Quality control for BAL pool: capture1
Anson Wong
2026-04-23
Last updated: 2026-04-23
Checks: 7 0
Knit directory: cf-eti-bal-scrna/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20260422) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 64553ed. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: ._.DS_Store
Ignored: code/._bal_RecM.integration_clustering.R
Ignored: data/._README.md
Ignored: data/AM.markers.csv
Ignored: data/DE/AM-CCL/CFB_vs_C/
Ignored: data/DE/AM-CCL/CFN_vs_C/
Ignored: data/DE/AM-CCL/CFN_vs_CFB/GO_BP/
Ignored: data/DE/AM-CCL/CFN_vs_CFB/GO_CC/
Ignored: data/DE/AM-CCL/CFN_vs_CFB/GO_MF/
Ignored: data/DE/AM-CCL/CFN_vs_CFB/Reactome/
Ignored: data/DE/AM-CCL/CFN_vs_CFB/WikiPathway/
Ignored: data/DE/Prolif M/CFB_vs_C/GO_BP/
Ignored: data/DE/Prolif M/CFB_vs_C/GO_CC/
Ignored: data/DE/Prolif M/CFB_vs_C/GO_MF/
Ignored: data/DE/Prolif M/CFB_vs_C/Reactome/
Ignored: data/DE/Prolif M/CFB_vs_C/WikiPathway/
Ignored: data/DE/Prolif M/CFN_vs_C/
Ignored: data/DE/ruv/
Ignored: data/Fig3A.stat.csv
Ignored: data/Li.stat.csv
Ignored: data/Liao.stat.csv
Ignored: data/Major.markers.csv
Ignored: data/Morrell.stat.csv
Ignored: data/Present.stat.csv
Ignored: data/RecM.markers.csv
Ignored: data/SCEs
Ignored: data/Wendisch.stat.csv
Ignored: data/cellbender
Ignored: data/cytokine
Ignored: data/plots
Ignored: data/sample_sheets
Ignored: data/vireo
Ignored: output/plots/combined/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/02.qc_capture1.Rmd) and
HTML (docs/02.qc_capture1.html) files. If you’ve configured
a remote Git repository (see ?wflow_git_remote), click on
the hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | e9cb32b | tcwong1994 | 2026-04-23 | Commit all analysis Rmd sources |
| html | d219a80 | tcwong1994 | 2026-04-23 | Add demux and QC reports (batches 1-4) |
| Rmd | 29ed4f7 | tcwong1994 | 2026-04-22 | Initial import of workflowr site content and analysis files |
| html | 29ed4f7 | tcwong1994 | 2026-04-22 | Initial import of workflowr site content and analysis files |
knitr::opts_chunk$set(warning = FALSE, message = FALSE)
xaringanExtra::use_panelset()1 Load packages
suppressPackageStartupMessages({
library(DropletUtils)
library(here)
library(ggplot2)
library(Seurat)
library(cowplot)
library(patchwork)
library(scater)
library(dplyr)
library(forcats)
library(janitor)
library(stringr)
library(AnnotationHub)
library(ensembldb)
library(msigdbr)
library(Homo.sapiens)
})
set.seed(1990)2 Set names for this pool
# Specify batch name
batch_name <- "capture1"
num_of_captures <- 4
# Specify capture name
capture_names <- c(paste0(batch_name, "-",1:num_of_captures))
capture_names <- setNames(capture_names, capture_names)
# Assign sample ID to HTO ID
# Manually list sample names matching HTO id (in alphabetical order)
samples <- c("M1C188", #HTO3
"M1C188B", #HTO6
"M1C160(1)", #HTO7
"M1C160F", #HTO8
"M1C170C", #HTO10
"M1C170D", #HTO12
"M1C176", #HTO13
"M1C176C", #HTO14
"M1N092", #HTO15
"M1N075" #HTO16
)3 Filter out ambiguous droplets and assign droplets to samples
# read demultiplexing outputs
sce <- readRDS(here("data",
"SCEs",
"demux",
paste0(batch_name,".cellbender.demux.SCE.rds")))
# number of droplets before removal
dim(sce)[1] 36601 143049# define non-longitudinal samples
nl.samples <- c("M1N092", "M1N075")
l.samples <- setdiff(samples, nl.samples)
# Step 1. remove droplets with unmatched genetic and best HTO classification
# in non-longitudinal samples
unmatched <- sce$sampleID.genetics %in% nl.samples &
(sce$sampleID.HTO != sce$sampleID.genetics & !(sce$sampleID.HTO %in% c("Doublet","Negative")))
sce <- sce[,!unmatched]
dim(sce)[1] 36601 142893# Step 2. remove droplets classified as "Doublet" and
# that are not longitudinal samples
genetic.doublets <- sce$sampleID.genetics == "Doublet" &
!(sce$sampleID.HTO %in% l.samples)
sce <- sce[,!genetic.doublets]
dim(sce)[1] 36601 128747# Step 3. remove droplets classified as "Negative" by genetics and
# "Doublet" or "Negative" by HTO
negatives <- sce$sampleID.genetics == "Negative" &
sce$sampleID.HTO %in% c("Doublet","Negative")
sce <- sce[,!negatives]
dim(sce)[1] 36601 116665# Step 4. remove droplets with posterior counts of zero called by CellBender
