recomb_size_founder_prop
Hao He
2019-09-23
Last updated: 2019-09-23
Checks: 6 1
Knit directory: csna_workflow/
This reproducible R Markdown analysis was created with workflowr (version 1.4.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190918) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /projects/heh/csna_workflow/output/prop_across_generation_chr | output/prop_across_generation_chr |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: output/permu/
Untracked files:
Untracked: analysis/01_geneseek2qtl2.R
Untracked: analysis/01_geneseek2qtl2.Rout
Untracked: analysis/01_geneseek2qtl2.script
Untracked: analysis/01_geneseek2qtl2.stderr
Untracked: analysis/01_geneseek2qtl2.stdout
Untracked: analysis/02_geneseek2intensity.R
Untracked: analysis/02_geneseek2intensity.Rout
Untracked: analysis/02_geneseek2intensity.script
Untracked: analysis/02_geneseek2intensity.stderr
Untracked: analysis/02_geneseek2intensity.stdout
Untracked: analysis/03_firstgm2genoprobs.R
Untracked: analysis/03_firstgm2genoprobs.Rout
Untracked: analysis/03_firstgm2genoprobs.script
Untracked: analysis/03_firstgm2genoprobs.stderr
Untracked: analysis/03_firstgm2genoprobs.stdout
Untracked: analysis/04_diagnosis_qc_gigamuga_nine_batches.R
Untracked: analysis/04_diagnosis_qc_gigamuga_nine_batches.Rout
Untracked: analysis/04_diagnosis_qc_gigamuga_nine_batches.script
Untracked: analysis/04_diagnosis_qc_gigamuga_nine_batches.stderr
Untracked: analysis/04_diagnosis_qc_gigamuga_nine_batches.stdout
Untracked: analysis/05_after_diagnosis_qc_gigamuga_nine_batches.R
Untracked: analysis/05_after_diagnosis_qc_gigamuga_nine_batches.Rout
Untracked: analysis/05_after_diagnosis_qc_gigamuga_nine_batches.script
Untracked: analysis/05_after_diagnosis_qc_gigamuga_nine_batches.stderr
Untracked: analysis/05_after_diagnosis_qc_gigamuga_nine_batches.stdout
Untracked: analysis/06_final_pr_apr_69K.R
Untracked: analysis/06_final_pr_apr_69K.Rout
Untracked: analysis/06_final_pr_apr_69K.script
Untracked: analysis/06_final_pr_apr_69K.stderr
Untracked: analysis/06_final_pr_apr_69K.stdout
Untracked: analysis/07.1_html_founder_prop.R
Untracked: analysis/07.1_html_founder_prop.Rout
Untracked: analysis/07.1_html_founder_prop.script
Untracked: analysis/07.1_html_founder_prop.stderr
Untracked: analysis/07.1_html_founder_prop.stdout
Untracked: analysis/07_recomb_size_founder_prop.R
Untracked: analysis/07_recomb_size_founder_prop.Rout
Untracked: analysis/07_recomb_size_founder_prop.script
Untracked: analysis/07_recomb_size_founder_prop.stderr
Untracked: analysis/07_recomb_size_founder_prop.stdout
Untracked: analysis/08_gcta_herit.R
Untracked: analysis/09_qtlmapping.R
Untracked: analysis/10_qtl_permu.R
Untracked: analysis/Novelty_resids_datarelease_07302918.gcta.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918.gcta.script
Untracked: analysis/Novelty_resids_datarelease_07302918.gcta.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918.gcta.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m1.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m1.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m1.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m1.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m2.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m2.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m2.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m2.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m3.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m3.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_1.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_1.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_1.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_1.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_2.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_2.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_2.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_2.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_3.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_3.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_3.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_3.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_4.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_4.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_4.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_4.stdout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_5.Rout
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_5.script
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_5.stderr
Untracked: analysis/Novelty_resids_datarelease_07302918_m3_5.stdout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918.gcta.Rout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918.gcta.script
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918.gcta.stderr
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918.gcta.stdout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m1.Rout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m1.script
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m1.stderr
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m1.stdout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m2.Rout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m2.script
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m2.stderr
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m2.stdout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m3.Rout
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m3.script
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m3.stderr
Untracked: analysis/Novelty_residuals_RankNormal_datarelease_07302918_m3.stdout
Untracked: analysis/run_01_geneseek2qtl2.R
Untracked: analysis/run_02_geneseek2intensity.R
Untracked: analysis/run_03_firstgm2genoprobs.R
Untracked: analysis/run_04_diagnosis_qc_gigamuga_nine_batches.R
Untracked: analysis/run_05_after_diagnosis_qc_gigamuga_nine_batches.R
Untracked: analysis/run_06_final_pr_apr_69K.R
Untracked: analysis/run_07.1_html_founder_prop.R
Untracked: analysis/run_07_recomb_size_founder_prop.R
Untracked: analysis/run_08_gcta_herit.R
Untracked: analysis/run_09_qtlmapping.R
Untracked: analysis/run_10_qtl_permu.R
Untracked: code/reconst_utils.R
Untracked: data/FinalReport/
Untracked: data/GCTA/
Untracked: data/GM/
Untracked: data/Jackson_Lab_Bubier_MURGIGV01/
Untracked: data/cc_variants.sqlite
Untracked: data/marker_grid_0.02cM_plus.txt
Untracked: data/mouse_genes_mgi.sqlite
Untracked: data/pheno/
Untracked: output/AfterQC_Percent_missing_genotype_data.pdf
Untracked: output/AfterQC_Proportion_matching_genotypes_after_removal_of_bad_samples.pdf
Untracked: output/AfterQC_Proportion_matching_genotypes_before_removal_of_bad_samples.pdf
Untracked: output/AfterQC_number_crossover.pdf
Untracked: output/Percent_genotype_errors_obs_vs_predicted.pdf
Untracked: output/Percent_missing_genotype_data.pdf
Untracked: output/Percent_missing_genotype_data_per_marker.pdf
Untracked: output/Proportion_matching_genotypes_after_removal_of_bad_samples.pdf
Untracked: output/Proportion_matching_genotypes_before_removal_of_bad_samples.pdf
Untracked: output/genotype_error_marker.pdf
Untracked: output/genotype_frequency_marker.pdf
Untracked: output/m1.Novelty_resids_datarelease_07302918.RData
Untracked: output/m1.Novelty_residuals_RankNormal_datarelease_07302918.RData
Untracked: output/m2.Novelty_resids_datarelease_07302918.RData
Untracked: output/m2.Novelty_residuals_RankNormal_datarelease_07302918.RData
Untracked: output/m3.Novelty_resids_datarelease_07302918.RData
Untracked: output/m3.Novelty_residuals_RankNormal_datarelease_07302918.RData
Untracked: output/num.geno.pheno.in.Novelty_resids_datarelease_07302918.csv
Untracked: output/num.geno.pheno.in.Novelty_residuals_RankNormal_datarelease_07302918.csv
Untracked: output/number_crossover.pdf
Untracked: output/prop_across_generation_chr_p.RData
Unstaged changes:
Modified: README.md
Modified: _workflowr.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | fb74e74 | xhyuo | 2019-09-23 | 07_recomb_size_founder_prop.Rmd |
| html | 97188bc | xhyuo | 2019-09-23 | Build site. |
| Rmd | e3f5af2 | xhyuo | 2019-09-23 | 07_recomb_size_founder_prop.Rmd |
| html | 0d00279 | xhyuo | 2019-09-23 | Build site. |
| Rmd | 504d14d | xhyuo | 2019-09-23 | 07_recomb_size_founder_prop.Rmd |
This script will plot the recombination block size and founder props across all generations
library
library(qtl2)
library(abind)
library(tidyverse)
library(plotly)
require(vcd)
require(MASS)
options(stringsAsFactors = F)
# Reformat for tidyverse.
reformat_probs = function(probs) {
mat = matrix(0, nrow = nrow(probs) * dim(probs)[3], ncol = 8,
dimnames = list(rep(rownames(probs), dim(probs)[3]),
names(CCcolors)))
for(i in 1:dim(probs)[3]) {
st = (i - 1) * nrow(probs) + 1
en = i * nrow(probs)
mat[st:en,] = probs[,,i]
} # for(i)
return(data.frame(chr = rep(markers$chr, each = nrow(probs)),
pos = rep(markers$pos, each = nrow(probs)), mat))
} # reformat_probs()
# NOTE: I'm using a lower case L as the beginning of 129 becuase
# the tidy functions won't allow a number or a "_" at the
# beginning of a variable name.
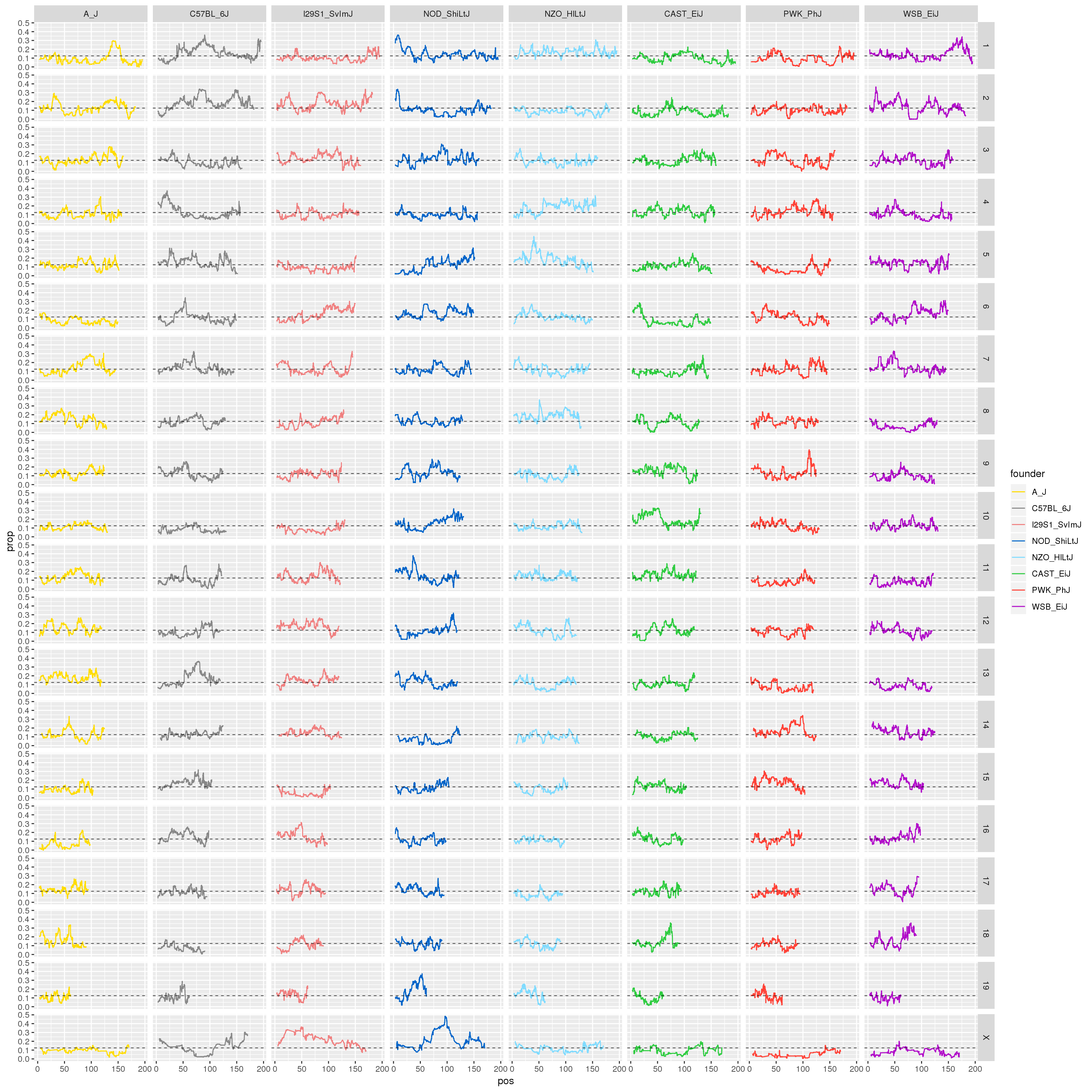
names(CCcolors) = c("A_J", "C57BL_6J", "l29S1_SvImJ", "NOD_ShiLtJ",
"NZO_HlLtJ", "CAST_EiJ", "PWK_PhJ", "WSB_EiJ")plot founder_proportions
#allele probs
load("data/Jackson_Lab_Bubier_MURGIGV01/apr_DO2437.RData")
#geno probs
load("data/Jackson_Lab_Bubier_MURGIGV01/pr_DO2437.RData")
#cross infor
load("data/Jackson_Lab_Bubier_MURGIGV01/gm_DO2437_qc.RData")
#combine all chrs into one 3d array
apr.3d.all <- do.call("abind",list(apr,along = 3))
rm(apr)
# Load in the markers.
load(url("ftp://ftp.jax.org/MUGA/GM_snps.Rdata"))
GM_snps <- GM_snps[!is.na(GM_snps$chr),]
GM_snps$chr <- factor(GM_snps$chr)
markers = GM_snps[intersect(dimnames(apr.3d.all)[[3]],GM_snps$marker),1:4]
markers$chr = factor(markers$chr, levels = c(1:19, "X"))
# subset to the markers
apr.3d.all <- apr.3d.all[,,markers$marker]
#subset to each generation
for(g in unique(gm_DO2437_qc$covar$ngen)){
#g = "21"
print(g)
apr.3d <- apr.3d.all[gm_DO2437_qc$covar[gm_DO2437_qc$covar$ngen == g,"id"],,]
#reformat
probs = reformat_probs(apr.3d)
gc()
