallel_1.20.1 matrixStats_0.56.0
[31] Biobase_2.46.0 GenomicRanges_1.38.0
[33] GenomeInfoDb_1.22.1 IRanges_2.20.2
[35] S4Vectors_0.24.4 BiocGenerics_0.32.0
[37] here_0.1 workflowr_1.6.1
loaded via a namespace (and not attached):
[1] rappdirs_0.3.1
[2] rtracklayer_1.44.4
[3] R.methodsS3_1.7.1
[4] pkgmaker_0.27
[5] tidyr_1.0.2
[6] acepack_1.4.1
[7] bit64_0.9-7
[8] knitr_1.28
[9] R.utils_2.9.0
[10] data.table_1.12.8
[11] rpart_4.1-15
[12] RCurl_1.95-4.12
[13] GEOquery_2.54.1
[14] AnnotationFilter_1.8.0
[15] GenomicFeatures_1.36.4
[16] preprocessCore_1.48.0
[17] RSQLite_2.1.2
[18] bit_1.1-14
[19] xml2_1.3.1
[20] httpuv_1.5.2
[21] assertthat_0.2.1
[22] IlluminaHumanMethylation450kmanifest_0.4.0
[23] viridis_0.5.1
[24] IlluminaHumanMethylationEPICanno.ilm10b4.hg19_0.6.0
[25] xfun_0.13
[26] hms_0.5.3
[27] evaluate_0.14
[28] promises_1.1.0
[29] scrime_1.3.5
[30] progress_1.2.2
[31] readxl_1.3.1
[32] DBI_1.0.0
[33] htmlwidgets_1.3
[34] reshape_0.8.8
[35] purrr_0.3.3
[36] backports_1.1.6
[37] permute_0.9-5
[38] annotate_1.62.0
[39] biomaRt_2.42.1
[40] vctrs_0.2.4
[41] ensembldb_2.8.0
[42] withr_2.1.2
[43] Gviz_1.28.3
[44] BSgenome_1.52.0
[45] checkmate_1.9.4
[46] GenomicAlignments_1.20.1
[47] prettyunits_1.0.2
[48] mclust_5.4.6
[49] cluster_2.1.0
[50] GenomicDataCommons_1.10.0
[51] lazyeval_0.2.2
[52] crayon_1.3.4
[53] genefilter_1.68.0
[54] edgeR_3.26.8
[55] pkgconfig_2.0.3
[56] labeling_0.3
[57] nlme_3.1-147
[58] ProtGenerics_1.16.0
[59] palr_0.2.0
[60] nnet_7.3-12
[61] pals_1.6
[62] rlang_0.4.5
[63] lifecycle_0.2.0
[64] registry_0.5-1
[65] dichromat_2.0-0
[66] cellranger_1.1.0
[67] rprojroot_1.3-2
[68] rngtools_1.4
[69] IlluminaHumanMethylation450kanno.ilmn12.hg19_0.6.0
[70] base64_2.0
[71] Matrix_1.2-18
[72] Rhdf5lib_1.6.1
[73] base64enc_0.1-3
[74] whisker_0.4
[75] viridisLite_0.3.0
[76] oompaBase_3.2.9
[77] bitops_1.0-6
[78] R.oo_1.22.0
[79] blob_1.2.0
[80] DelayedMatrixStats_1.8.0
[81] doRNG_1.7.1
[82] stringr_1.4.0
[83] nor1mix_1.3-0
[84] readr_1.3.1
[85] IlluminaHumanMethylationEPICmanifest_0.3.0
[86] scales_1.1.0
[87] memoise_1.1.0
[88] magrittr_1.5
[89] plyr_1.8.6
[90] bibtex_0.4.2
[91] zlibbioc_1.30.0
[92] compiler_3.6.1
[93] RColorBrewer_1.1-2
[94] illuminaio_0.28.0
[95] Rsamtools_2.0.1
[96] DSS_2.34.0
[97] htmlTable_1.13.2
[98] Formula_1.2-3
[99] MASS_7.3-51.5
[100] tidyselect_0.2.5
[101] stringi_1.4.6
[102] yaml_2.2.1
[103] askpass_1.1
[104] latticeExtra_0.6-28
[105] grid_3.6.1
[106] VariantAnnotation_1.30.1
[107] tools_3.6.1
[108] ruv_0.9.7.1
[109] rstudioapi_0.11
[110] foreign_0.8-72
[111] git2r_0.26.1
[112] bsseq_1.22.0
[113] gridExtra_2.3
[114] farver_2.0.3
[115] digest_0.6.25
[116] BiocManager_1.30.10
[117] shiny_1.3.2
[118] quadprog_1.5-8
[119] Rcpp_1.0.4.6
[120] siggenes_1.60.0
[121] BiocVersion_3.10.1
[122] later_1.0.0
[123] org.Hs.eg.db_3.8.2
[124] httr_1.4.1
[125] AnnotationDbi_1.46.1
[126] biovizBase_1.32.0
[127] colorspace_1.4-1
[128] rvest_0.3.5
[129] XML_3.98-1.20
[130] fs_1.4.1
[131] splines_3.6.1
[132] statmod_1.4.32
[133] rematch2_2.1.0
[134] multtest_2.40.0
[135] mapproj_1.2.6
[136] jcolors_0.0.4
[137] xtable_1.8-4
[138] jsonlite_1.6.1
[139] scico_1.1.0
[140] R6_2.4.1
[141] Hmisc_4.2-0
[142] pillar_1.4.3
[143] htmltools_0.4.0
[144] mime_0.9
[145] interactiveDisplayBase_1.22.0
[146] beanplot_1.2
[147] codetools_0.2-16
[148] maps_3.3.0
[149] lattice_0.20-41
[150] curl_4.3
[151] BiasedUrn_1.07
[152] gtools_3.8.1
[153] GO.db_3.8.2
[154] openssl_1.4.1
[155] survival_2.44-1.1
[156] rmarkdown_2.1
[157] methylumi_2.30.0
[158] munsell_0.5.0
[159] GenomeInfoDbData_1.2.1
[160] HDF5Array_1.14.3
[161] gtable_0.3.0