# see: https://github.com/broadinstitute/CellBender/issues/111
sce <- sce[, colSums(counts(sce)) != 0]
# number of droplets after removing droplets with zero counts
dim(sce)[1] 36601 115132# assign HTO result to longitudinal samples,
# genetics results to non-longitudinal samples
sampleID <- setNames(factor(case_when(
sce$sampleID.HTO %in% l.samples ~ sce$sampleID.HTO,
sce$sampleID.genetics == "Negative" ~ sce$sampleID.HTO,
TRUE ~ sce$sampleID.genetics),levels = samples),
colnames(sce))
sce$sampleID <- sampleID4 Visualize results after filtering
Double-check the figure of cell proportion after filtering. If the assignment is correct, the following should be observed: - genetic negatives should comprise HTO ID only without “Doublet” and “Negative” - genetic doublet should exist and comprise HTO ID from longitudinal samples only - non-longitudinal samples should comprise the best HTO, “Doublet”, or “Negative”
# number of droplets assigned by HTO method
p1 <- ggcells(sce) +
geom_bar(aes(x = sampleID.HTO, fill = sampleID.HTO)) +
geom_text(stat='count', aes(x = sampleID.HTO, label=..count..), hjust=1, size=2) +
coord_flip() +
ggtitle("By HTO") +
ylab("Number of droplets") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,40000,8000),limits=c(0,40000)) +
scale_fill_manual(values = sce$colours$sampleID.HTO_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=guide_legend(title="sampleID"))
p1.facet <- ggcells(sce) +
geom_bar(aes(x = sampleID.HTO, fill = sampleID.HTO)) +
geom_text(stat='count', aes(x = sampleID.HTO, label=..count..),
hjust=1, size=2) +
#ggtitle("By HTO") +
ylab("Number of droplets") +
facet_grid(~Capture, scales = "fixed", space = "fixed") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,8000,2000), limits = c(0,10000)) +
scale_fill_manual(values = sce$colours$sampleID.HTO_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=guide_legend(title="sampleID")) +
coord_flip()
# number of droplets assigned by genetic method
p2 <- ggcells(sce) +
geom_bar(aes(x = sampleID.genetics, fill = sampleID.genetics)) +
geom_text(stat='count', aes(x = sampleID.genetics, label=..count..), hjust=1, size=2) +
coord_flip() +
ggtitle("By genetics") +
ylab("Number of droplets") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,40000,8000), limits = c(0,40000)) +
scale_fill_manual(values = sce$colours$sampleID.genetics_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=FALSE)
p2.facet <- ggcells(sce) +
geom_bar(aes(x = sampleID.genetics, fill = sampleID.genetics)) +
geom_text(stat='count', aes(x = sampleID.genetics, label=..count..),
hjust=1, size=2) +
ylab("Number of droplets") +
facet_grid(~Capture, scales = "fixed", space = "fixed") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,8000,2000), limits = c(0,10000)) +
scale_fill_manual(values = sce$colours$sampleID.genetics_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=guide_legend(title="sampleID")) +
coord_flip()
(p1+p1.facet+plot_layout(width=c(1,2))) /
(p2+p2.facet+plot_layout(width=c(1,2))) +
plot_layout(guides="collect")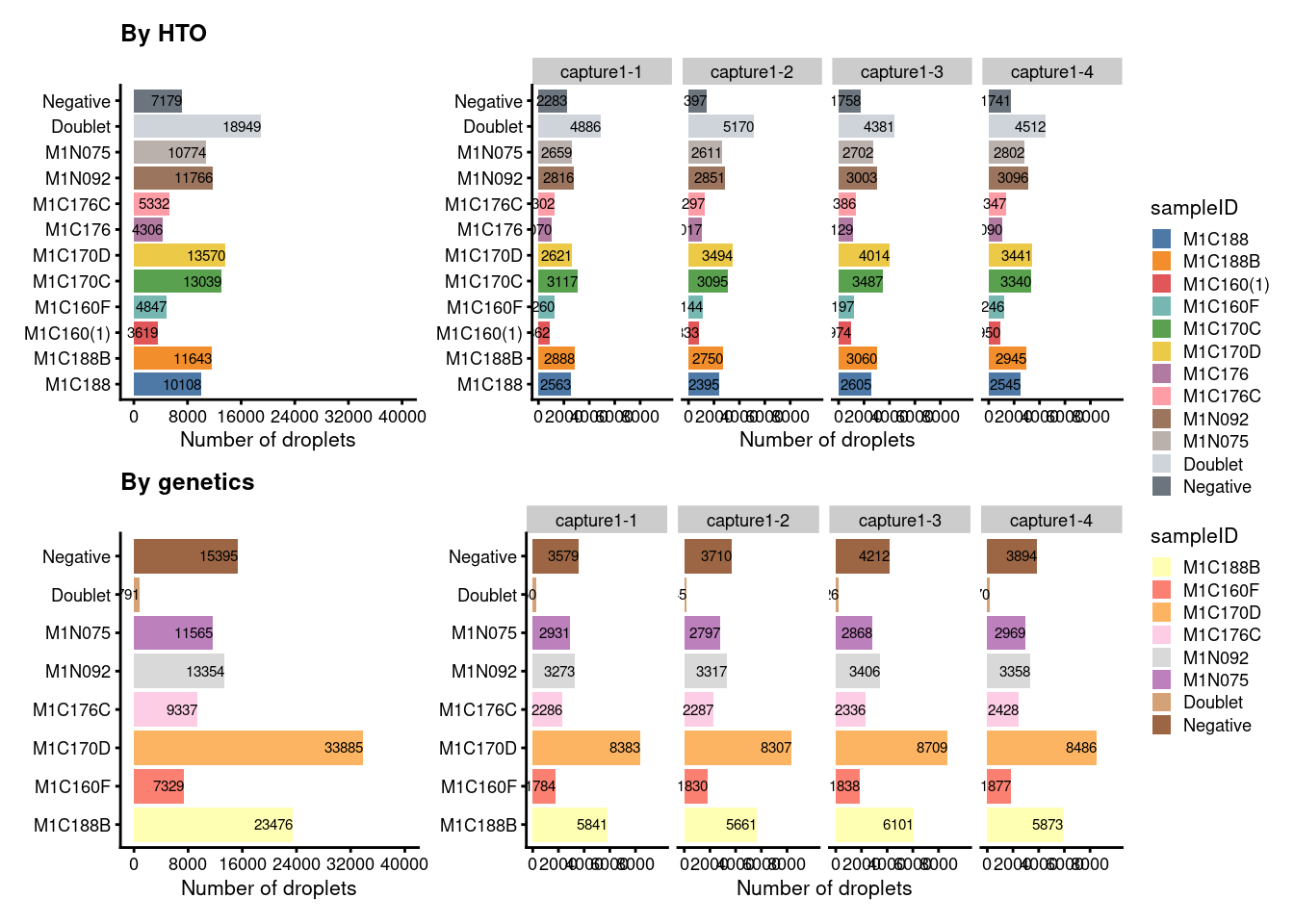
# proportion of genetically assigned droplets in each HTO
p3 <- ggcells(sce) +
geom_bar(
aes(x = sampleID.HTO, fill = sampleID.genetics),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
theme_cowplot(font_size = 8) +
scale_fill_manual(values = sce$colours$sampleID.genetics_colours)
# proportion of HTO assigned droplets in each genetic donor
p4 <- ggcells(sce) +
geom_bar(
aes(x = sampleID.genetics, fill = sampleID.HTO),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
theme_cowplot(font_size = 8) +
scale_fill_manual(values = sce$colours$sampleID.HTO_colours)
((p3 + ggtitle("By HTO")) +
p3 + facet_grid(~Capture) + plot_layout(widths = c(1, 2))) /
((p4 + ggtitle("By genetics")) +
p4 + facet_grid(~Capture) + plot_layout(widths = c(1, 2))) +
plot_layout(guides = "collect")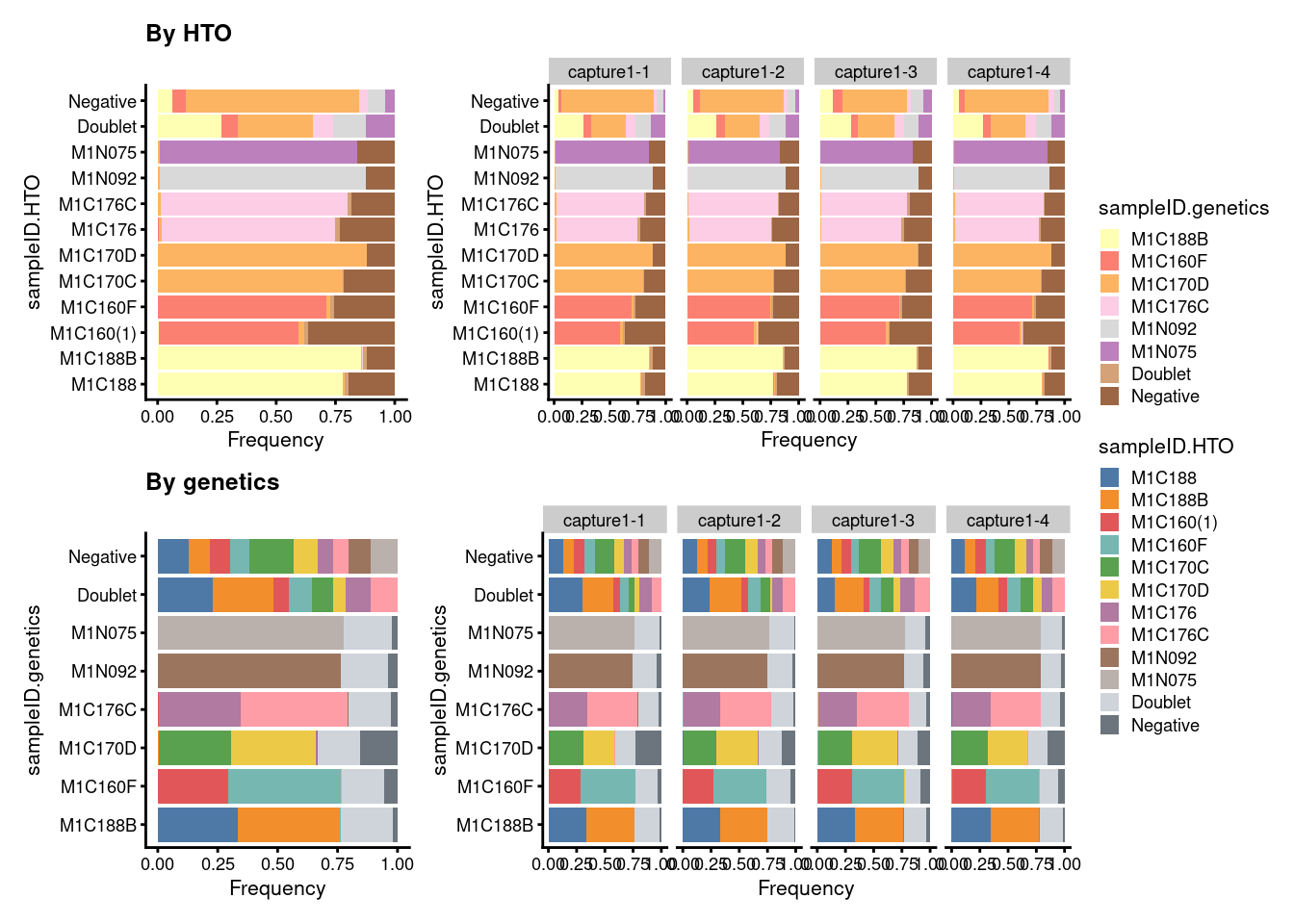
5 Add metadata and sex check
# read metadata.csv
metadata <- read.csv(here("data",
"sample_sheets",
paste0(batch_name,".metadata.csv")))
i <- match(sce$sampleID, metadata$sampleID)
# add patient demographics
colData(sce) <- cbind(
colData(sce),
metadata[i,c("Age","Sex","Condition","Bronchiectasis")]
)