# Summarize founder proportion by chromosome.
fp = probs %>% group_by(chr, pos) %>%
summarize_all(mean) %>%
gather(founder, prop, 3:10)
fp$founder = factor(fp$founder, levels = names(CCcolors))
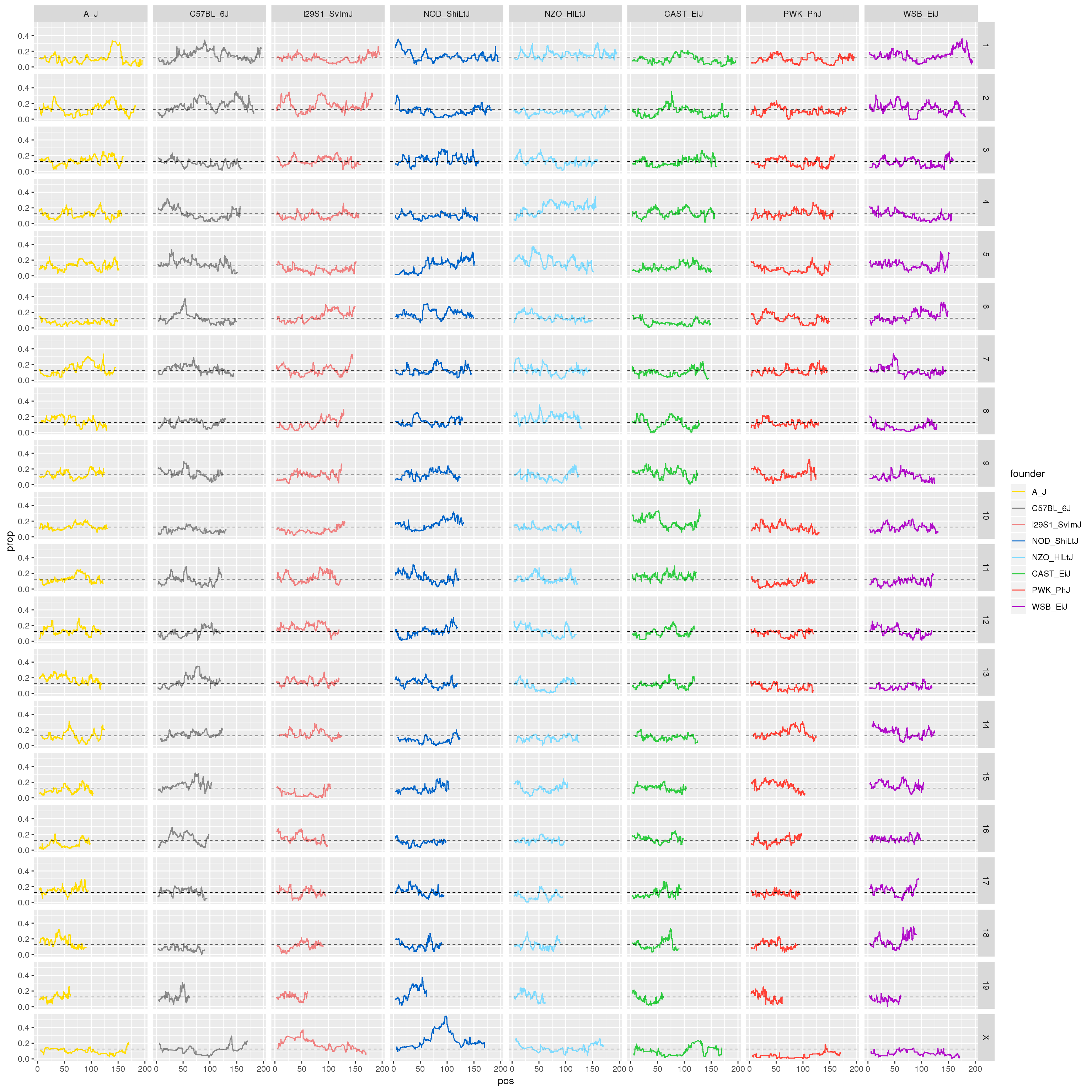
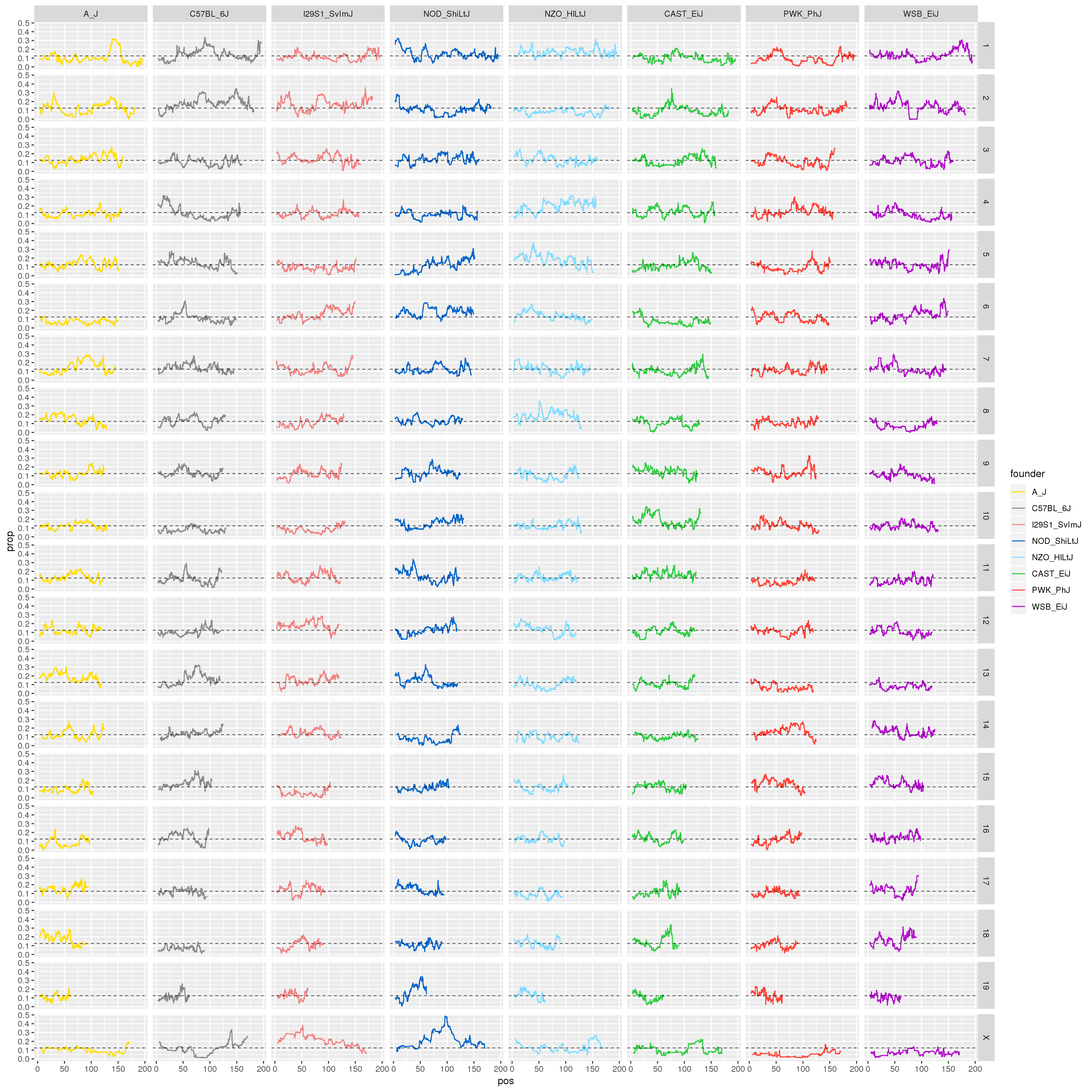
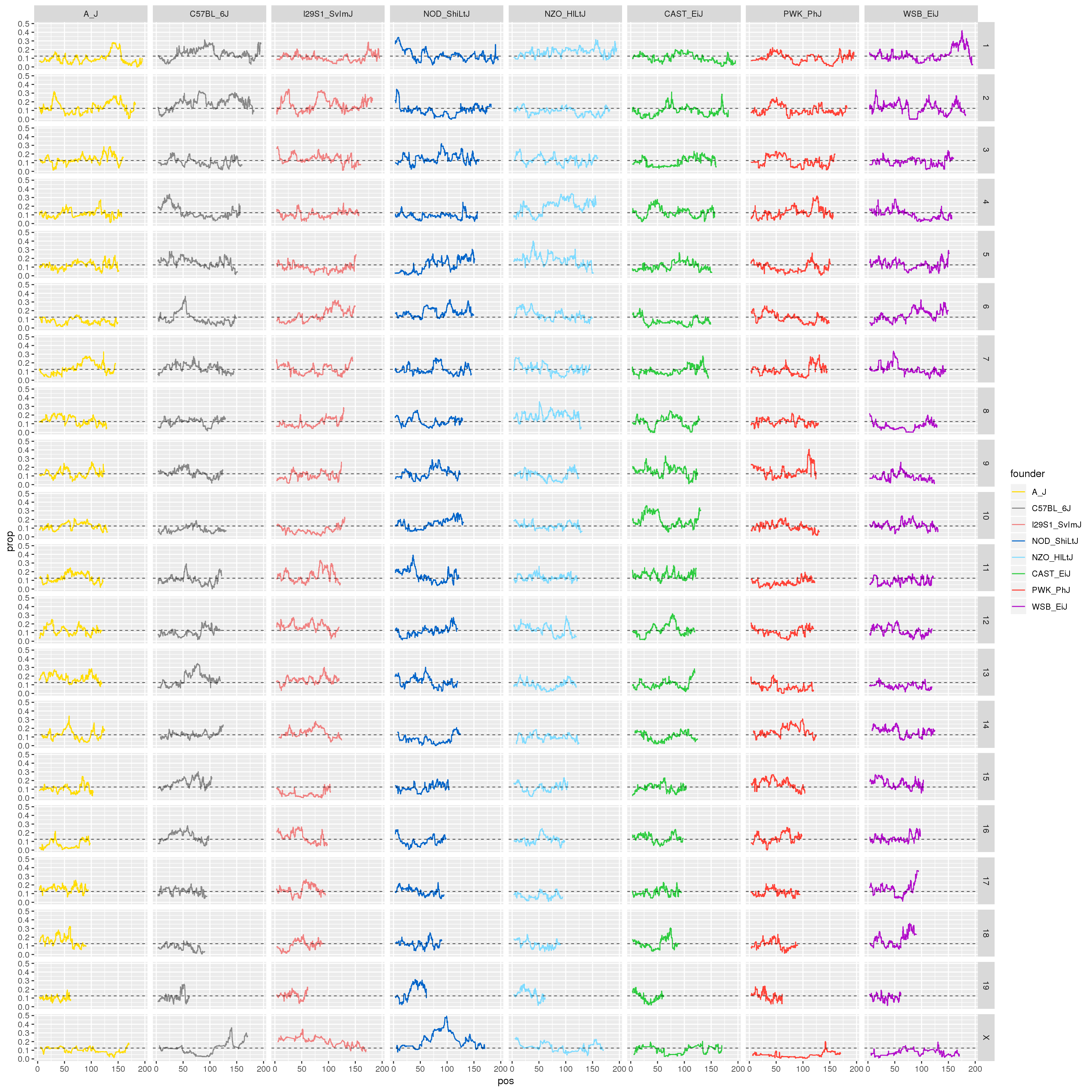
#plot for all the chromosomes
png(paste0("output/DO_Gigamuga_founder_proportions_G", g, ".png"), width = 3600,
height = 2000, res = 128)
p1 <- ggplot(fp, aes(pos, prop, color = founder)) +
geom_line() +
geom_hline(yintercept=0.125, linetype="dashed", color = "black", size = 0.25) +
scale_color_manual(values = CCcolors) +
facet_grid(chr~founder)
print(p1)
dev.off()
print(p1)