# sex check by the expression of XIST, a female-specific gene.
# this detects certain types of sample-mix-ups.
plotExpression(
sce,
"XIST",
x = "sampleID",
colour_by = "Sex",
exprs_values = "counts",
swap_rownames = "Symbol") +
scale_colour_manual(
values = c(
"Female" = "deeppink3",
"Male" = "deepskyblue"),
name = "Sex") +
theme_cowplot() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1))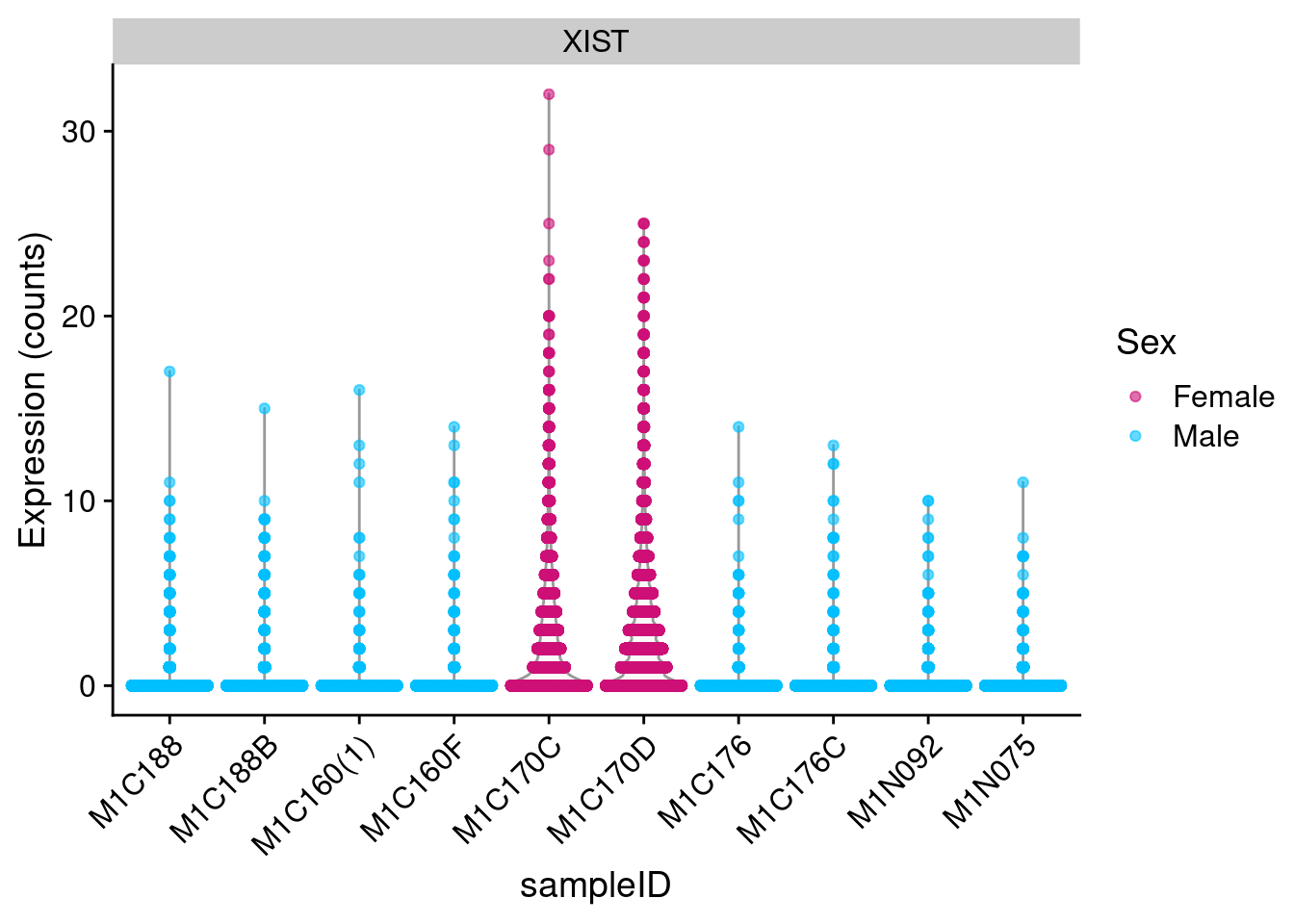
6 Add feature-based annotation
# make feature names unique
rownames(sce) <- uniquifyFeatureNames(rowData(sce)$ID, rowData(sce)$Symbol)
# prepare ensembl v98 database
ah <- AnnotationHub(cache="/group/canc2/anson/.cache/R/AnnotationHub", ask=FALSE)
EnsDb.Hsapiens.v98 <- query(ah, c("EnsDb", "Homo Sapiens", 98))[[1]]
ensdb_columns <- setNames(c("GENEBIOTYPE", "SEQNAME"),
paste0("ENSEMBL.",
c("GENEBIOTYPE", "SEQNAME")))
stopifnot(all(ensdb_columns %in% columns(EnsDb.Hsapiens.v98)))
ensdb_df <- DataFrame(
lapply(ensdb_columns, function(column) {
mapIds(
x = EnsDb.Hsapiens.v98,
keys = rowData(sce)$ID,
keytype = "GENEID",
column = column,
multiVals = "first")
}),
row.names = rowData(sce)$ID)
# prepare ncbi database
ncbi_columns <- setNames(c("ALIAS", "ENTREZID", "GENENAME"),
paste0("NCBI.", c("ALIAS", "ENTREZID", "GENENAME")))
stopifnot(all(ncbi_columns %in% columns(Homo.sapiens)))
ncbi_df <- DataFrame(
lapply(ncbi_columns, function(column) {
mapIds(
x = Homo.sapiens,
keys = rowData(sce)$ID,
keytype = "ENSEMBL",
column = column,
multiVals = "CharacterList")
}),
row.names = rowData(sce)$ID)
rowData(sce) <- cbind(rowData(sce), ensdb_df, ncbi_df)7 QC
Some code in this section are derived from Dr. Jovana Maksimovic’s work for (Maksimovic et al. 2022)
7.1 Calculate metrics
# prepare gene sets
## mitochondrial gene set
mito_set <- rownames(sce)[which(rowData(sce)$ENSEMBL.SEQNAME == "MT")]
is_mito <- rownames(sce) %in% mito_set
summary(is_mito) Mode FALSE TRUE
logical 36588 13 ## ribosomal gene set
ribo_set <- grep("^RP(S|L)", rownames(sce), value = TRUE)
c2_sets <- msigdbr(species = "Homo sapiens", category = "C2")
ribo_set <- union(
ribo_set,
c2_sets[c2_sets$gs_name == "KEGG_RIBOSOME", ]$human_gene_symbol)
is_ribo <- rownames(sce) %in% ribo_set
summary(is_ribo) Mode FALSE TRUE