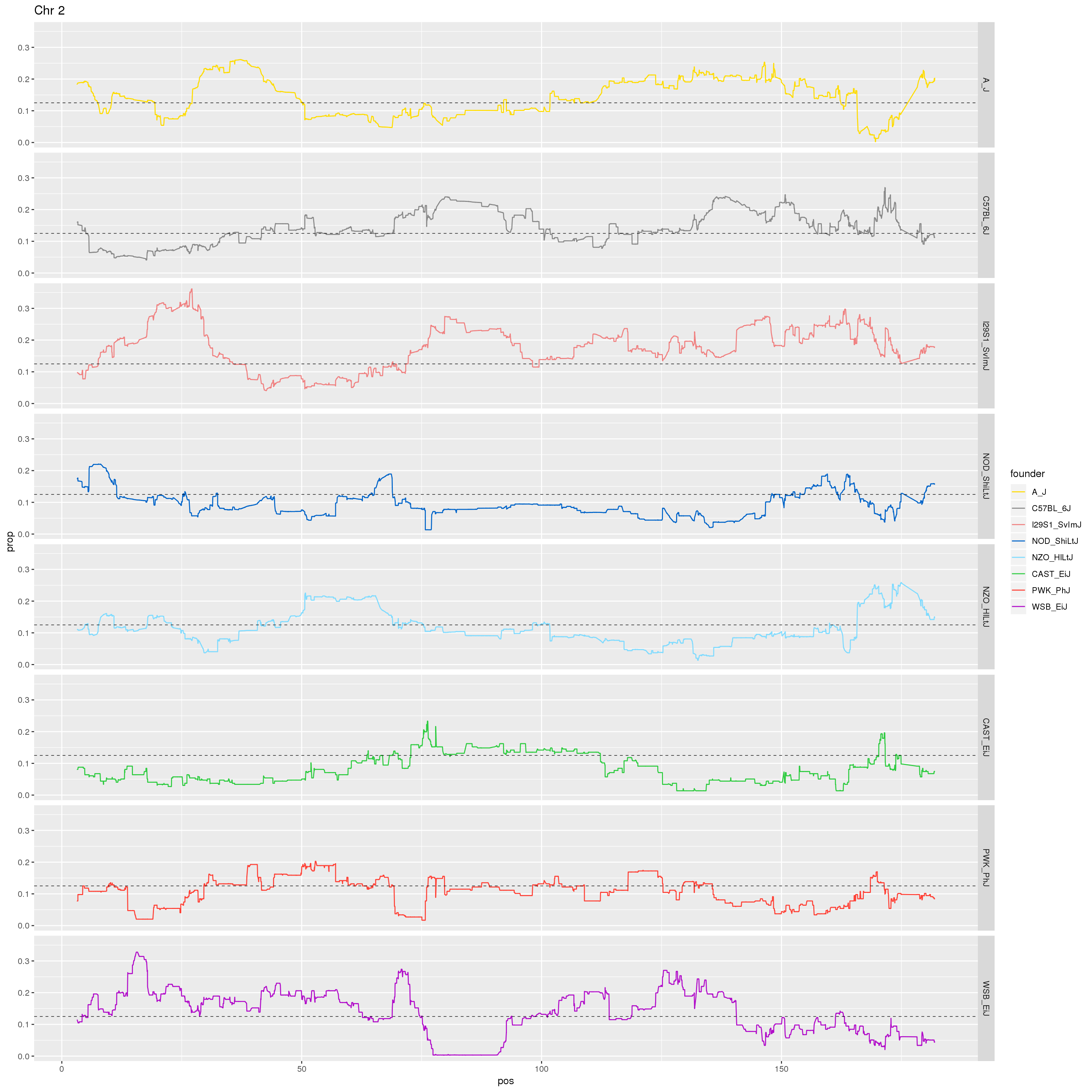
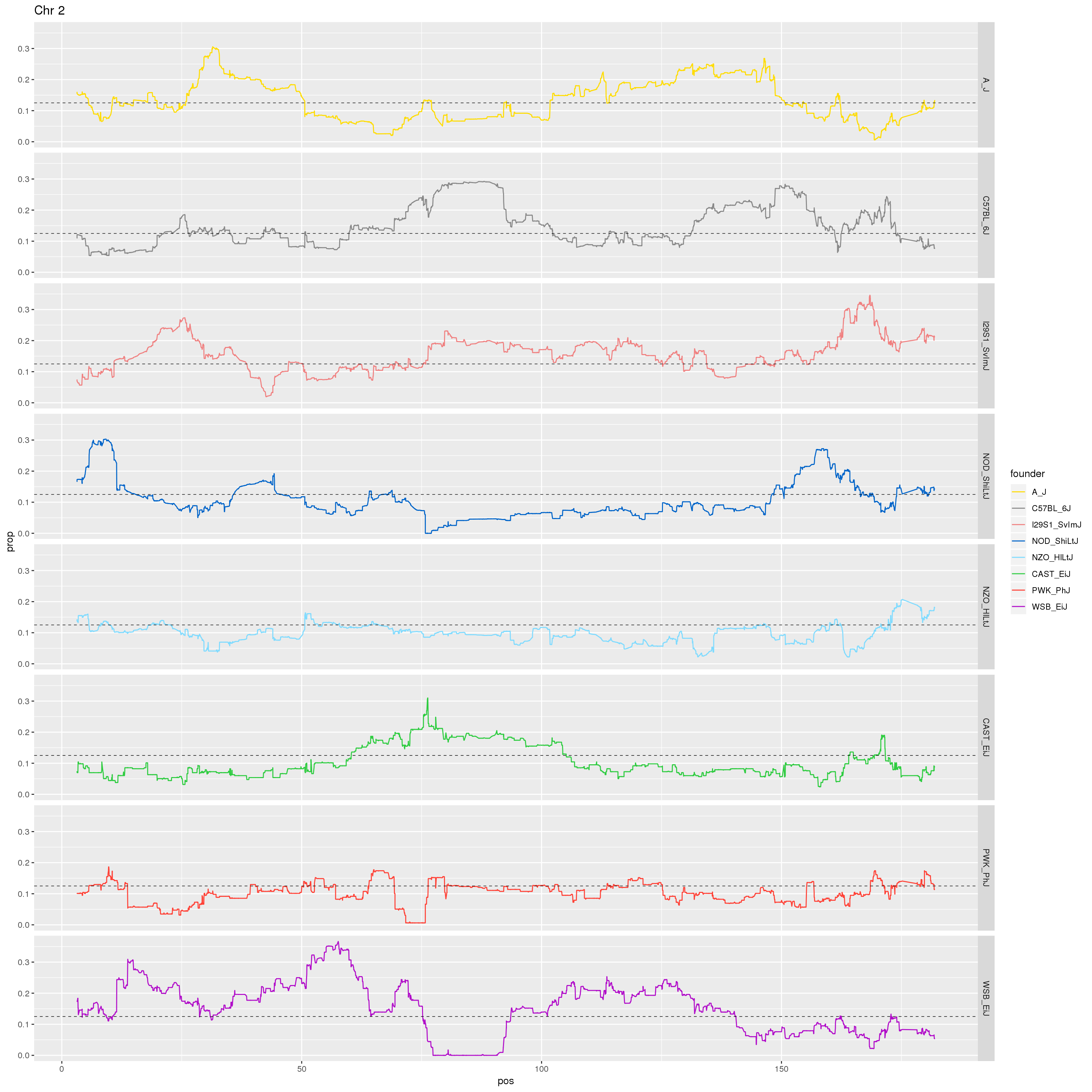
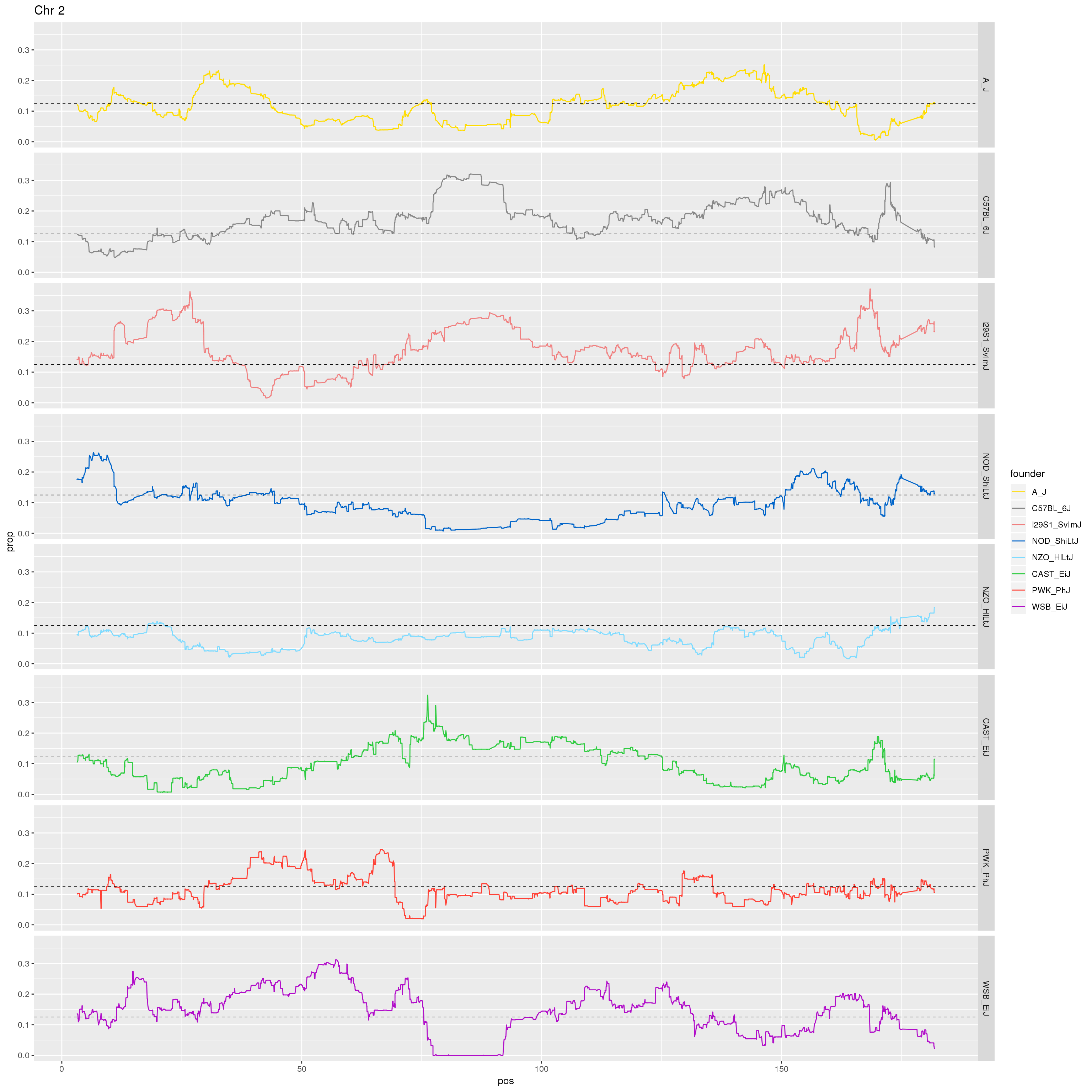
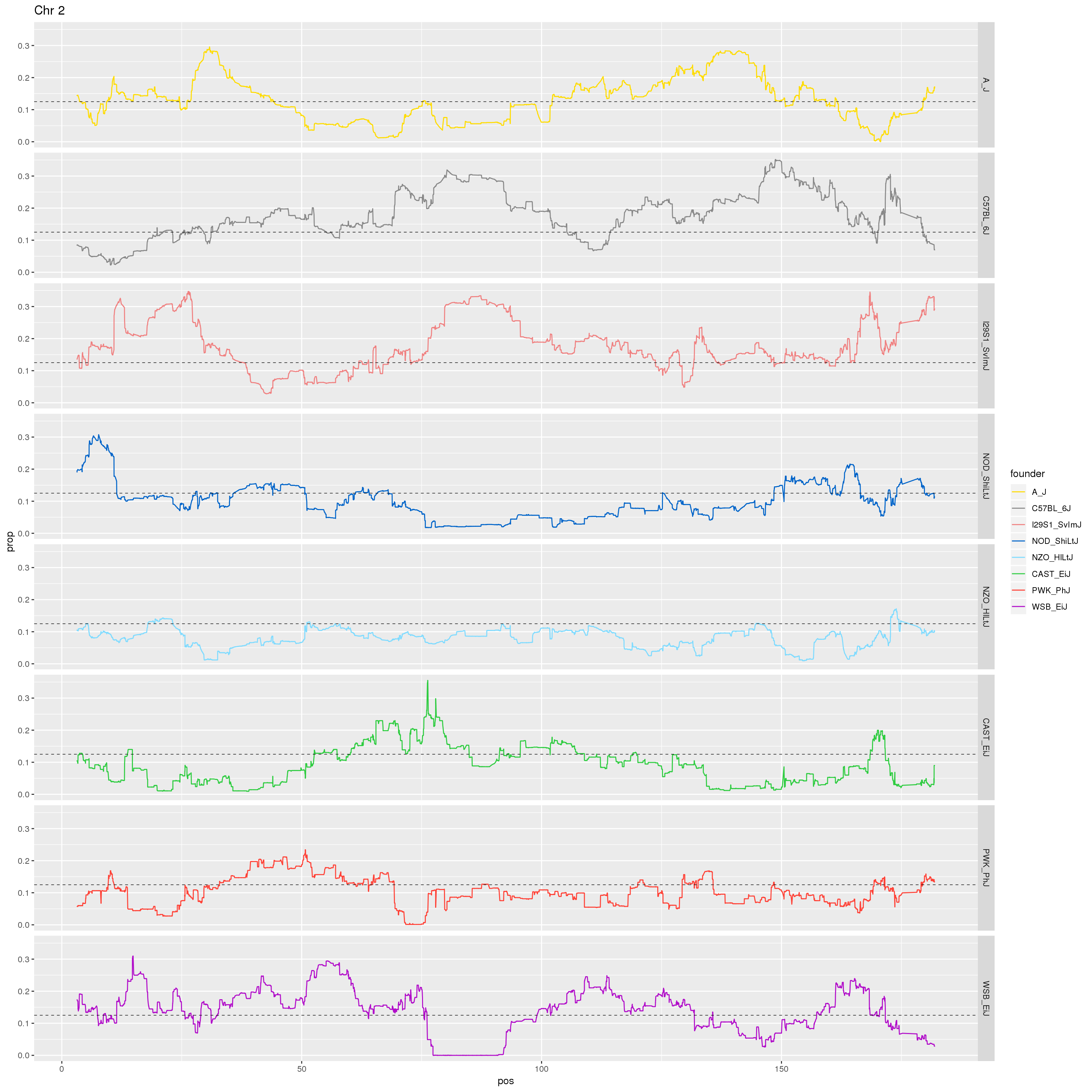
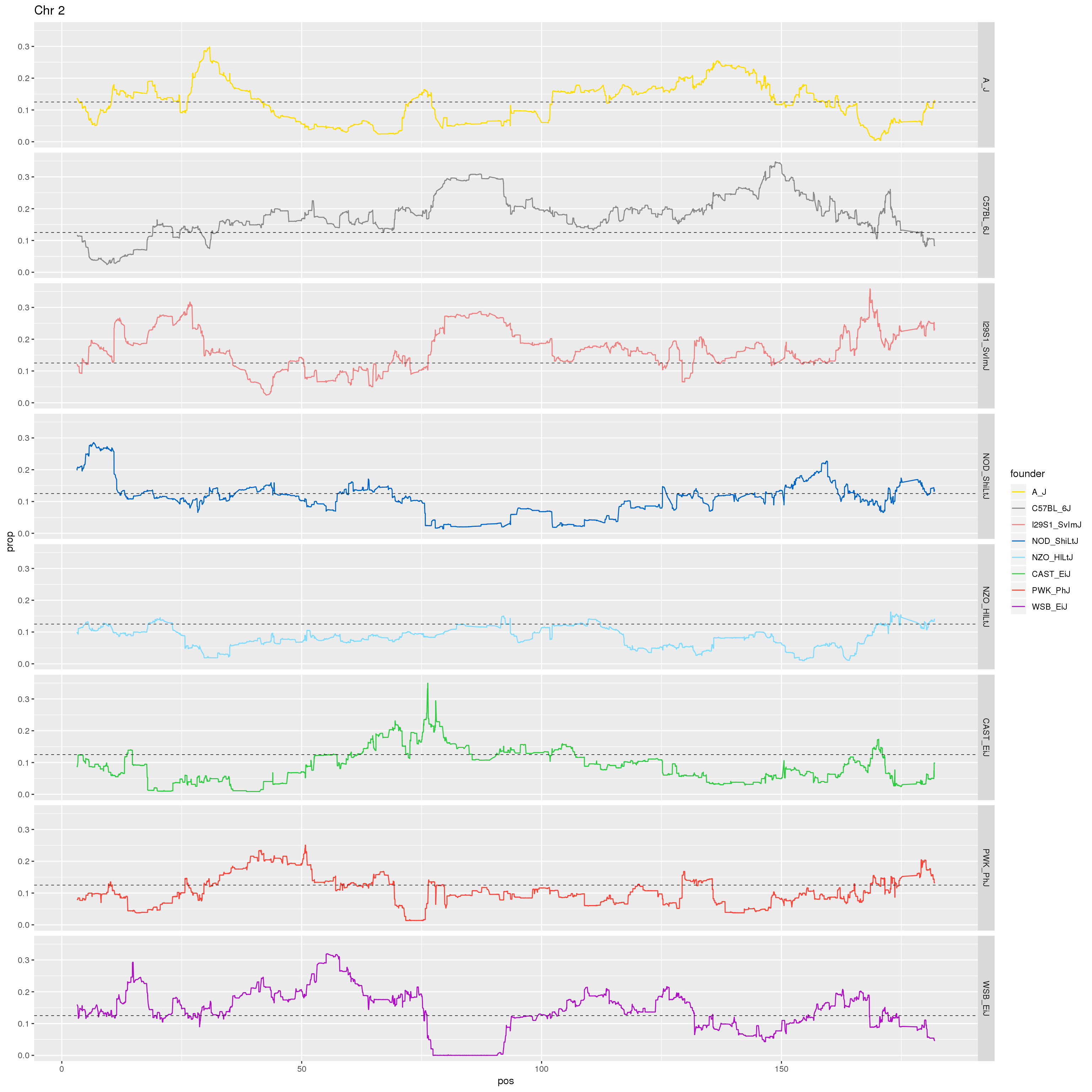
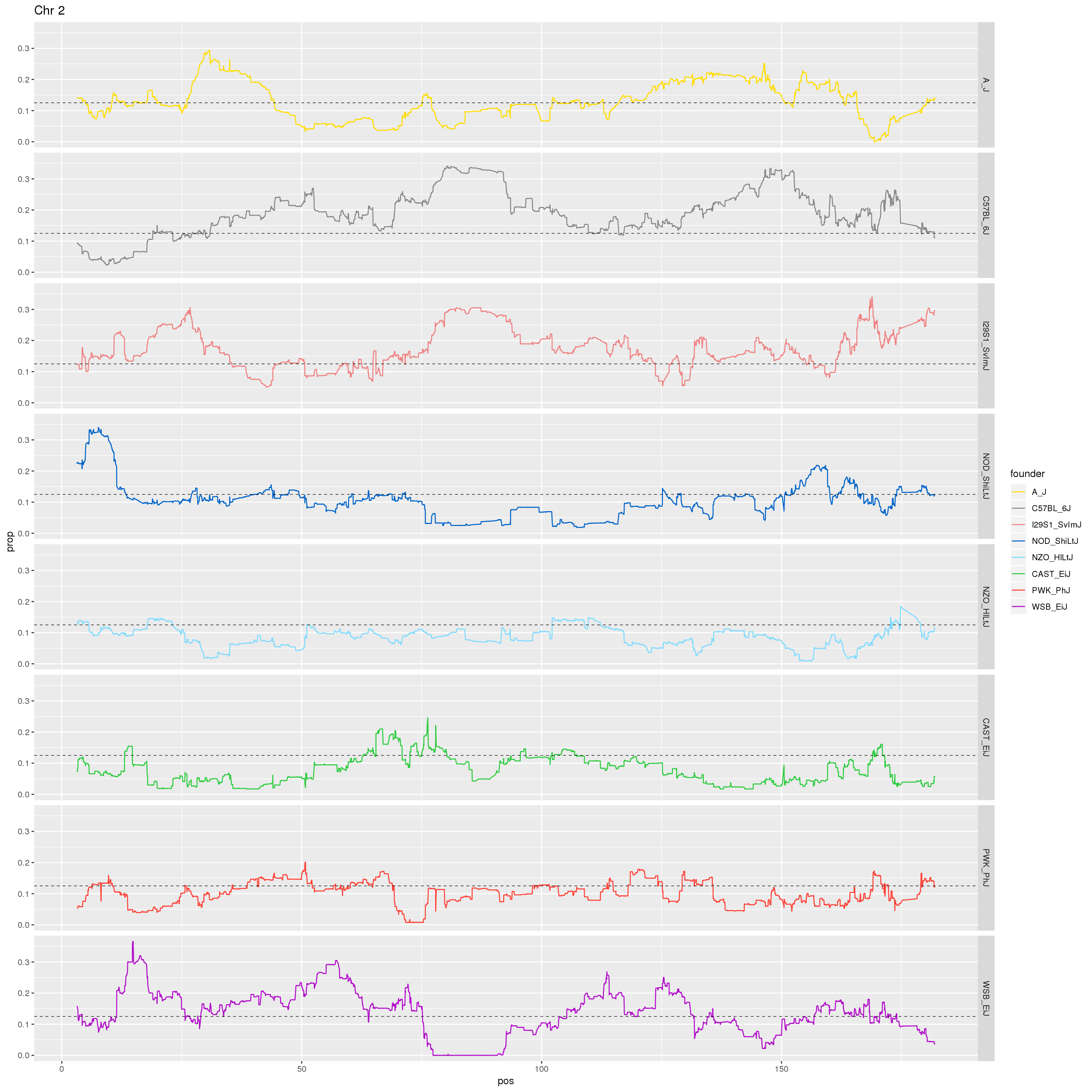
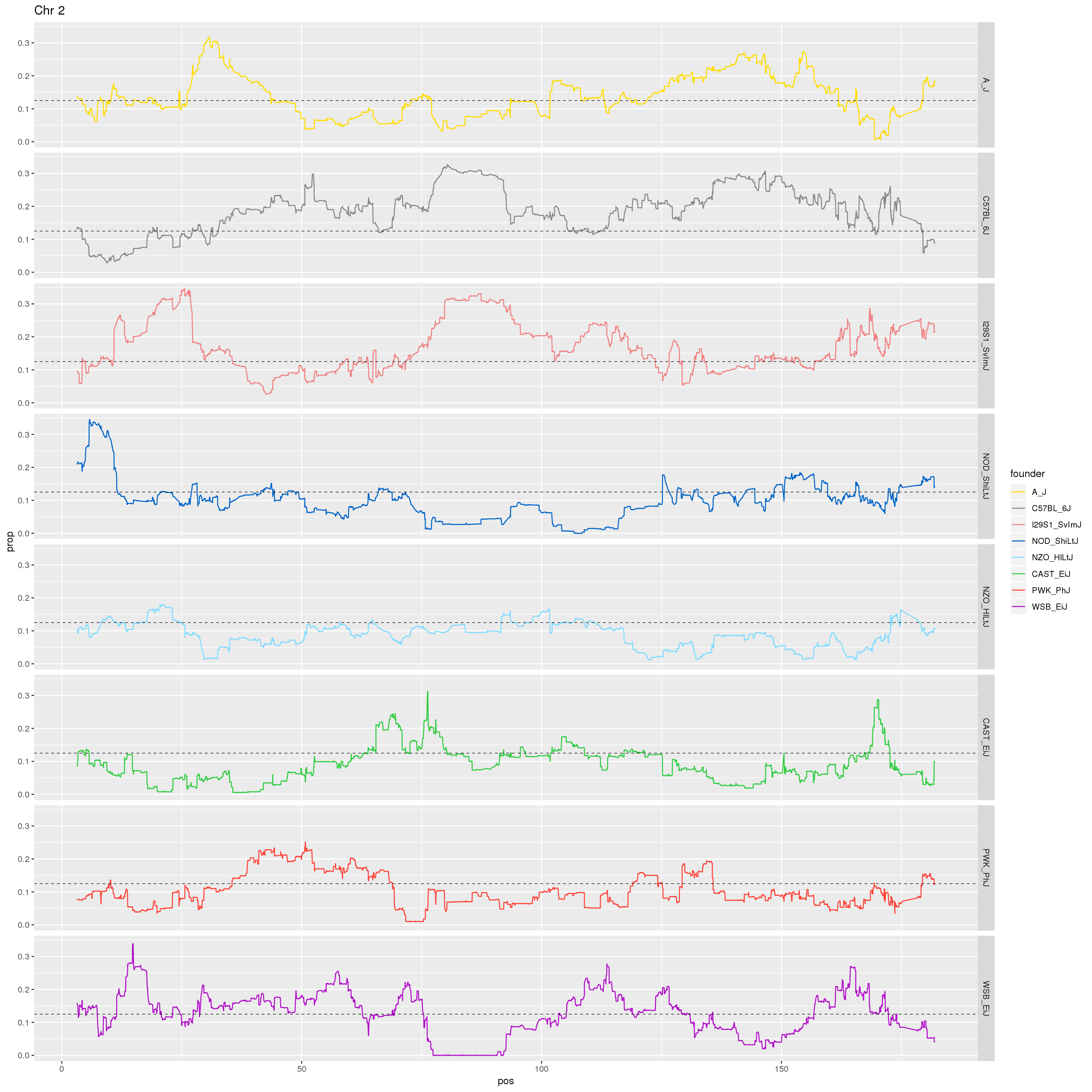
# Make a plot of just Chr 2.
png(paste0("output/DO_Gigamuga_founder_proportions_chr2_G", g, ".png"), width = 1200,
height = 800, res = 128)
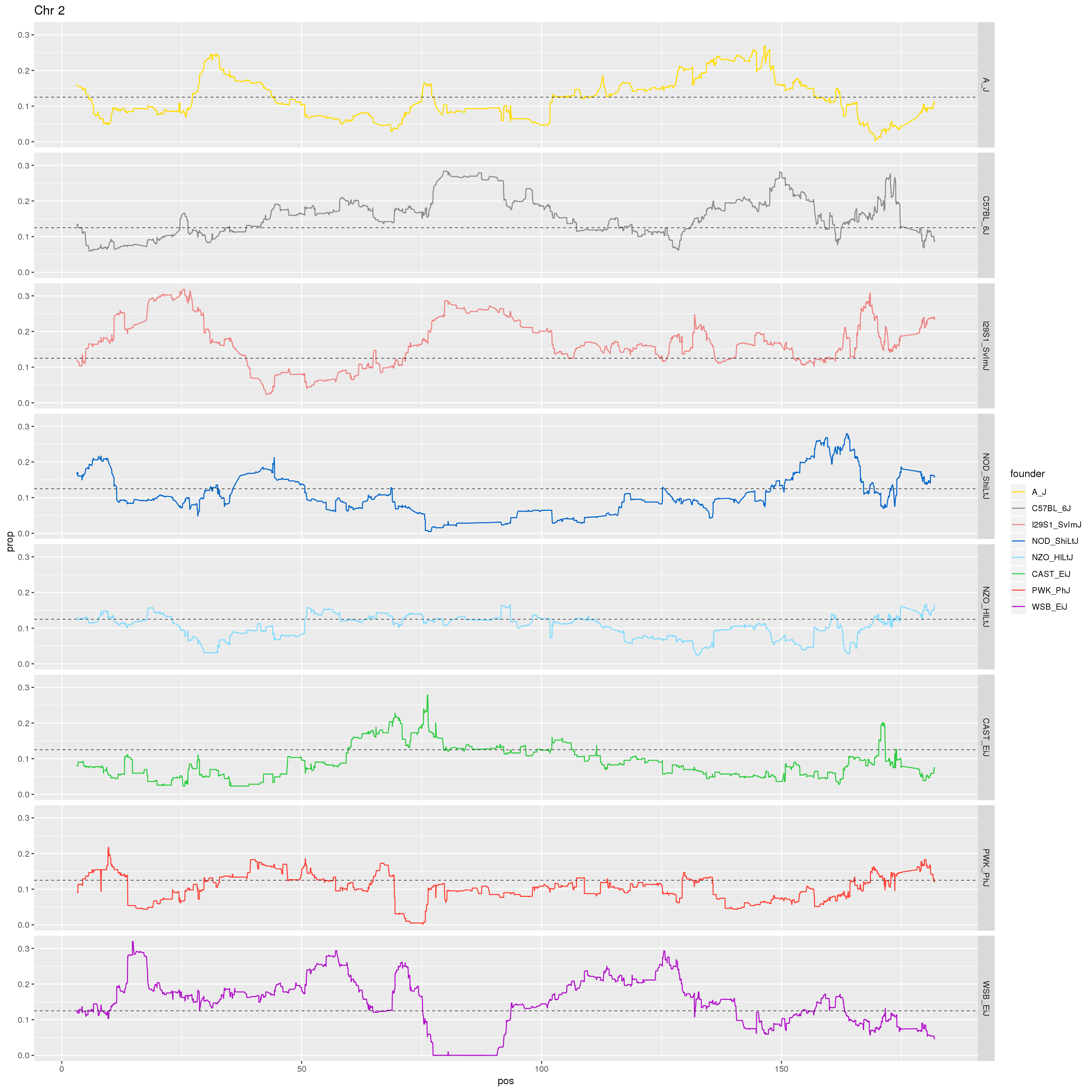
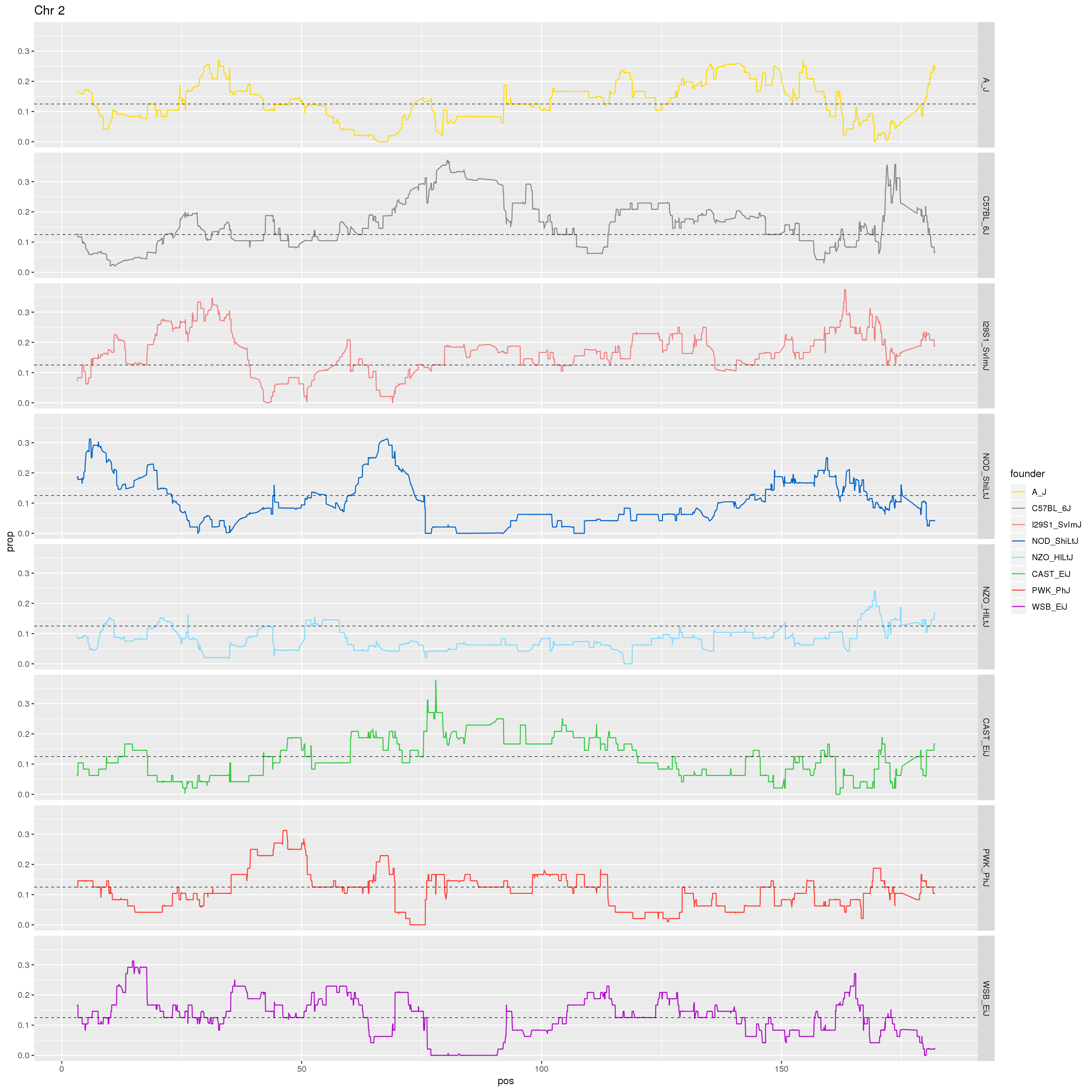
p2 <- fp %>% filter(chr == 2) %>%
ggplot(aes(pos, prop, color = founder)) +
geom_line() +
geom_hline(yintercept=0.125, linetype="dashed", color = "black", size = 0.25) +
scale_color_manual(values = CCcolors) +
facet_grid(founder~.)+
labs(title = "Chr 2")
print(p2)
dev.off()
print(p2)
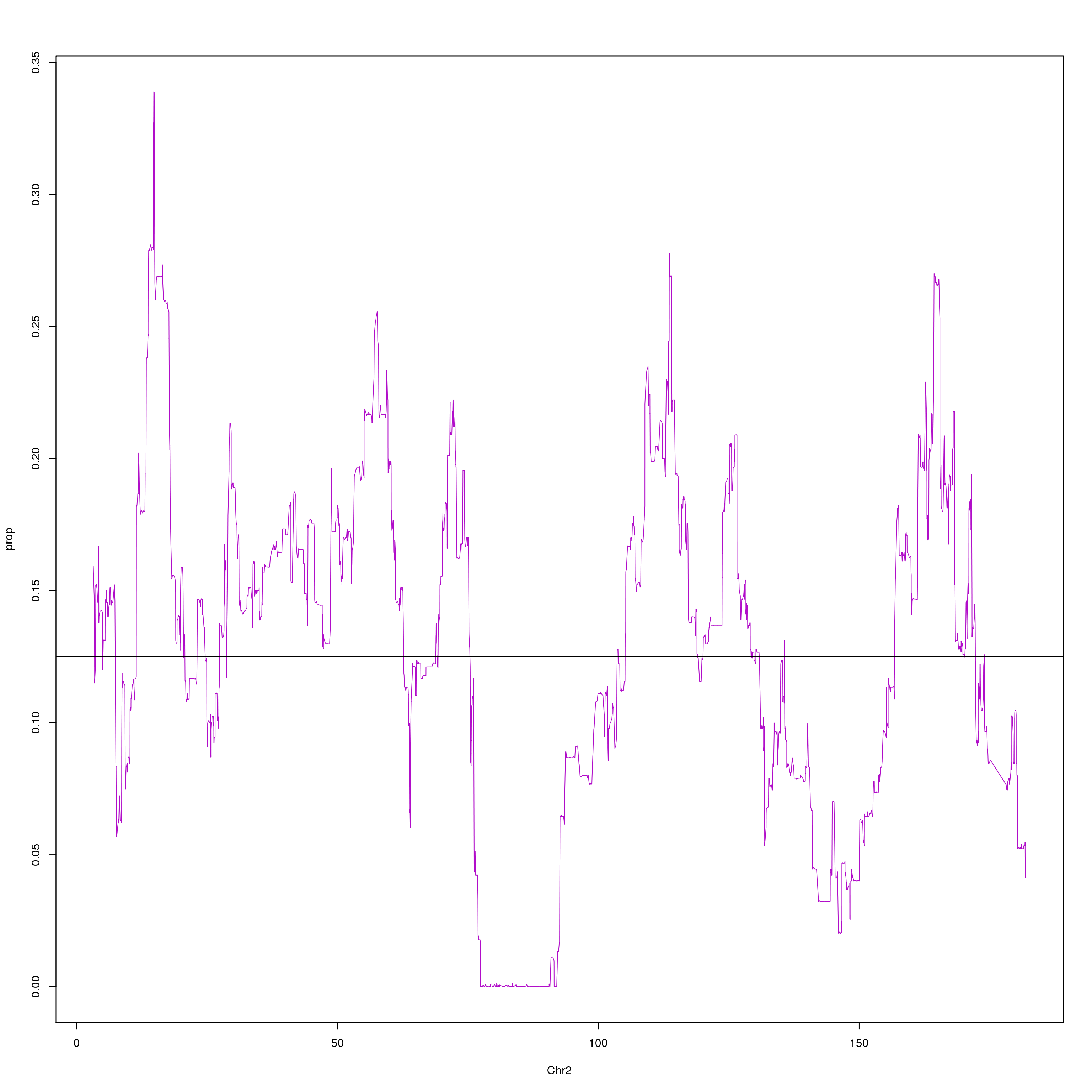
#plot on chr2 for WSB
png(paste0("output/DO_Gigamuga_chr2_WSB_G", g, ".png"), width = 1200,
height = 800, res = 128)
chr2 = probs[probs$chr == 2, c(1,2,10)]
agg = aggregate(chr2$WSB_EiJ, list(chr2$pos), mean)
plot(agg, type = "l", ylab = "prop", xlab = "Chr2", col = "#B10DC9")
abline(h=0.125, col="black")
dev.off()
chr2 = probs[probs$chr == 2, c(1,2,10)]
agg = aggregate(chr2$WSB_EiJ, list(chr2$pos), mean)
plot(agg, type = "l", ylab = "prop", xlab = "Chr2", col = "#B10DC9")
abline(h=0.125, col="black")
}[1] "21"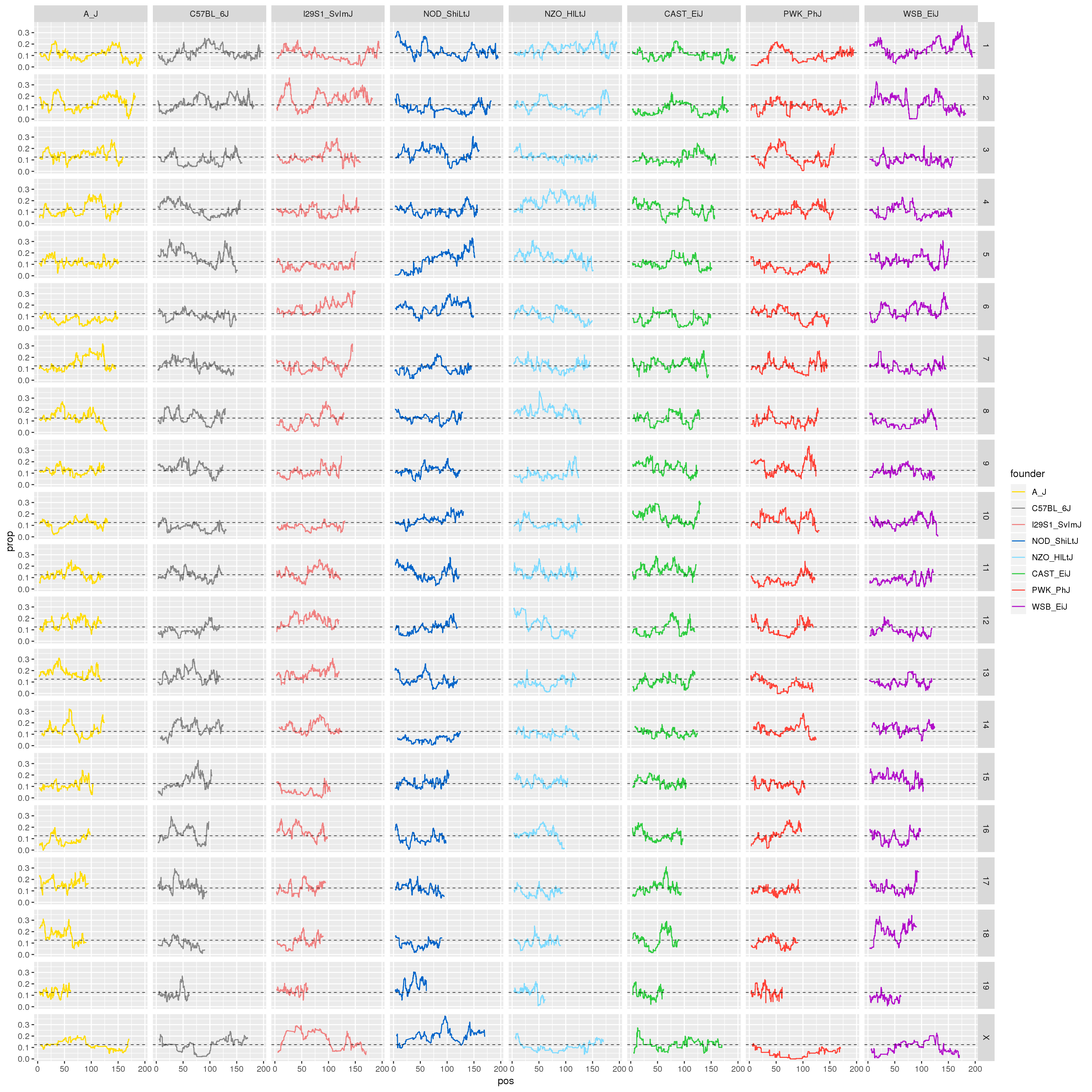