logical 36494 107 ## sex-linked genes
sex_set <- rownames(sce)[rowData(sce)$ENSEMBL.SEQNAME %in% c("X", "Y")]
## pseudogenes
pseudogene_set <- rownames(sce)[grepl("pseudogene", rowData(sce)$ENSEMBL.GENEBIOTYPE)]
# calculate QC metrics
sce <- addPerCellQCMetrics(
sce,
subsets = list(Mito = which(is_mito),
Ribo = which(is_ribo)),
use.altexps = NULL)7.2 QC: nCount and nFeature
7.2.1 Visualize metrics before removal
# library size
p1 <- plotColData(
sce,
"sum",
x = "sampleID",
colour_by = "sampleID",
point_size = 0.5) +
scale_y_log10() +
scale_colour_manual(values = sce$colours$sample_colours, name = "sampleID") +
theme(axis.text.x = element_blank())
# number of genes detected
p2 <- plotColData(
sce,
"detected",
x = "sampleID",
colour_by = "sampleID",
point_size = 0.5) +
scale_colour_manual(values = sce$colours$sample_colours, name = "sampleID") +
theme(axis.text.x = element_blank())
((p1 + NoLegend()) + p1 + facet_grid(~sce$Capture) + plot_layout(widths=c(1, length(capture_names))))/
((p2 + NoLegend()) + p2 + facet_grid(~sce$Capture) + plot_layout(widths=c(1, length(capture_names)))) +
plot_layout(guides="collect")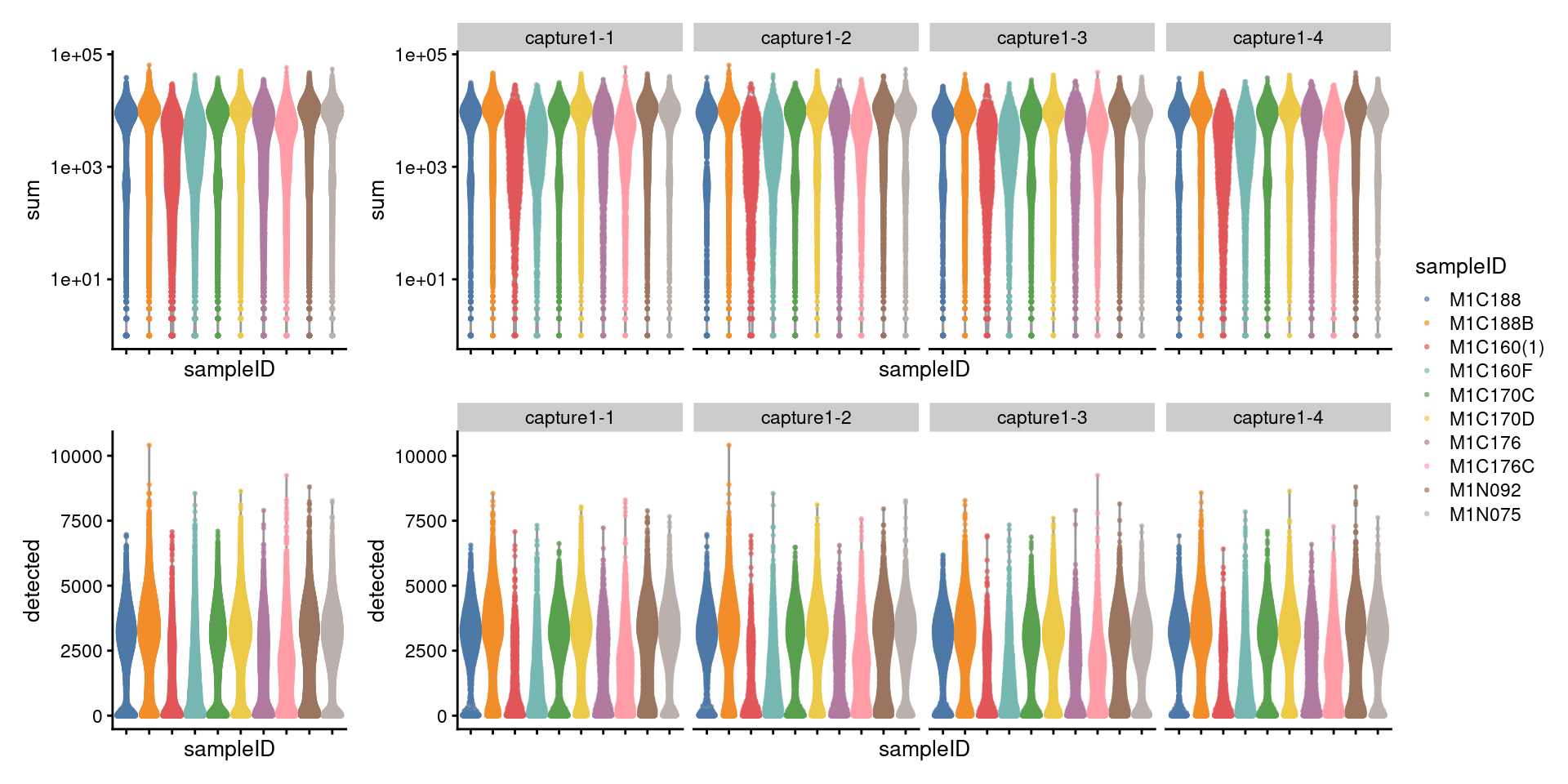
7.2.2 Remove cells with features < 200
sce$batch <- interaction(
sce$Capture,
sce$sampleID,
drop = TRUE,
lex.order = FALSE)
feature_drop <- sce$detected < 200
sce_pre_QC_outlier_removal <- sce
keep <- !feature_drop
sce_pre_QC_outlier_removal$keep <- keep
sce <- sce[, keep]
data.frame(
ByFeature = tapply(
feature_drop,
sce_pre_QC_outlier_removal$batch,
sum,
na.rm = TRUE),
Remaining = as.vector(unname(table(sce$batch))),
PercRemaining = round(
100 * as.vector(unname(table(sce$batch))) /
as.vector(
unname(
table(sce_pre_QC_outlier_removal$batch))), 1)) |>
tibble::rownames_to_column("batch") |>
dplyr::arrange(dplyr::desc(PercRemaining)) |>
DT::datatable(
caption = "Number of droplets removed by each QC step and the number of droplets remaining.",
rownames = FALSE) |>
DT::formatRound("PercRemaining", 1)7.2.3 Visualize metrics after removal
p3 <- plotColData(
sce_pre_QC_outlier_removal,
"sum",
x = "sampleID",
colour_by = "keep",
point_size = 0.5) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
p4 <- plotColData(
sce_pre_QC_outlier_removal,
"detected",
x = "sampleID",
colour_by = "keep",