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
[1] "22"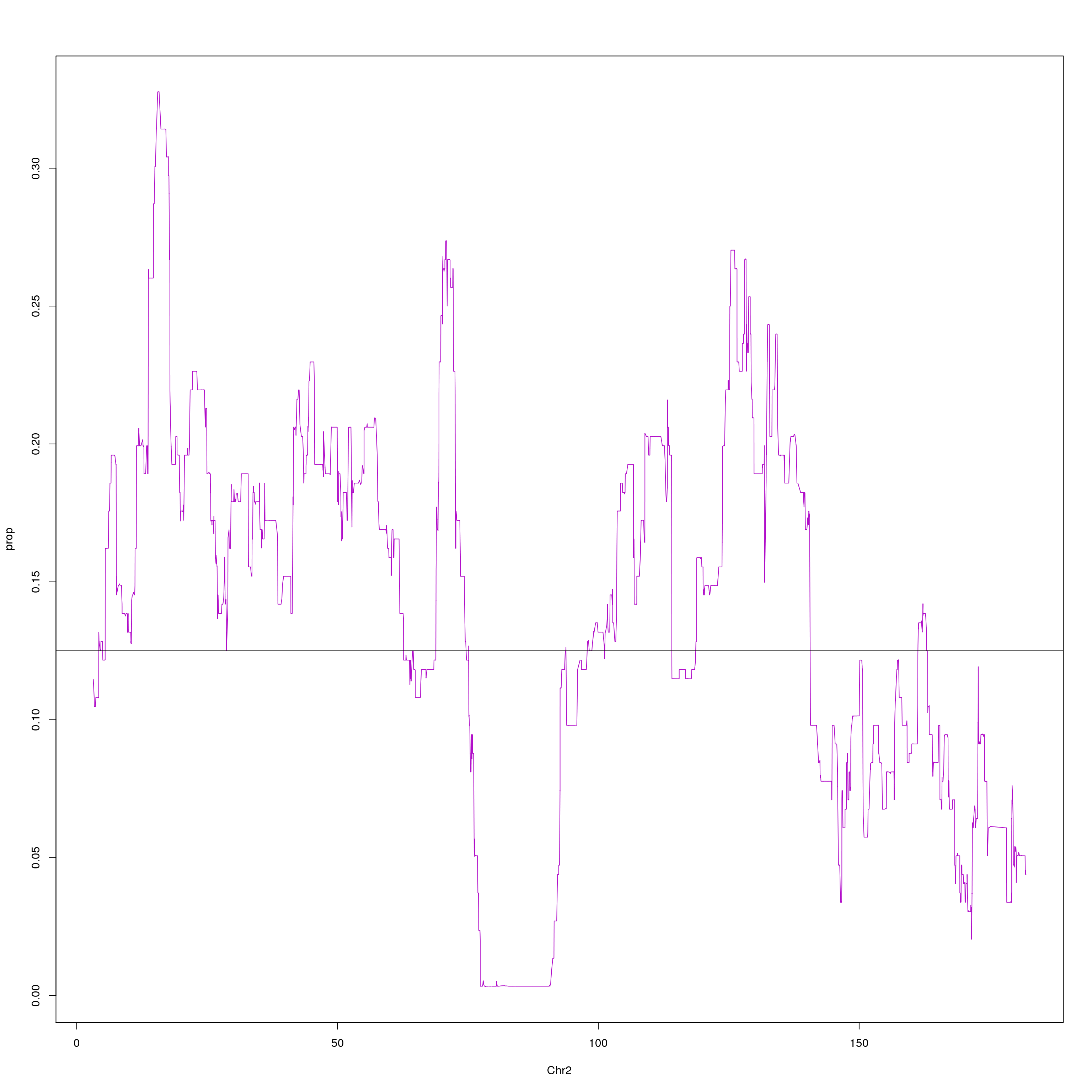
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |

| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
[1] "23"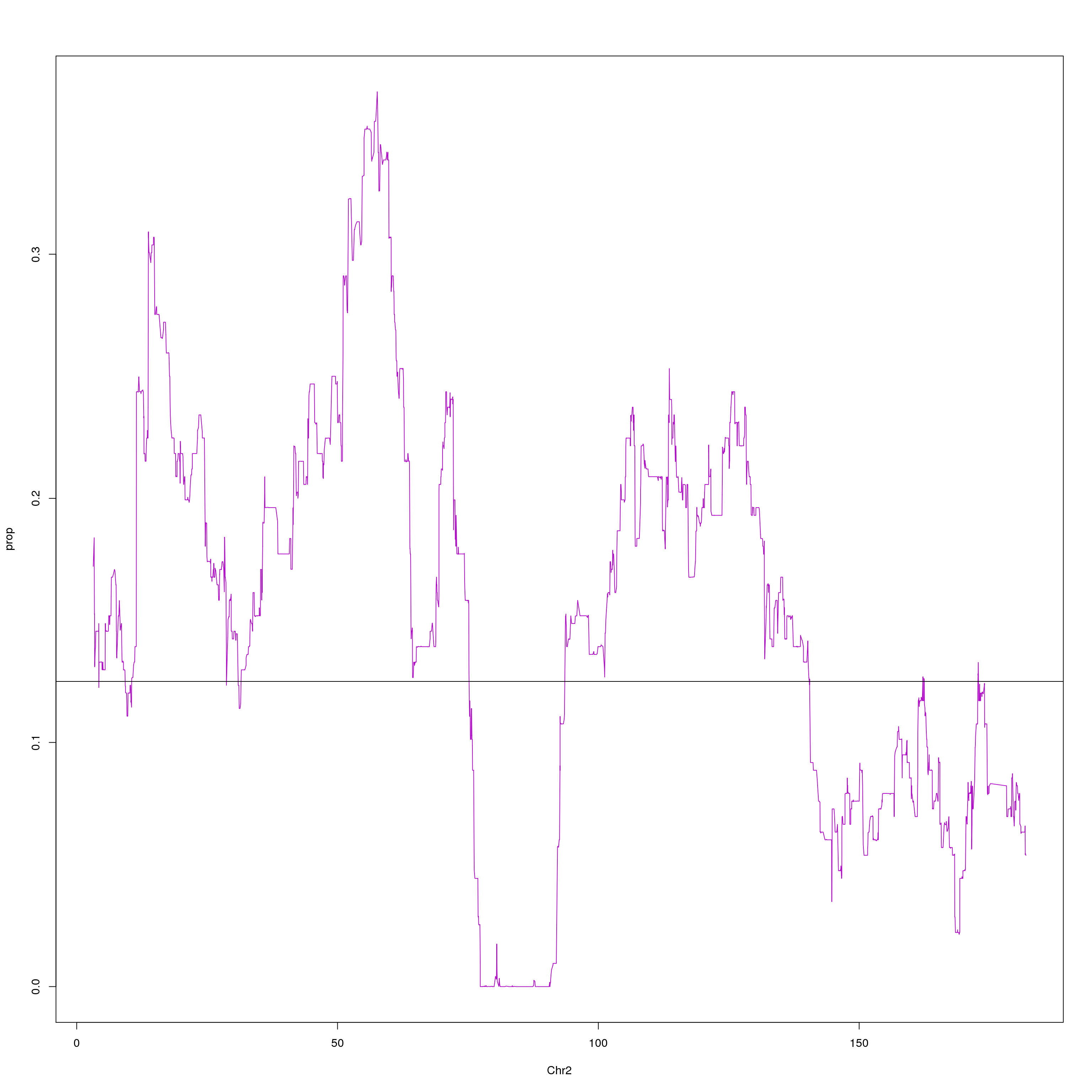
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |

| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
[1] "25"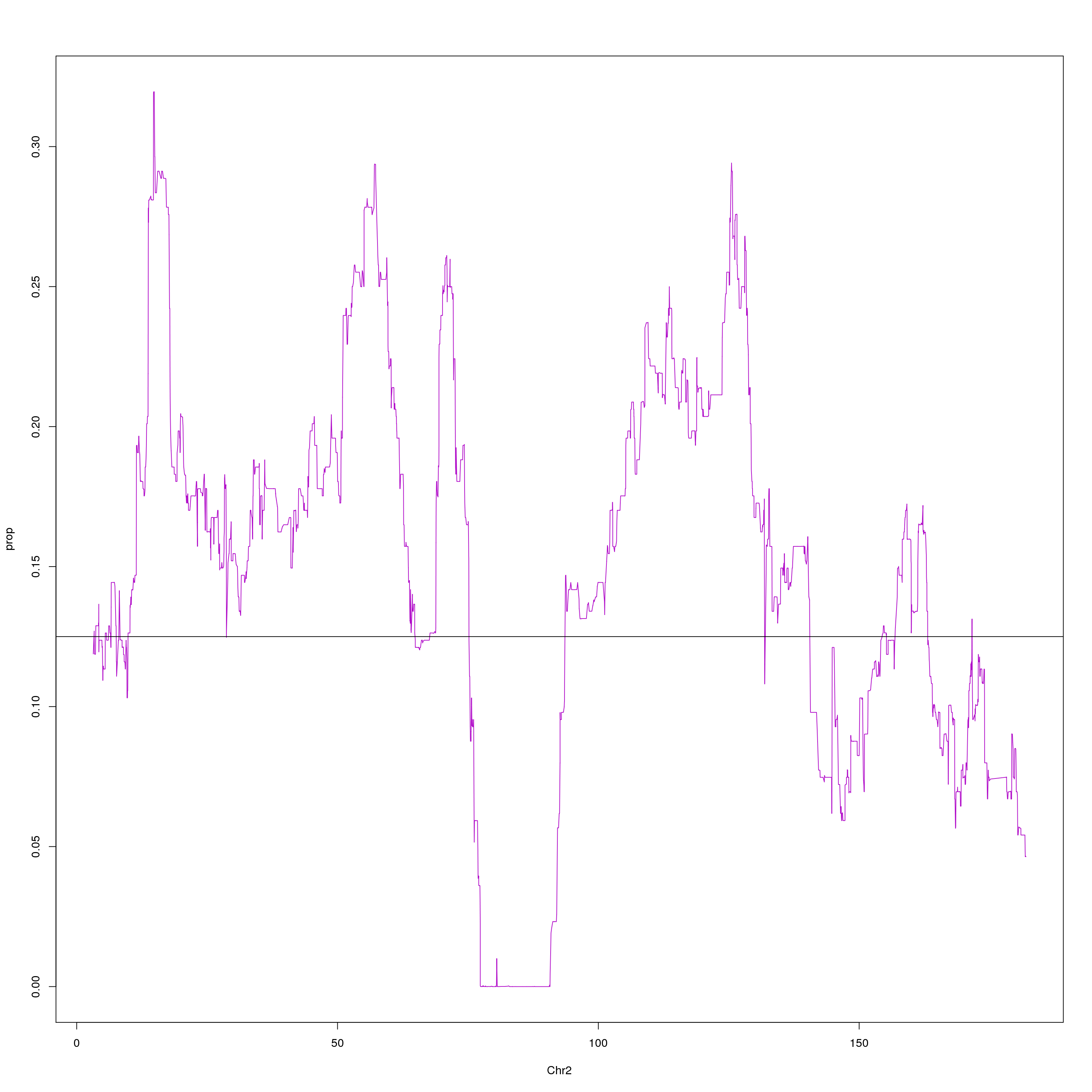
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |

| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
[1] "29"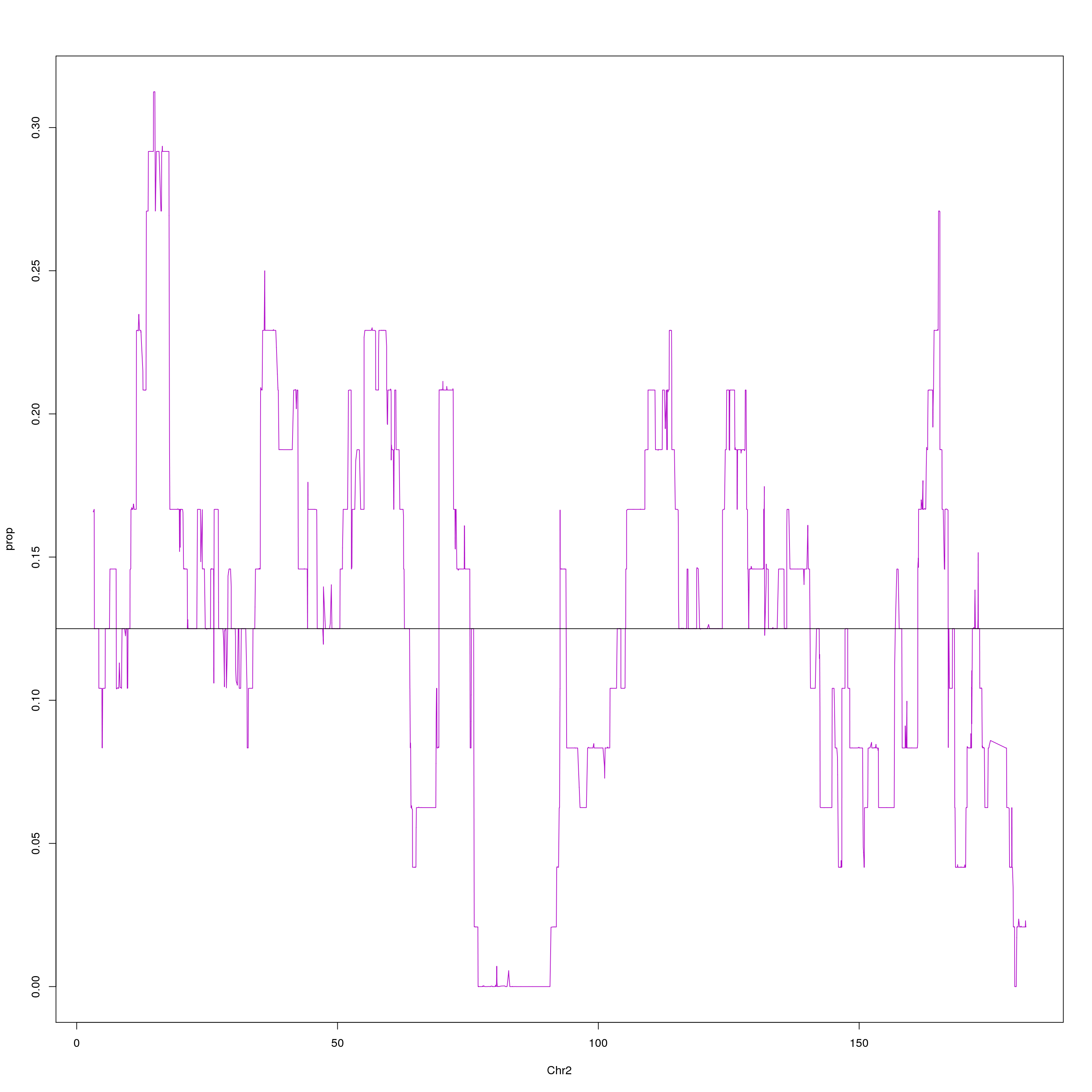
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |

| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
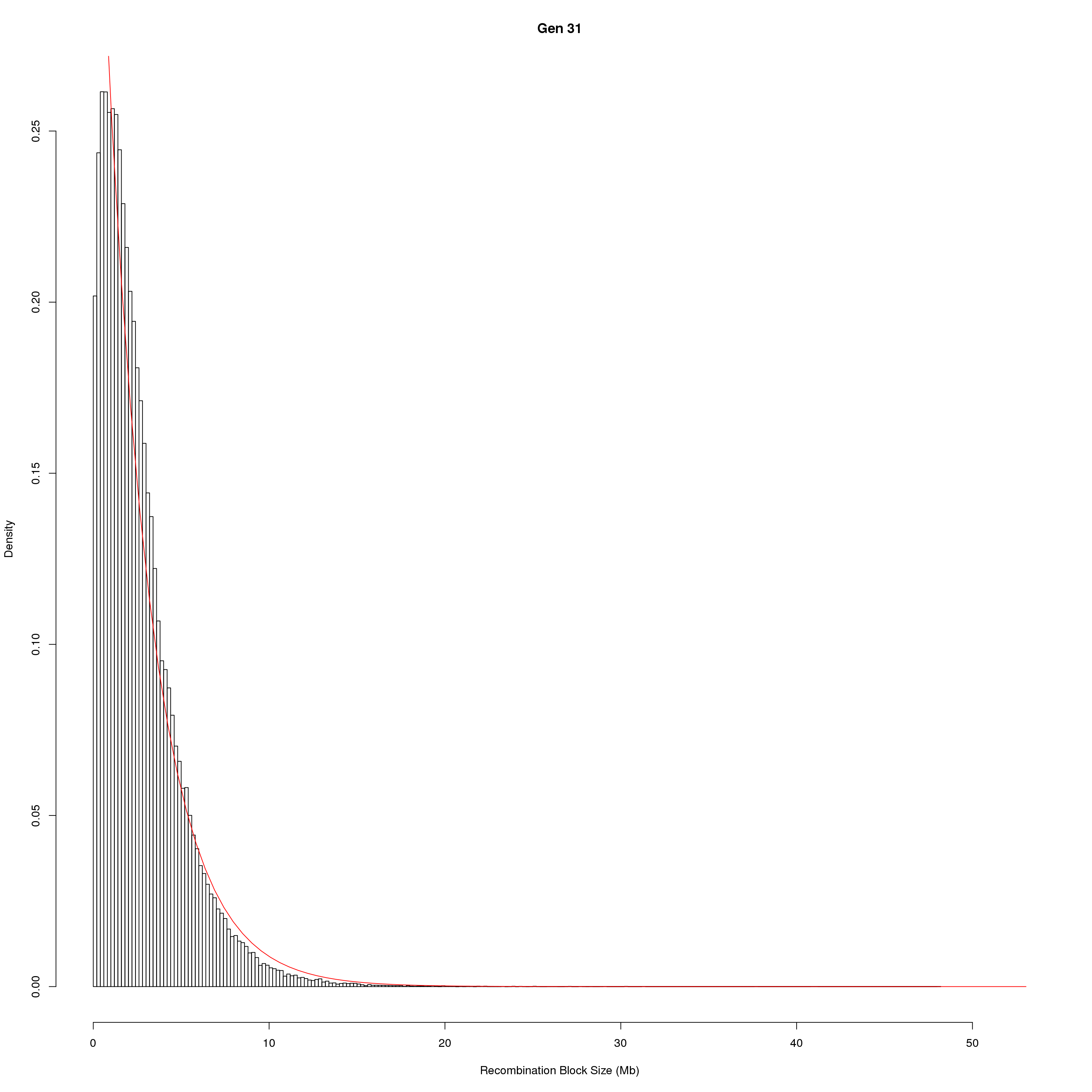
[1] "31"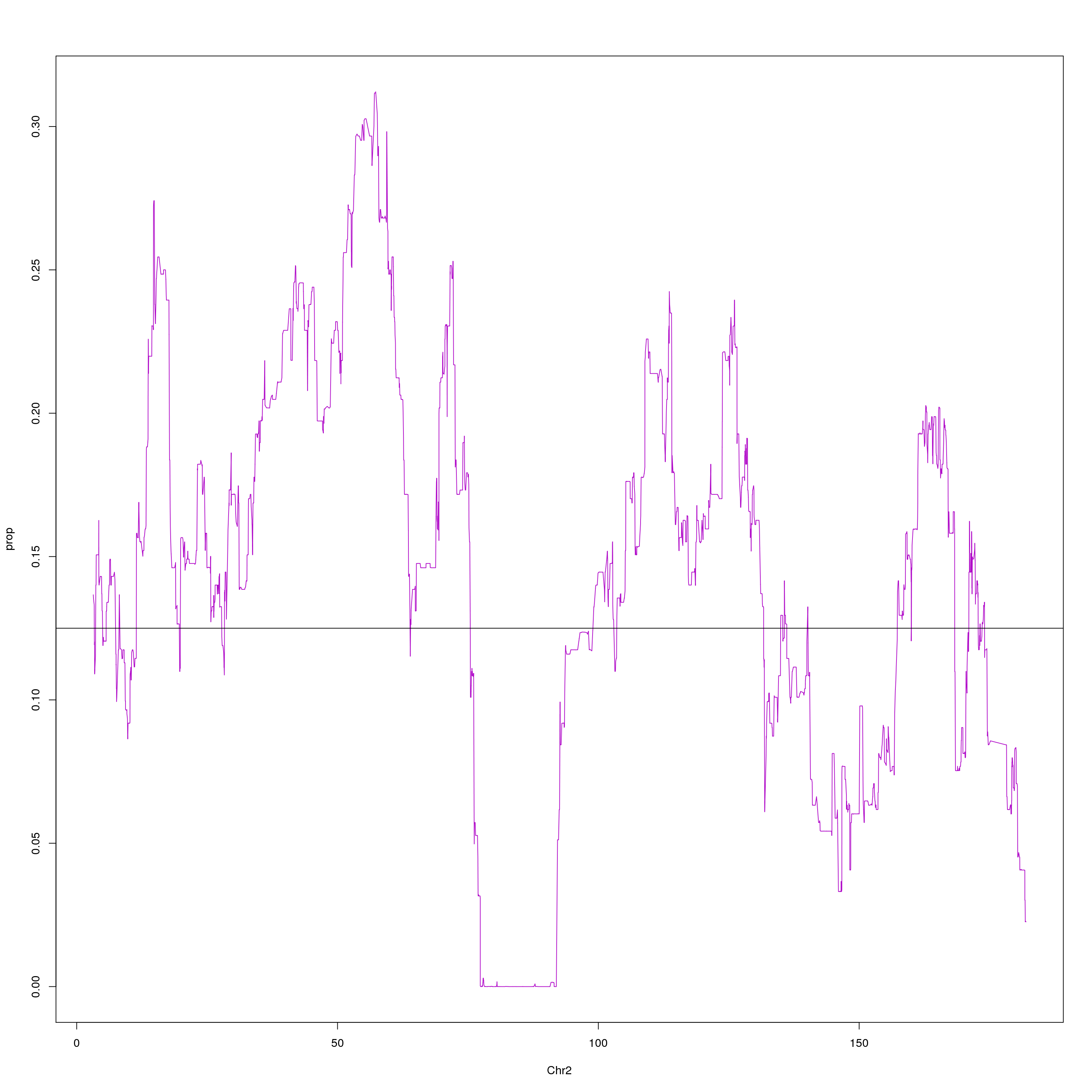
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
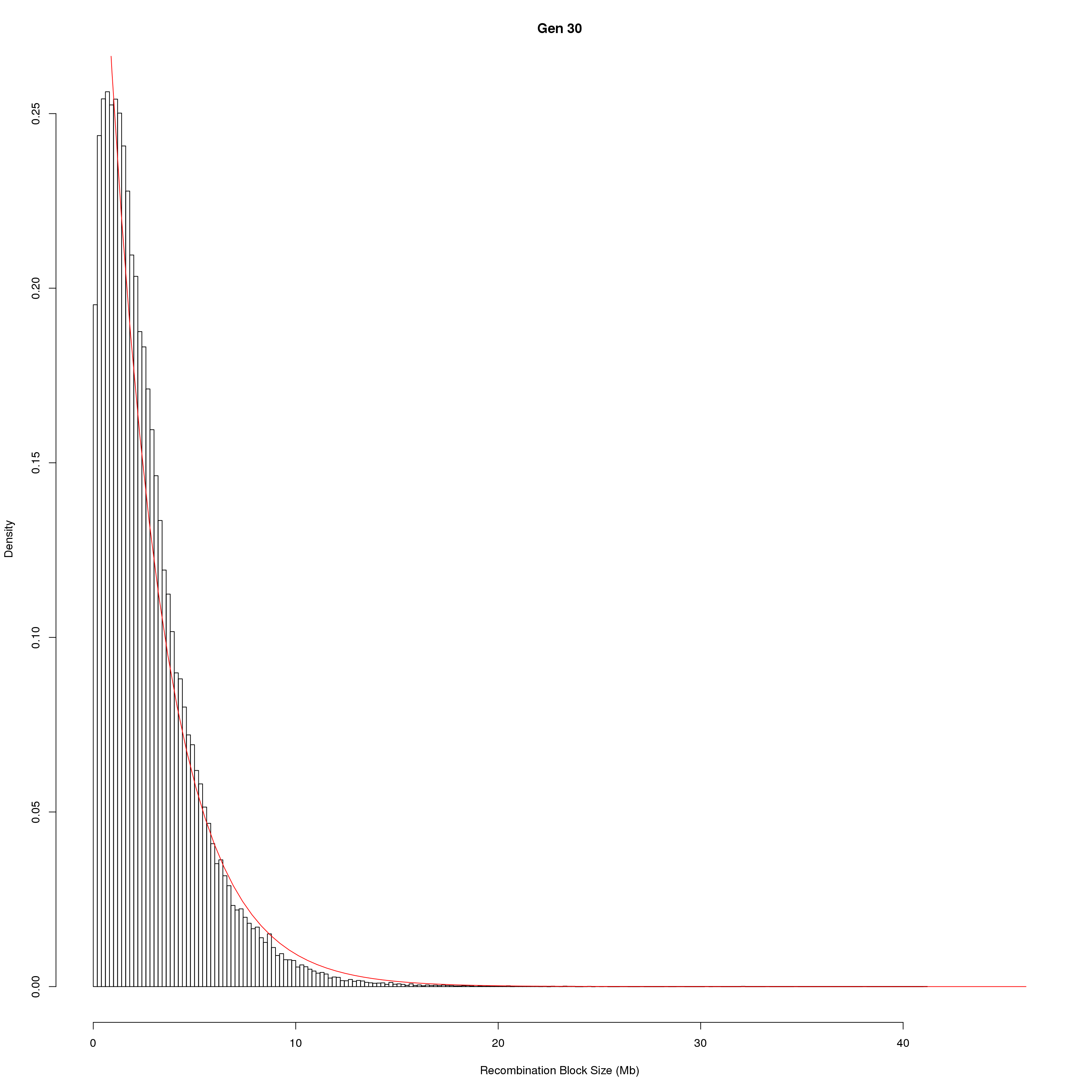
[1] "30"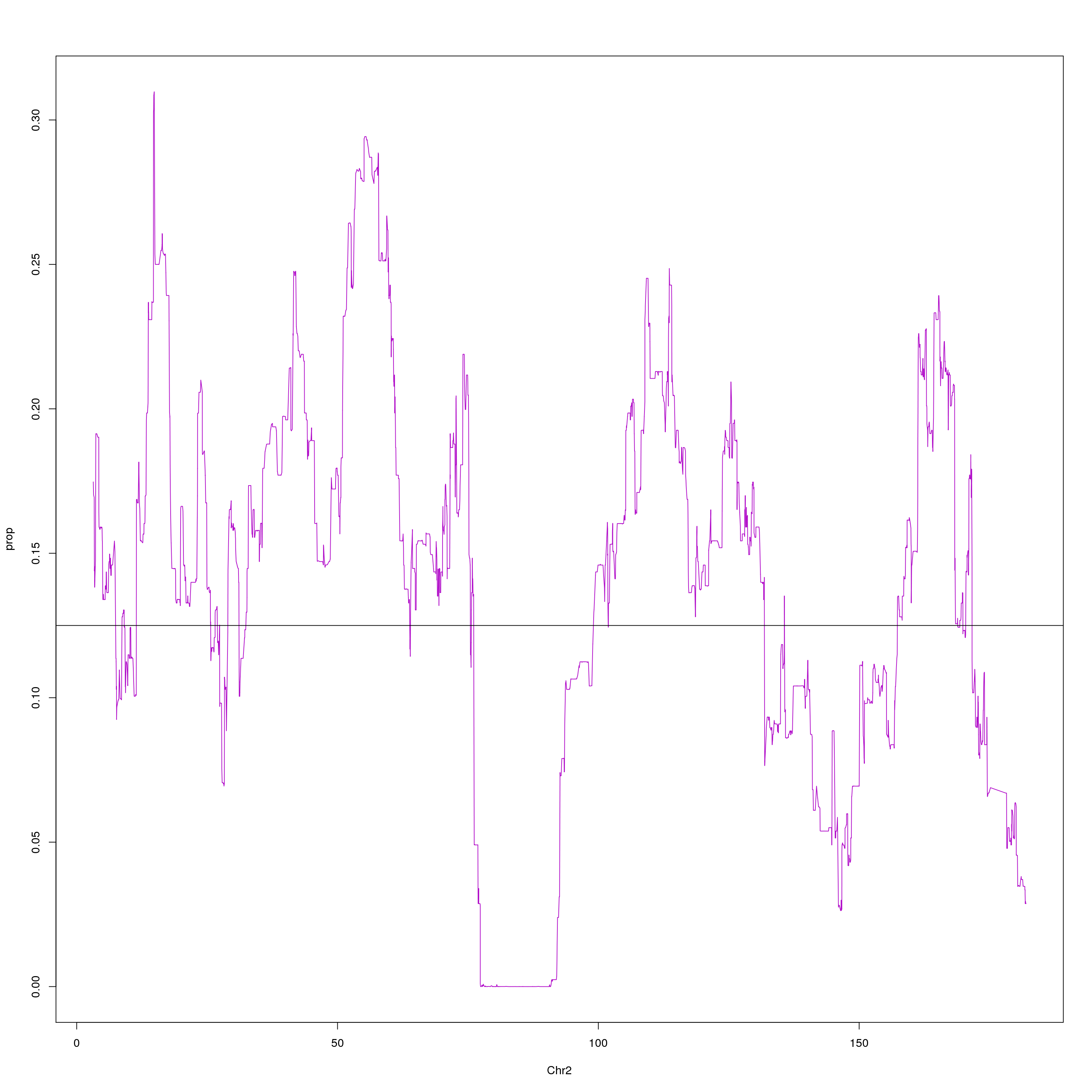
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
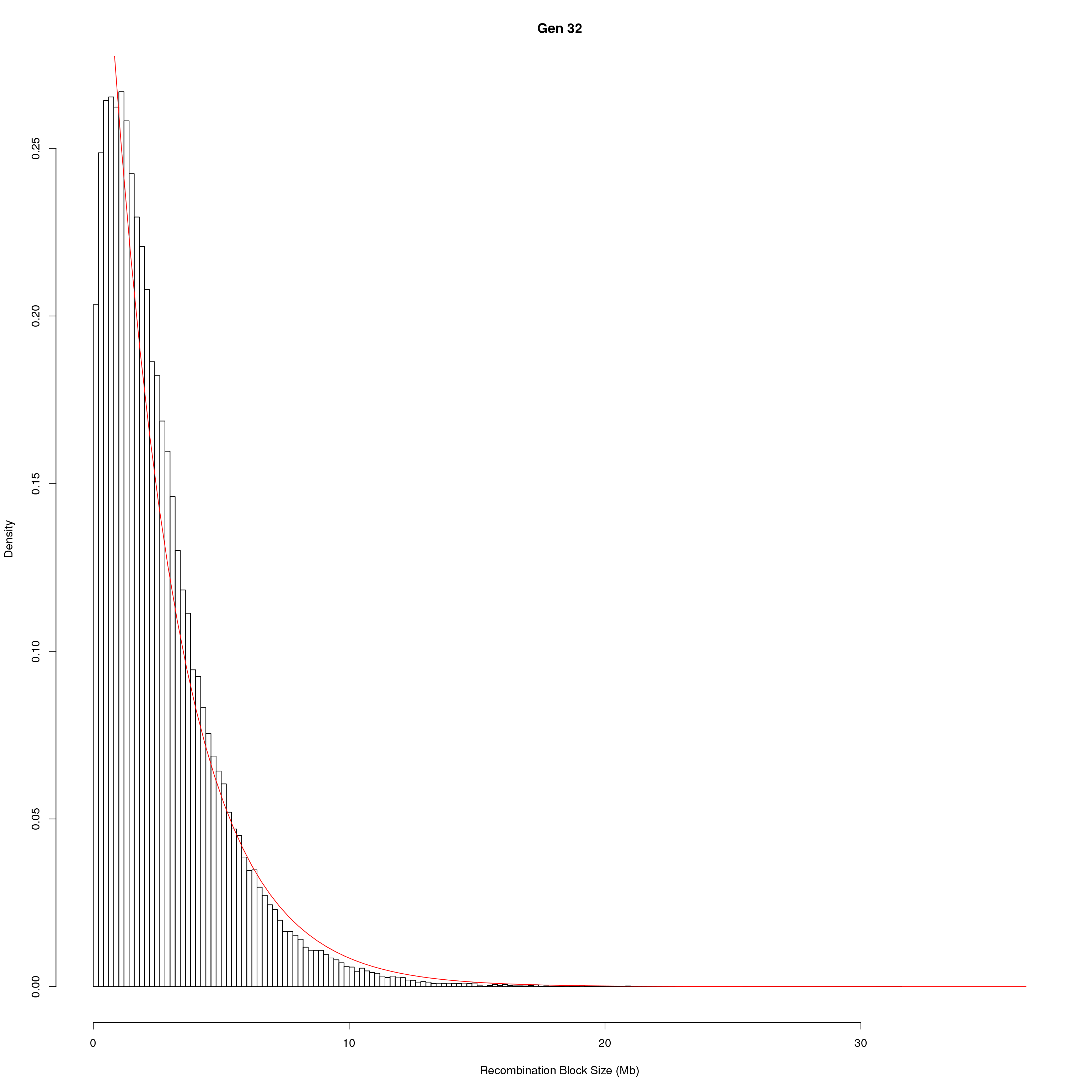
[1] "32"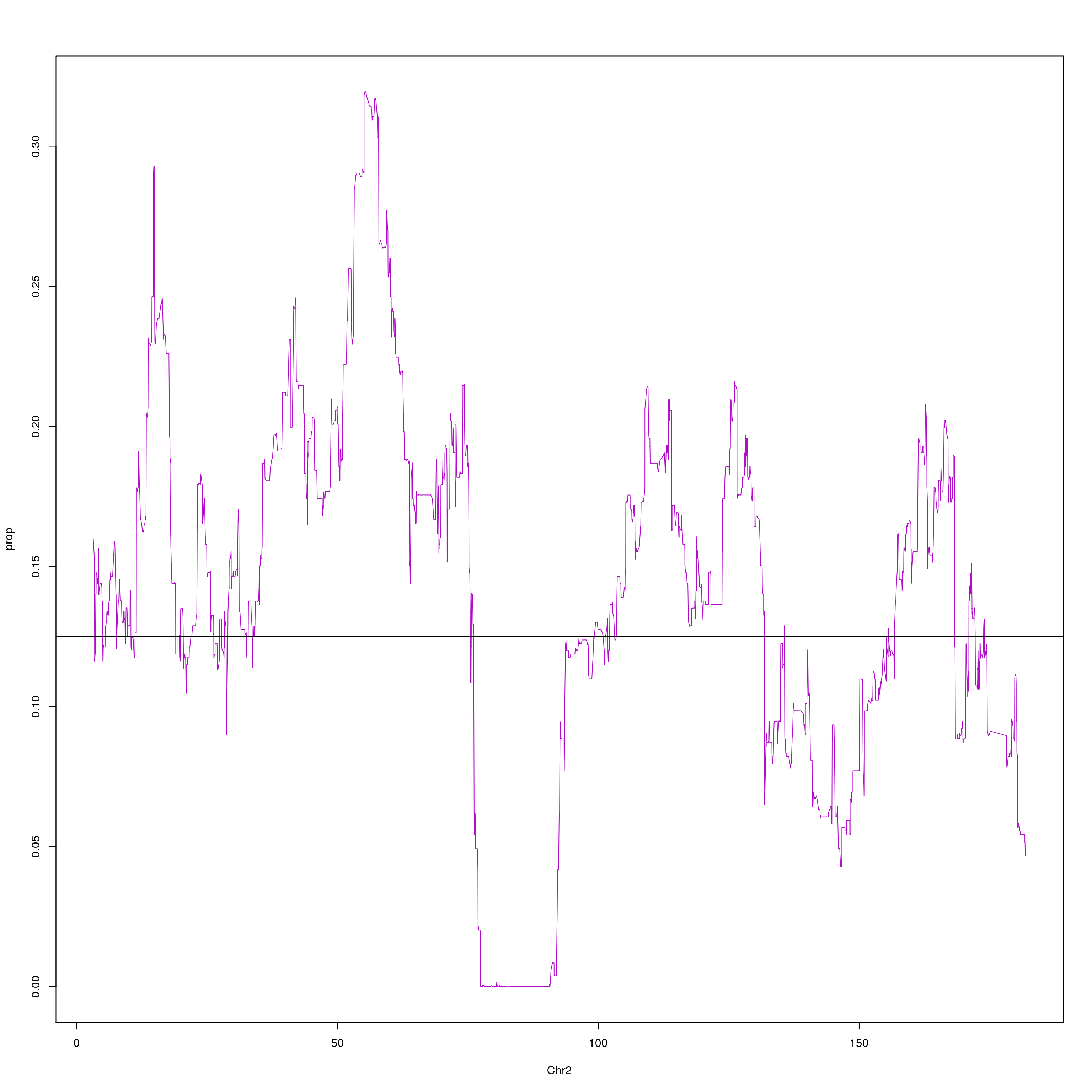
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
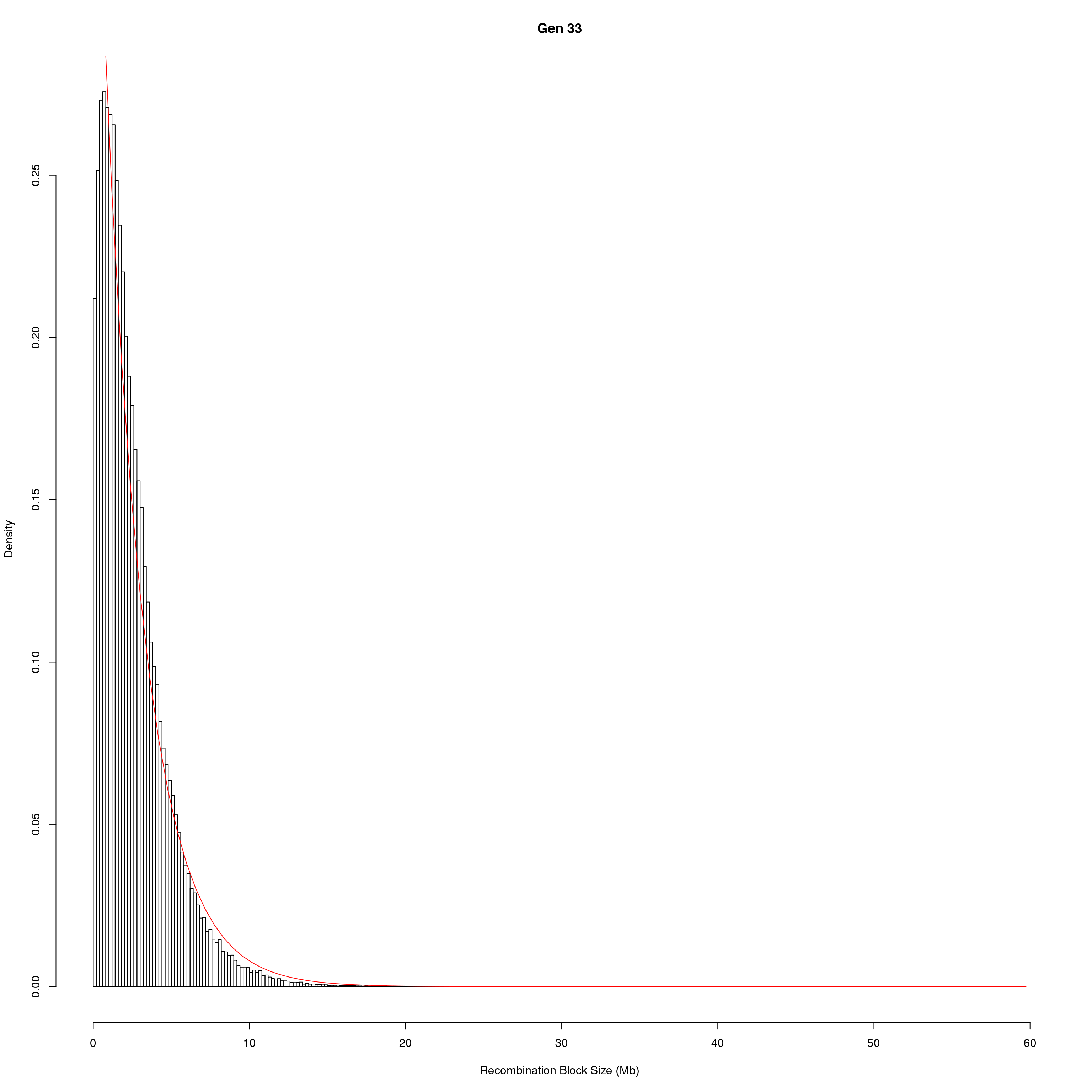
[1] "33"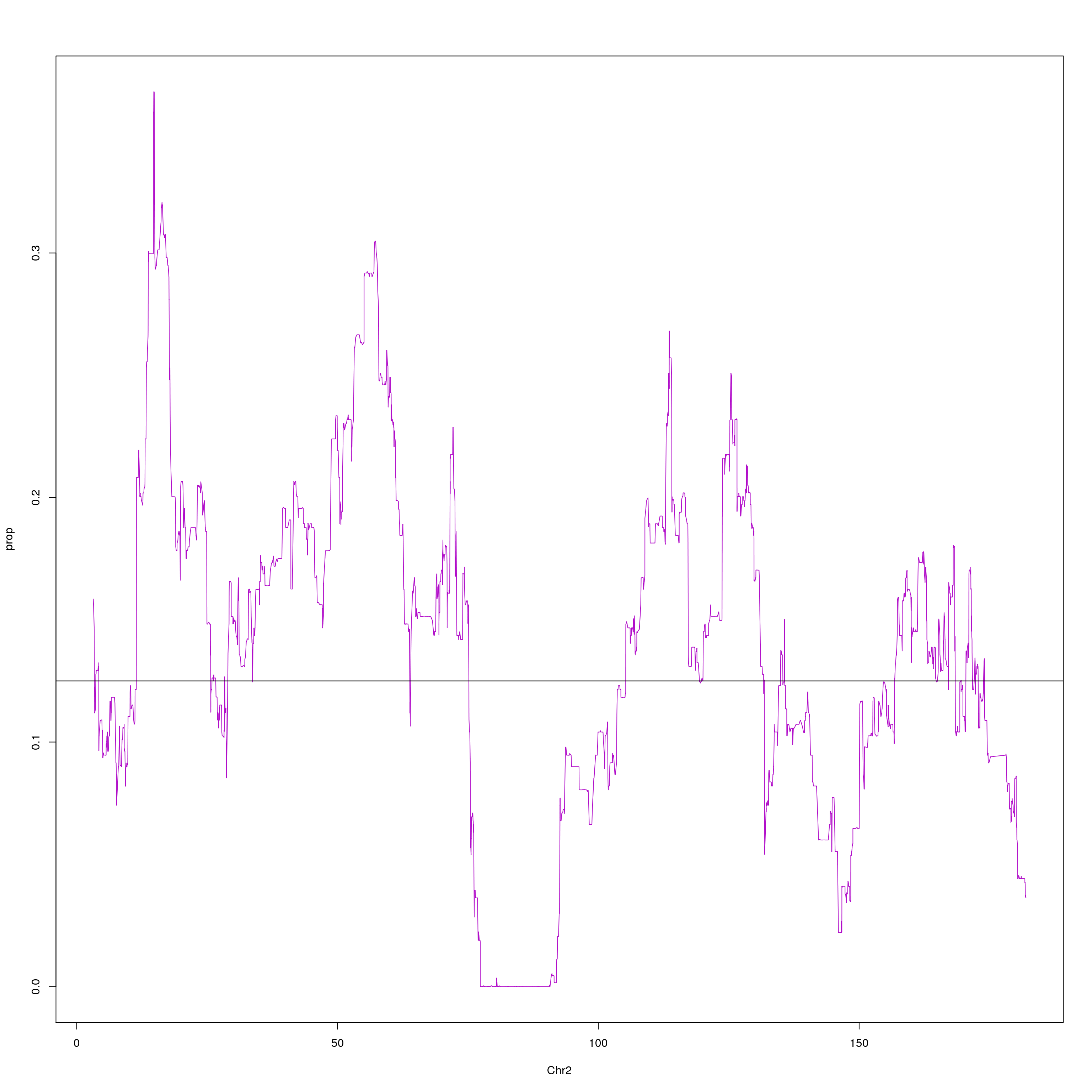
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
#locate xo pos
g <- maxmarg(pr, cores = 20)
pos <- locate_xo(g, gm_DO2437_qc$gmap, cores = 20)
pos_ind <- list()
for(i in ind_ids(gm_DO2437_qc)){