point_size = 0.5) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
((p3 + NoLegend()) + p3 + facet_grid(~sce_pre_QC_outlier_removal$Capture) + plot_layout(widths=c(1, length(capture_names))))/
((p4 + NoLegend()) + p4 + facet_grid(~sce_pre_QC_outlier_removal$Capture) + plot_layout(widths=c(1, length(capture_names)))) +
plot_layout(guides="collect")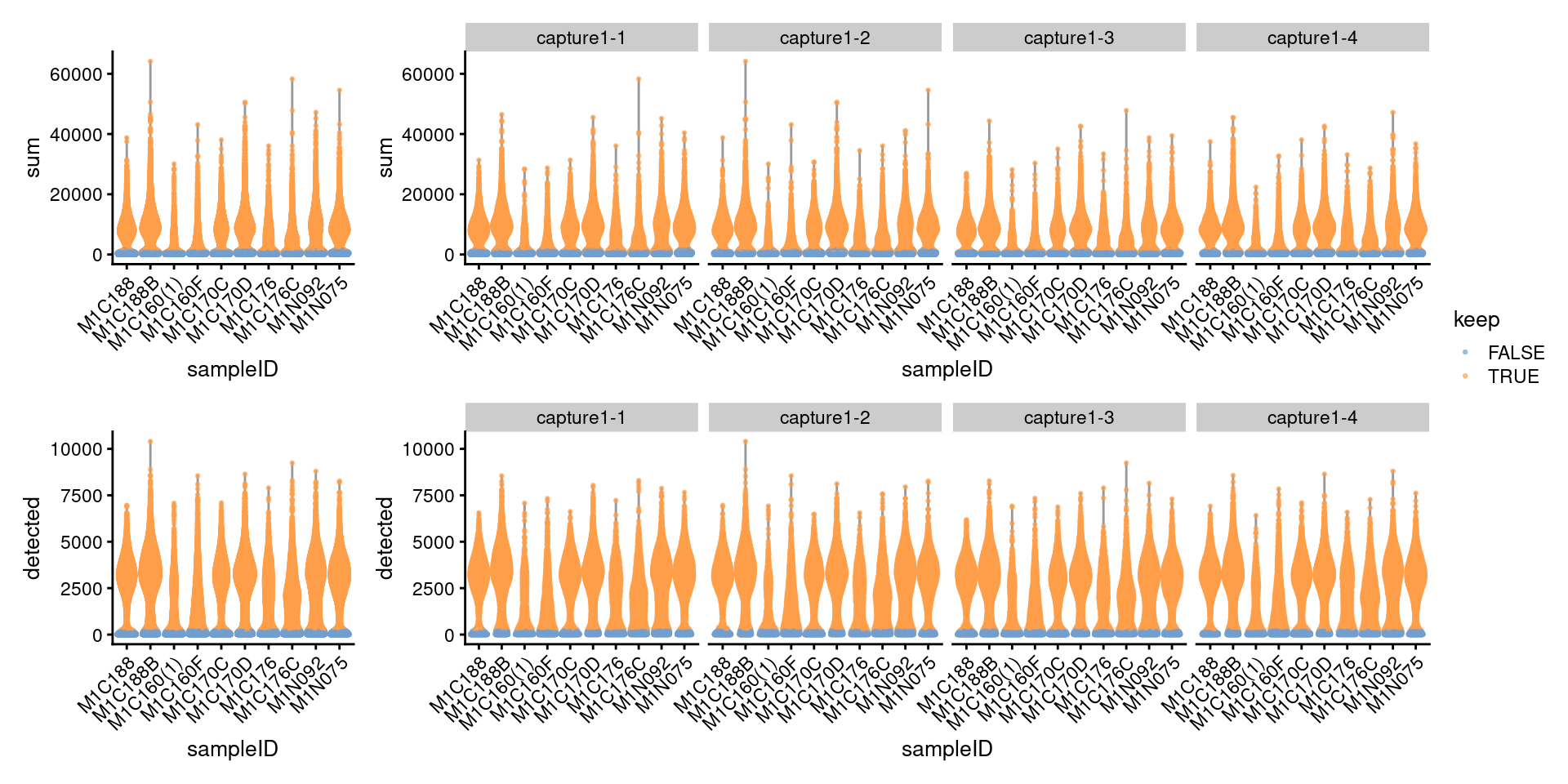
7.3 QC: mitochondrial percent
7.3.1 Visualize metrics before removal
# subsets_Mito_percent
p1 <- plotColData(
sce,
"subsets_Mito_percent",
x = "sampleID",
colour_by = "sampleID",
point_size = 0.5) +
scale_y_log10() +
scale_colour_manual(values = sce$colours$sample_colours, name = "sampleID") +
theme(axis.text.x = element_blank())
# subsets_Ribo_percent
p2 <- plotColData(
sce,
"subsets_Ribo_percent",
x = "sampleID",
colour_by = "sampleID",
point_size = 0.5) +
scale_colour_manual(values = sce$colours$sample_colours, name = "sampleID") +
theme(axis.text.x = element_blank())
((p1 + NoLegend()) + p1 + facet_grid(~sce$Capture) + plot_layout(widths=c(1, length(capture_names))))/
((p2 + NoLegend()) + p2 + facet_grid(~sce$Capture) + plot_layout(widths=c(1, length(capture_names)))) +
plot_layout(guides="collect")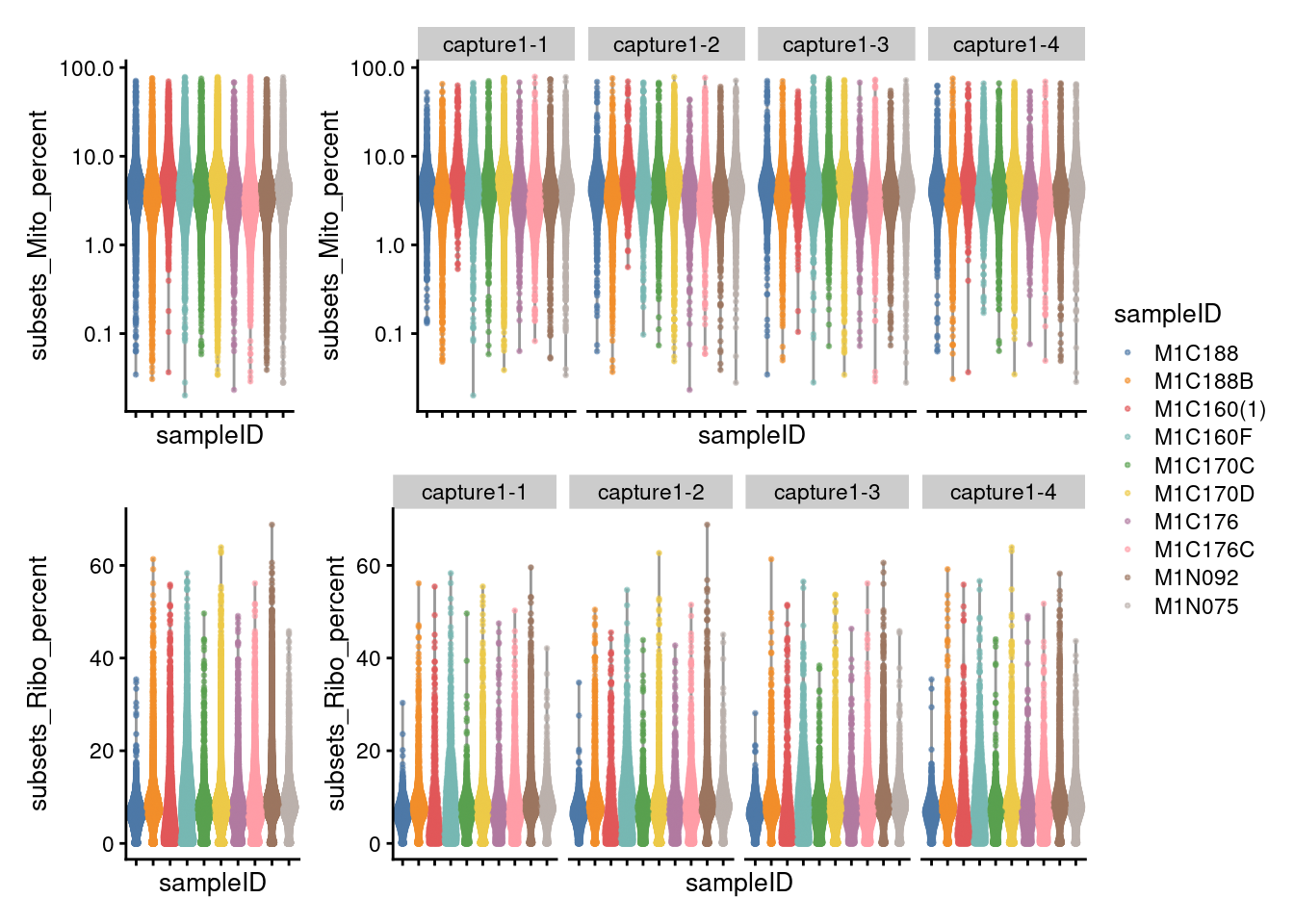
7.3.2 Remove mito% outlier
mito_drop <- isOutlier(
metric = sce$subsets_Mito_percent,
nmads = 3,
type = "higher",
batch = sce$batch)
sce_pre_QC_outlier_removal <- sce
keep <- !mito_drop
sce_pre_QC_outlier_removal$keep <- keep
sce <- sce[, keep]
data.frame(
ByMito = tapply(
mito_drop,
sce_pre_QC_outlier_removal$batch,
sum,
na.rm = TRUE),
Remaining = as.vector(unname(table(sce$batch))),
PercRemaining = round(
100 * as.vector(unname(table(sce$batch))) /
as.vector(
unname(
table(sce_pre_QC_outlier_removal$batch))), 1)) |>
tibble::rownames_to_column("batch") |>
dplyr::arrange(dplyr::desc(PercRemaining)) |>
DT::datatable(
caption = "Number of droplets removed by each QC step and the number of droplets remaining.",
rownames = FALSE) |>
DT::formatRound("PercRemaining", 1)7.3.1 Visualize metrics after removal
p3 <- plotColData(
sce_pre_QC_outlier_removal,
"subsets_Mito_percent",
x = "sampleID",
colour_by = "keep",
point_size = 0.5) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
p4 <- plotColData(