pos_ind[[i]] <- list()
for (j in c(1:19, "X")) {
pos_ind[[i]][[j]] <- diff(pos[[j]][[i]])
}
pos_ind[[i]] <- as.vector(unlist(pos_ind[[i]]))
}
# for each generation
#subset to each generation
pos_ind_gen <- list()
for(g in unique(gm_DO2437_qc$covar$ngen)){
#g = "21"
pos_ind_gen[[g]] <- as.vector(unlist(pos_ind[gm_DO2437_qc$covar[gm_DO2437_qc$covar$ngen == g,"id"]]))
}
save(pos_ind, pos_ind_gen, file = "data/Jackson_Lab_Bubier_MURGIGV01/recom_block_size.RData")plot Recombination Block Size
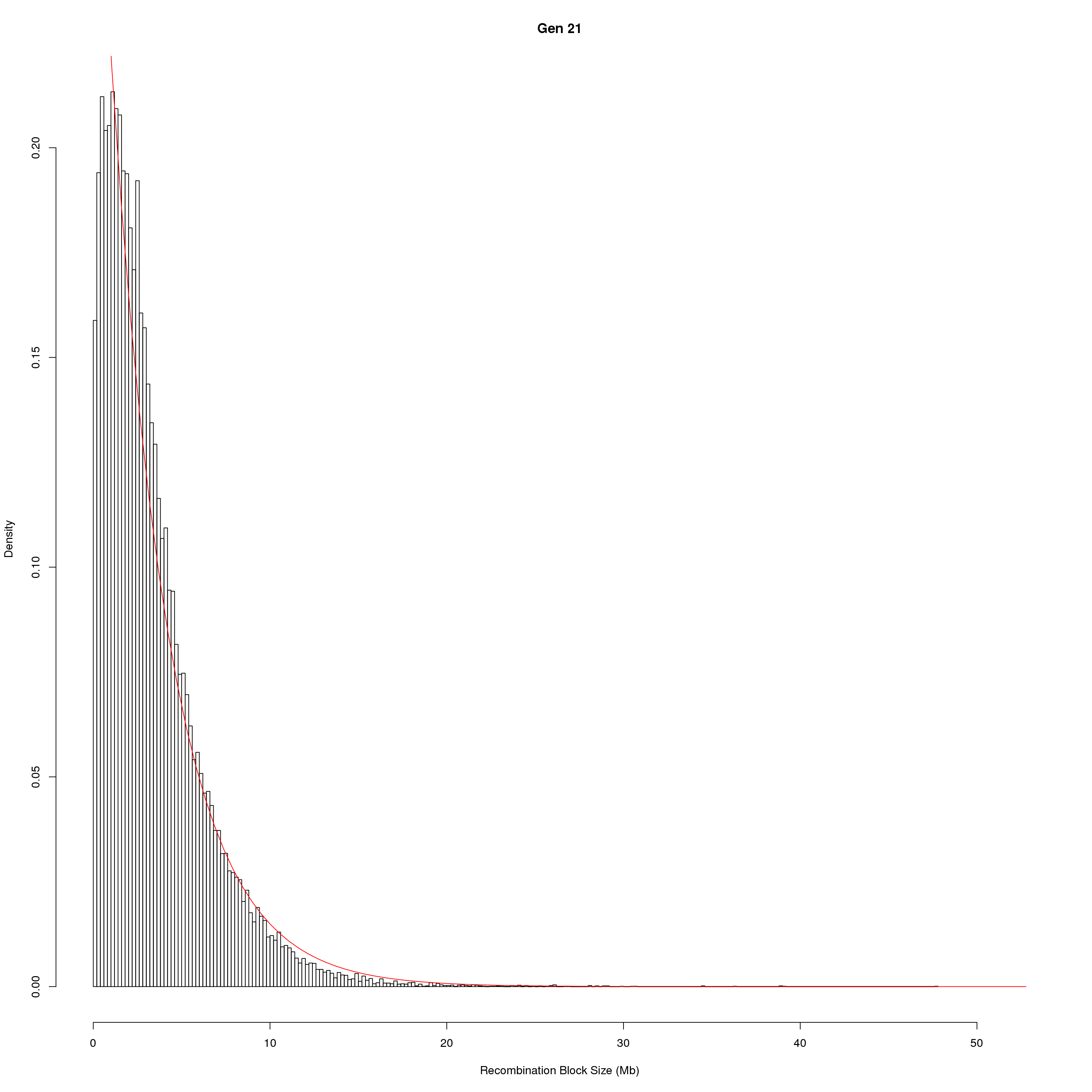
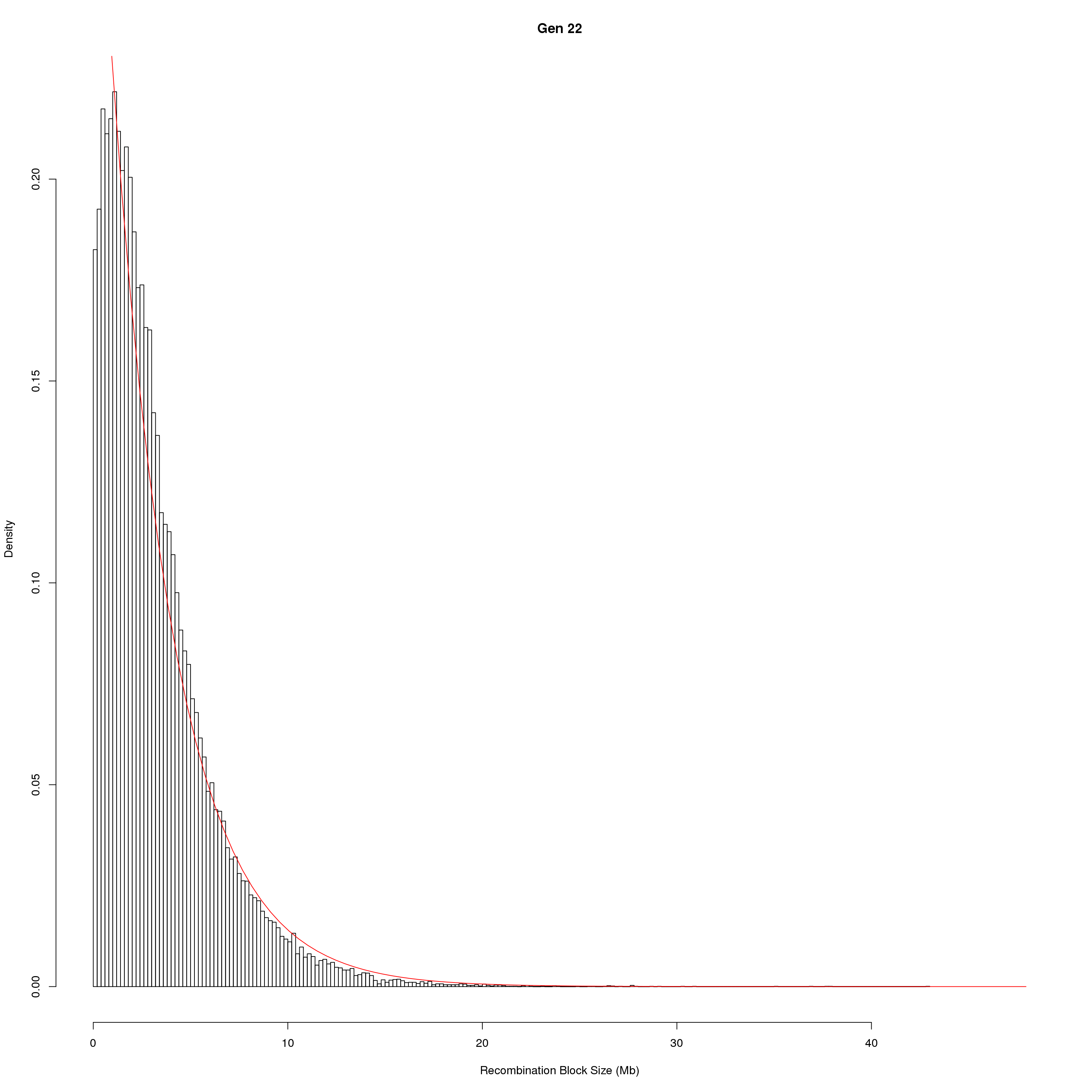
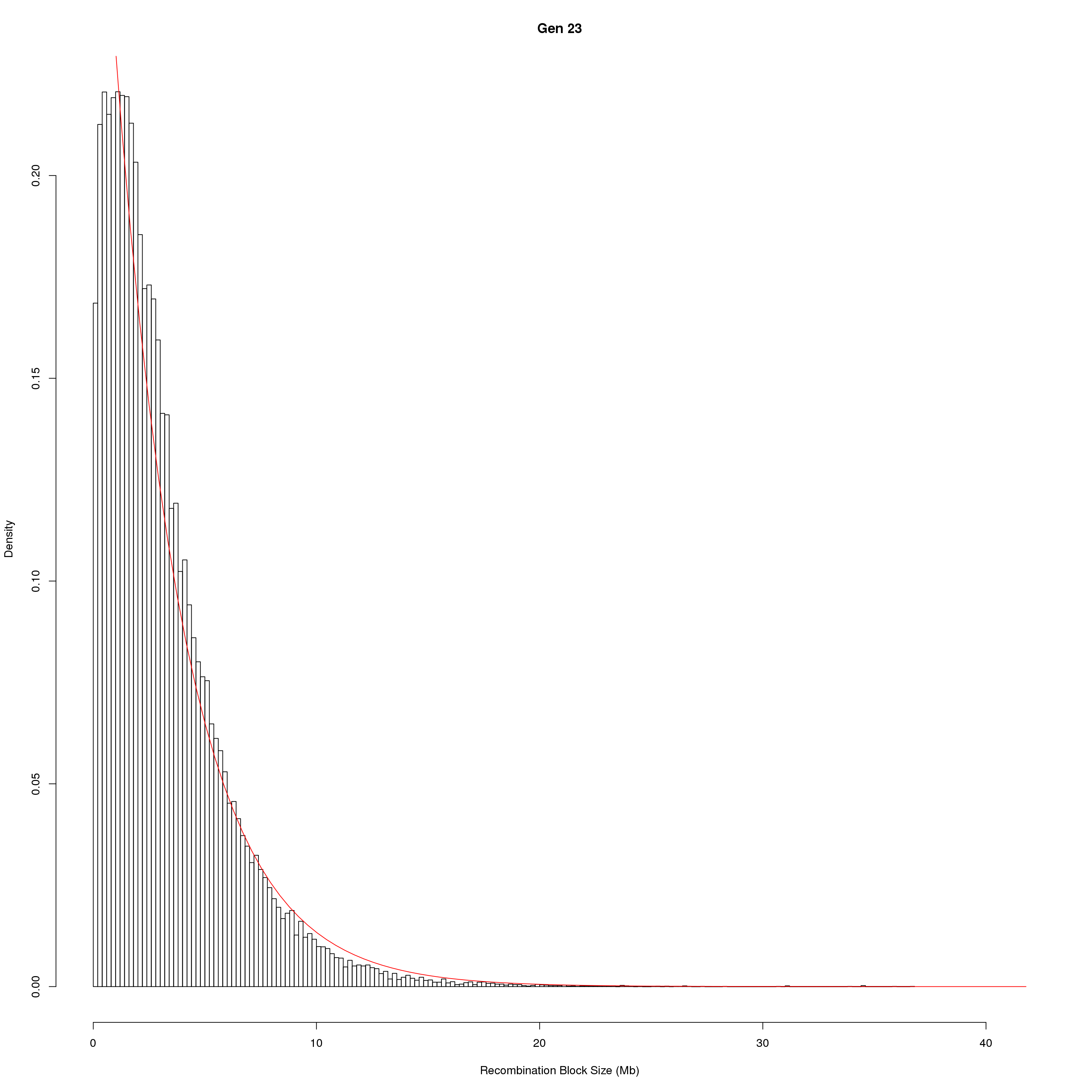
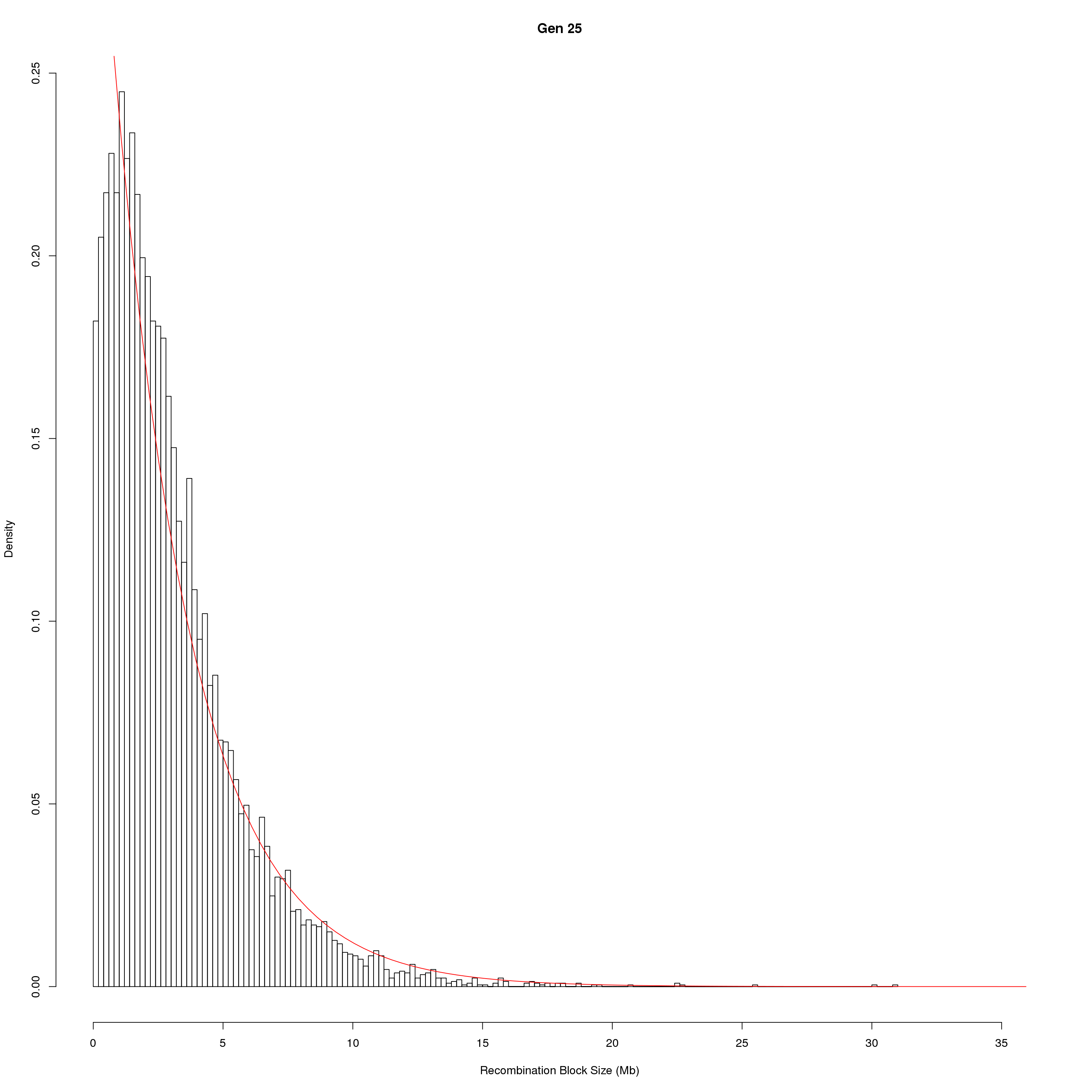
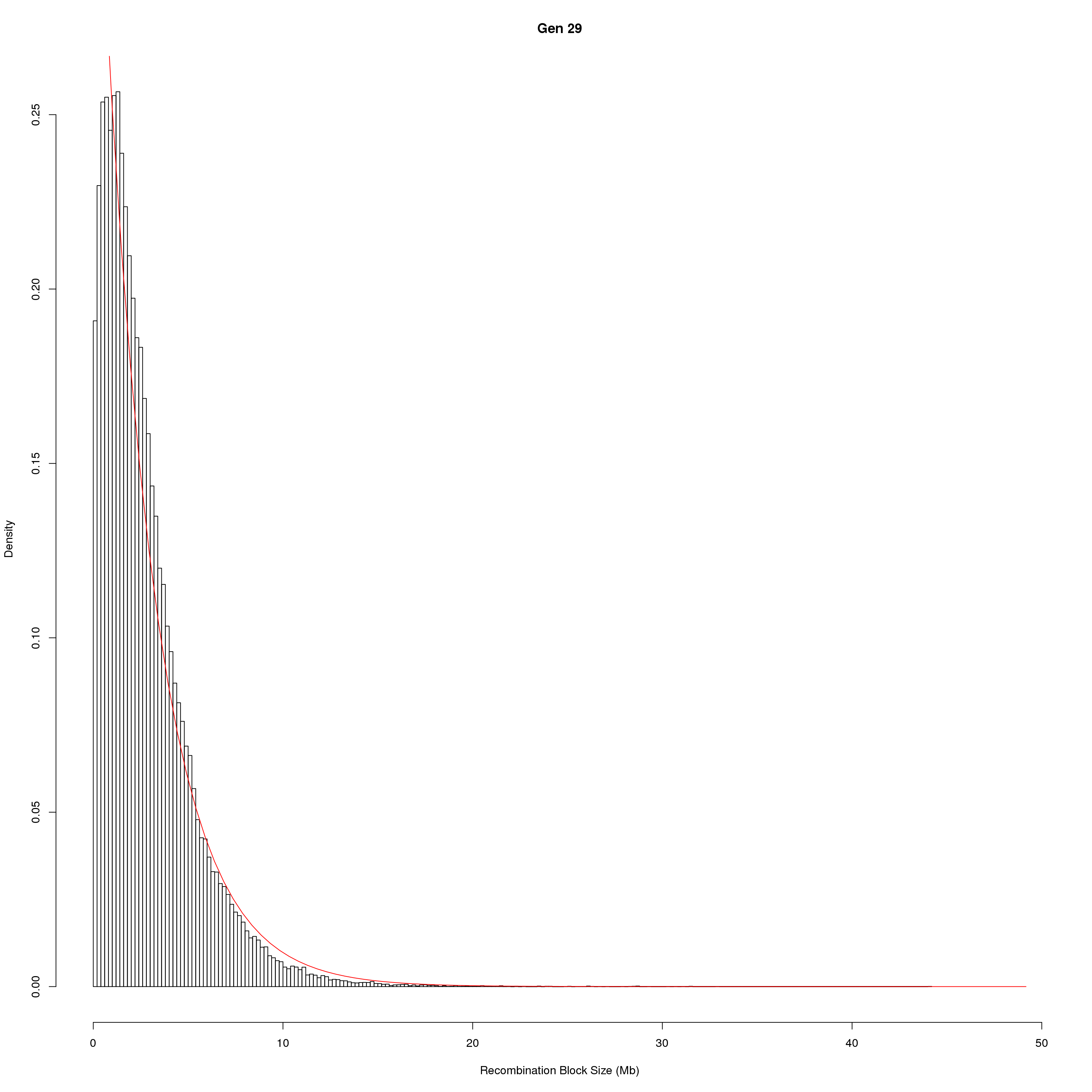
for(g in unique(gm_DO2437_qc$covar$ngen)){
#plot for recom block size
png(paste0("output/DO_recom_block_size_G", g, ".png"))
x <- pos_ind_gen[[g]][pos_ind_gen[[g]] != 0]
# estimate the parameters
fit1 <- fitdistr(x, "exponential")
# goodness of fit test
ks.test(x, "pexp", fit1$estimate) # p-value > 0.05 -> distribution not refused
# plot a graph
hist(x,
freq = FALSE,
breaks = 200,
xlim = c(0, 5+quantile(x, 1)),
#ylim = c(0,0.3),
xlab = "Recombination Block Size (Mb)",
main = paste0("Gen ", g))
curve(dexp(x, rate = fit1$estimate),
from = 0,
to = 5+quantile(x, 1),
col = "red",
add = TRUE)
dev.off()
}
for(g in unique(gm_DO2437_qc$covar$ngen)){
#plot for recom block size