sce_pre_QC_outlier_removal,
"subsets_Ribo_percent",
x = "sampleID",
colour_by = "keep",
point_size = 0.5) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
((p3 + NoLegend()) + p3 + facet_grid(~sce_pre_QC_outlier_removal$Capture) + plot_layout(widths=c(1, length(capture_names))))/
((p4 + NoLegend()) + p4 + facet_grid(~sce_pre_QC_outlier_removal$Capture) + plot_layout(widths=c(1, length(capture_names)))) +
plot_layout(guides="collect")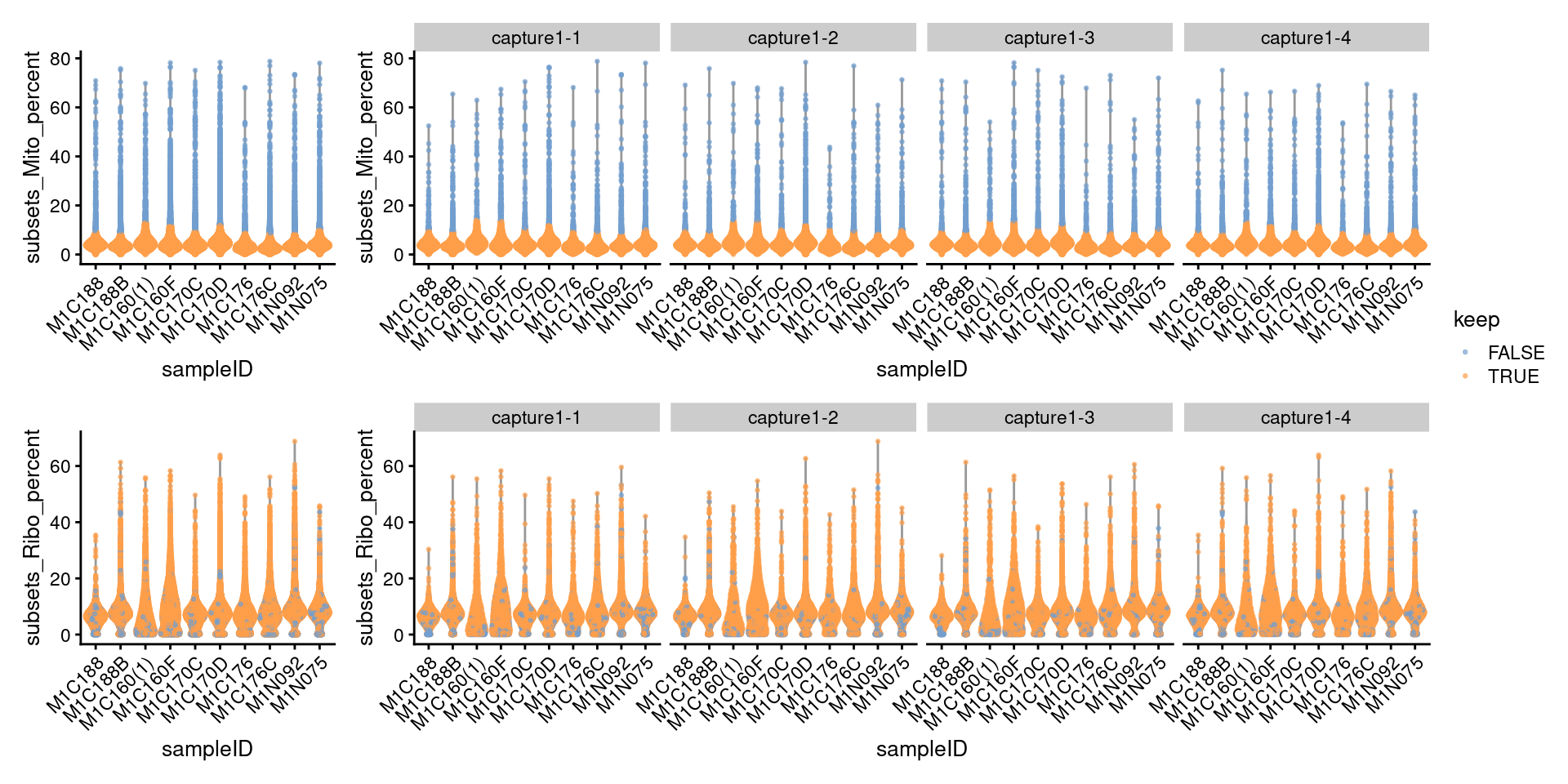
7.5.2 Number of droplets retained (named by sample ID)
# number of droplets assigned by HTO method
p1 <- ggcells(sce) +
geom_bar(aes(x = sampleID.HTO, fill = sampleID.HTO)) +
geom_text(stat='count', aes(x = sampleID.HTO, label=..count..), hjust=1, size=2) +
coord_flip() +
ggtitle("By HTO") +
ylab("Number of droplets") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,16000,4000),limits=c(0,16000)) +
scale_fill_manual(values = sce$colours$sampleID.HTO_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=guide_legend(title="sampleID"))
p1.facet <- ggcells(sce) +
geom_bar(aes(x = sampleID.HTO, fill = sampleID.HTO)) +
geom_text(stat='count', aes(x = sampleID.HTO, label=..count..),
hjust=1, size=2) +
#ggtitle("By HTO") +
ylab("Number of droplets") +
facet_grid(~Capture, scales = "fixed", space = "fixed") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,4000,1000), limits = c(0,4000)) +
scale_fill_manual(values = sce$colours$sampleID.HTO_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=guide_legend(title="sampleID")) +
coord_flip()
# number of droplets assigned by genetic method
p2 <- ggcells(sce) +
geom_bar(aes(x = sampleID.genetics, fill = sampleID.genetics)) +
geom_text(stat='count', aes(x = sampleID.genetics, label=..count..), hjust=1, size=2) +
coord_flip() +
ggtitle("By genetics") +
ylab("Number of droplets") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,16000,4000), limits = c(0,16000)) +
scale_fill_manual(values = sce$colours$sampleID.genetics_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=FALSE)
p2.facet <- ggcells(sce) +
geom_bar(aes(x = sampleID.genetics, fill = sampleID.genetics)) +
geom_text(stat='count', aes(x = sampleID.genetics, label=..count..),
hjust=1, size=2) +
ylab("Number of droplets") +