#png(paste0("output/DO_recom_block_size_G", g, ".png"))
x <- pos_ind_gen[[g]][pos_ind_gen[[g]] != 0]
# estimate the parameters
fit1 <- fitdistr(x, "exponential")
# goodness of fit test
ks.test(x, "pexp", fit1$estimate) # p-value > 0.05 -> distribution not refused
# plot a graph
hist(x,
freq = FALSE,
breaks = 200,
xlim = c(0, 5+quantile(x, 1)),
#ylim = c(0,0.3),
xlab = "Recombination Block Size (Mb)",
main = paste0("Gen ", g))
curve(dexp(x, rate = fit1$estimate),
from = 0,
to = 5+quantile(x, 1),
col = "red",
add = TRUE)
#dev.off()
}
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
| Version | Author | Date |
|---|---|---|
| 0d00279 | xhyuo | 2019-09-23 |
plot for each generation
fp <- list()
#subset to each generation
for(g in unique(gm_DO2437_qc$covar$ngen)){
#g = "21"
apr.3d <- apr.3d.all[gm_DO2437_qc$covar[gm_DO2437_qc$covar$ngen == g,"id"],,]
#reformat
probs = reformat_probs(apr.3d)
gc()
# Summarize founder proportion by chromosome.
fp[[g]] = probs %>% group_by(chr, pos) %>%
summarize_all(mean) %>%
gather(founder, prop, 3:10)
fp[[g]]$founder = factor(fp[[g]]$founder, levels = names(CCcolors))
}
save(fp, file = "data/Jackson_Lab_Bubier_MURGIGV01/fp.RData")
# add gen
for(g in unique(gm_DO2437_qc$covar$ngen)){
#g = "21"
fp[[g]]$gen <- as.factor(g)
}
fp_data <- do.call(rbind.data.frame,fp)
p <- list()
for(c in unique(names(gm_DO2437_qc$geno))){
print(c)
fp_subdata <- fp_data[fp_data$chr == c,]