facet_grid(~Capture, scales = "fixed", space = "fixed") +
theme_cowplot(font_size = 8) +
scale_y_continuous(breaks=seq(0,4000,1000), limits = c(0,4000)) +
scale_fill_manual(values = sce$colours$sampleID.genetics_colours) +
theme(axis.title.y = element_blank()) +
guides(fill=guide_legend(title="sampleID")) +
coord_flip()
(p1+p1.facet+plot_layout(width=c(1,2))) /
(p2+p2.facet+plot_layout(width=c(1,2))) +
plot_layout(guides="collect")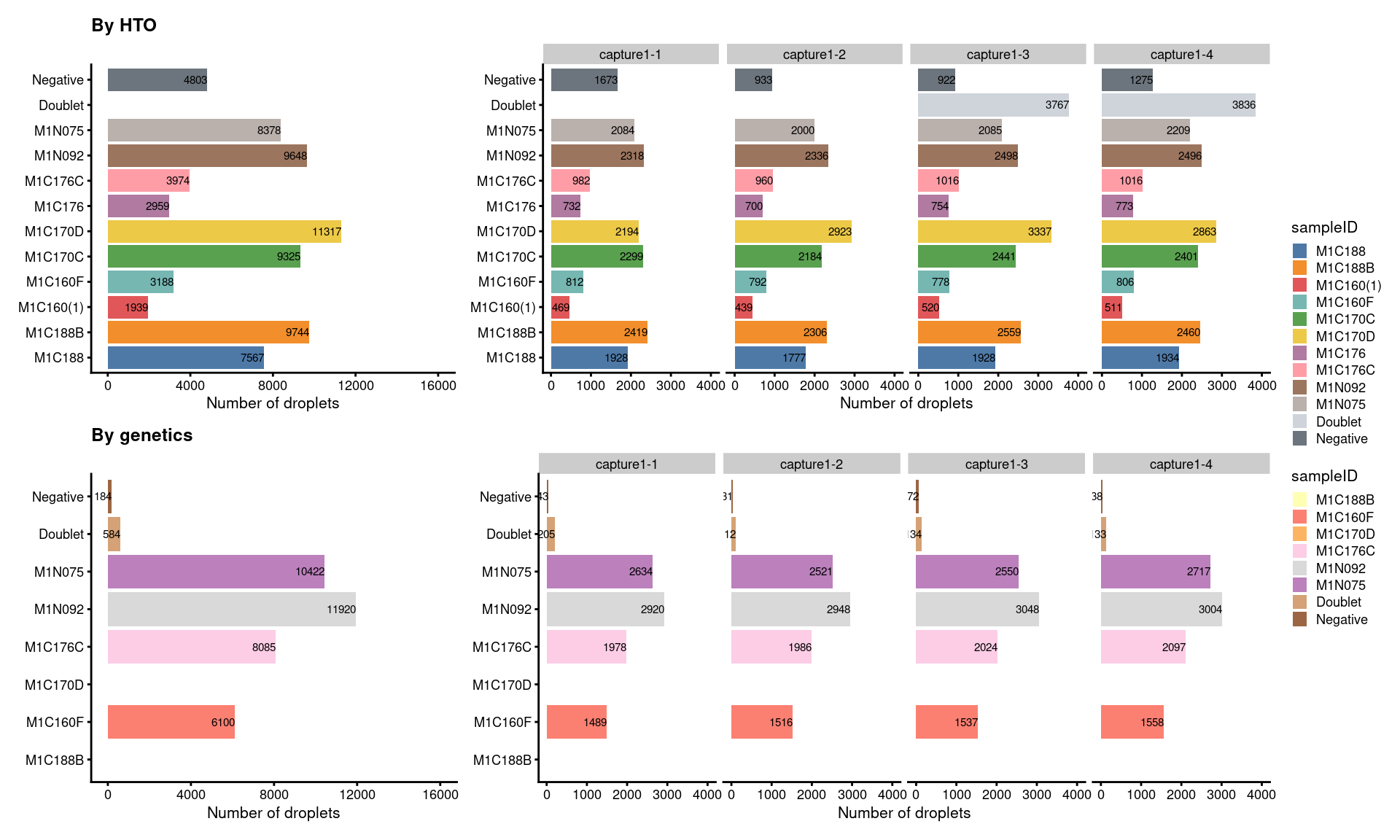
7.5.3 Proportion of droplets (named by sample ID)
# proportion of genetically assigned droplets in each HTO
p3 <- ggcells(sce) +
geom_bar(
aes(x = sampleID.HTO, fill = sampleID.genetics),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
theme_cowplot(font_size = 8) +
scale_fill_manual(values = sce$colours$sampleID.genetics_colours)
# proportion of HTO assigned droplets in each genetic donor
p4 <- ggcells(sce) +
geom_bar(
aes(x = sampleID.genetics, fill = sampleID.HTO),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
theme_cowplot(font_size = 8) +
scale_fill_manual(values = sce$colours$sampleID.HTO_colours)
((p3 + ggtitle("By HTO")) +
p3 + facet_grid(~Capture) + plot_layout(widths = c(1, 2))) /
((p4 + ggtitle("By genetics")) +
p4 + facet_grid(~Capture) + plot_layout(widths = c(1, 2))) +
plot_layout(guides = "collect")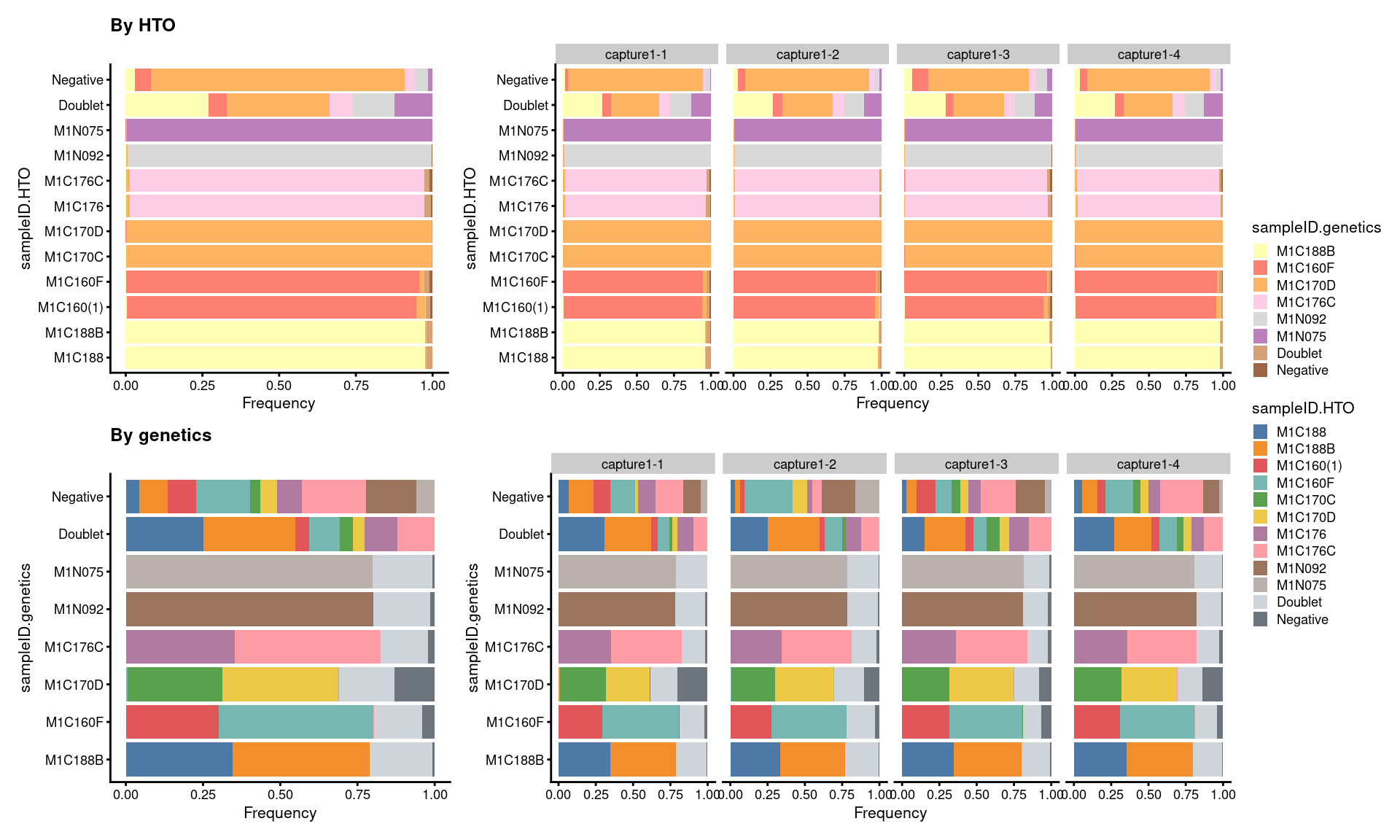
8 Remove undesired genes
# create uninformative gene sets
uninformative <- is_mito | is_ribo | rownames(sce) %in% sex_set | rownames(sce) %in% pseudogene_set
sum(uninformative)[1] 1608# remove uninformative genes
sce <- sce[!uninformative,]
# remove low-abundance genes
numCells <- nexprs(sce, byrow = TRUE)
keep <- numCells > 20
sum(keep)[1] 25676sce <- sce[keep,]
# number of genes retained
dim(sce)[1] 25676 889929 Save object
prep_dir <- here("data","SCEs","preprocessed")
if(!dir.exists(prep_dir)) {
dir.create(prep_dir, recursive = TRUE)
}
out <- paste0(prep_dir,'/',
paste0(batch_name,".preprocessed.SCE.rds"))
if(!file.exists(out)) saveRDS(sce, out)References: Maksimovic J, Shanthikumar S, Howitt G, Hickey PF, Ho W, Anttila C, et al. Single-cell atlas of bronchoalveolar lavage from preschool cystic fibrosis reveals new cell phenotypes. bioRxiv 2022: 2022.2006.2017.496207.