pp <- ggplot(data = fp_subdata,aes(pos, prop, group = gen, color = founder)) +
geom_line(aes(linetype=gen)) +
scale_linetype_manual(values=rep("solid",9)) +
geom_hline(yintercept=0.125, linetype="dashed", color = "black", size = 0.25) +
scale_color_manual(values = CCcolors) +
facet_grid(founder~.) +
labs(title = paste0("Chr ", c)) +
theme(legend.position='none')
p[[c]] <- ggplotly(pp, width = 1000, height = 1000)
#htmlwidgets::saveWidget(as_widget(p[[c]]), paste0("/projects/heh/csna_workflow/output/prop_across_generation_chr",c,".html"))
}[1] "1"
[1] "2"
[1] "3"
[1] "4"
[1] "5"
[1] "6"
[1] "7"
[1] "8"
[1] "9"
[1] "10"
[1] "11"
[1] "12"
[1] "13"
[1] "14"
[1] "15"
[1] "16"
[1] "17"
[1] "18"
[1] "19"
[1] "X"save(p, file = "output/prop_across_generation_chr_p.RData")
#as_widget(p[["1"]])
#as_widget(p[["2"]])
# as_widget(p[["3"]])
# as_widget(p[["4"]])
# as_widget(p[["5"]])
# as_widget(p[["6"]])
# as_widget(p[["7"]])
# as_widget(p[["8"]])
# as_widget(p[["9"]])
# as_widget(p[["10"]])
# as_widget(p[["11"]])
# as_widget(p[["12"]])
# as_widget(p[["13"]])
# as_widget(p[["14"]])
# as_widget(p[["15"]])
# as_widget(p[["16"]])
# as_widget(p[["17"]])
# as_widget(p[["18"]])
# as_widget(p[["19"]])
# as_widget(p[["X"]])
sessionInfo()R version 3.3.2 (2016-10-31)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS release 6.5 (Final)
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets base
other attached packages:
[1] MASS_7.3-50 vcd_1.4-4 plotly_4.9.0 forcats_0.4.0
[5] stringr_1.3.1 dplyr_0.8.3 purrr_0.3.2 readr_1.3.1
[9] tidyr_1.0.0 tibble_2.1.3 ggplot2_3.1.0 tidyverse_1.2.1
[13] abind_1.4-5 qtl2_0.18
loaded via a namespace (and not attached):
[1] httr_1.4.0 bit64_0.9-7 jsonlite_1.6
[4] viridisLite_0.3.0 modelr_0.1.4 shiny_1.3.2
[7] assertthat_0.2.1 highr_0.6 blob_1.1.1
[10] cellranger_1.1.0 yaml_2.2.0 pillar_1.3.1
[13] RSQLite_2.1.1 backports_1.1.2 lattice_0.20-35
[16] glue_1.3.1 digest_0.6.18 promises_1.0.1
[19] rvest_0.3.2 colorspace_1.4-0 htmltools_0.3.6
[22] httpuv_1.5.2 plyr_1.8.4 pkgconfig_2.0.1
[25] broom_0.5.2 haven_2.1.1 xtable_1.8-2
[28] scales_1.0.0 whisker_0.3-2 later_0.8.0
[31] git2r_0.23.0 generics_0.0.2 ellipsis_0.2.0.1
[34] withr_2.1.2 lazyeval_0.2.1 cli_1.1.0
[37] mime_0.6 magrittr_1.5 crayon_1.3.4
[40] readxl_1.3.1 memoise_1.1.0 evaluate_0.10
[43] methods_3.3.2 fs_1.2.6 nlme_3.1-128
[46] xml2_1.2.1 tools_3.3.2 data.table_1.11.4
[49] hms_0.5.1 lifecycle_0.1.0 munsell_0.5.0
[52] rlang_0.4.0 rstudioapi_0.10 htmlwidgets_1.3
[55] crosstalk_1.0.0 labeling_0.3 rmarkdown_1.11
[58] gtable_0.2.0 DBI_1.0.0 reshape2_1.4.3
[61] R6_2.4.0 zoo_1.8-6 lubridate_1.7.4
[64] knitr_1.20 bit_1.1-14 zeallot_0.1.0
[67] workflowr_1.4.0 rprojroot_1.3-2 stringi_1.2.4
[70] parallel_3.3.2 Rcpp_1.0.2 vctrs_0.2.0
[73] tidyselect_0.2.5 lmtest_0.9-36