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /hpc/software/installed/R/4.1.2/lib64/R/lib/libRblas.so
LAPACK: /hpc/software/installed/R/4.1.2/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] Homo.sapiens_1.3.1
[2] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
[3] org.Hs.eg.db_3.14.0
[4] GO.db_3.14.0
[5] OrganismDbi_1.36.0
[6] msigdbr_7.5.1
[7] ensembldb_2.18.4
[8] AnnotationFilter_1.22.0
[9] GenomicFeatures_1.46.4
[10] AnnotationDbi_1.56.2
[11] AnnotationHub_3.2.2
[12] BiocFileCache_2.6.1
[13] dbplyr_2.1.1
[14] stringr_1.5.1
[15] janitor_2.1.0
[16] forcats_1.0.1
[17] dplyr_1.1.4
[18] scater_1.22.0
[19] scuttle_1.4.0
[20] patchwork_1.2.0
[21] cowplot_1.1.3
[22] SeuratObject_5.0.1
[23] Seurat_4.4.0
[24] ggplot2_3.5.2
[25] here_1.0.1
[26] DropletUtils_1.14.2
[27] SingleCellExperiment_1.16.0
[28] SummarizedExperiment_1.24.0
[29] Biobase_2.54.0
[30] GenomicRanges_1.46.1
[31] GenomeInfoDb_1.30.1
[32] IRanges_2.28.0
[33] S4Vectors_0.32.4
[34] BiocGenerics_0.40.0
[35] MatrixGenerics_1.6.0
[36] matrixStats_1.1.0
[37] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] rappdirs_0.3.3 rtracklayer_1.54.0
[3] scattermore_1.2 R.methodsS3_1.8.1
[5] tidyr_1.3.1 bit64_4.6.0-1
[7] knitr_1.46 irlba_2.3.5.1
[9] DelayedArray_0.20.0 R.utils_2.12.2
[11] data.table_1.15.4 KEGGREST_1.34.0
[13] RCurl_1.98-1.9 generics_0.1.3
[15] ScaledMatrix_1.2.0 callr_3.7.3
[17] RSQLite_2.3.6 RANN_2.6.1
[19] future_1.33.2 bit_4.0.5
[21] spatstat.data_3.0-4 xml2_1.3.3
[23] lubridate_1.8.0 httpuv_1.6.15
[25] assertthat_0.2.1 viridis_0.6.2
[27] xfun_0.43 hms_1.1.4
[29] jquerylib_0.1.4 babelgene_22.9
[31] evaluate_0.23 promises_1.3.0
[33] restfulr_0.0.15 fansi_1.0.6
[35] progress_1.2.3 igraph_2.0.3
[37] DBI_1.2.2 htmlwidgets_1.6.4
[39] spatstat.geom_3.2-9 purrr_1.2.1
[41] crosstalk_1.2.1 biomaRt_2.50.3
[43] deldir_2.0-4 sparseMatrixStats_1.6.0
[45] vctrs_0.6.5 ROCR_1.0-11
[47] abind_1.4-8 cachem_1.0.8
[49] withr_3.0.0 progressr_0.14.0
[51] sctransform_0.4.1 GenomicAlignments_1.30.0
[53] prettyunits_1.2.0 xaringanExtra_0.7.0
[55] goftest_1.2-3 cluster_2.1.8.1
[57] dotCall64_1.1-1 lazyeval_0.2.2
[59] crayon_1.5.2 spatstat.explore_3.2-7
[61] labeling_0.4.3 edgeR_4.4.2
[63] pkgconfig_2.0.3 ProtGenerics_1.30.0
[65] nlme_3.1-155 vipor_0.4.5
[67] rlang_1.1.3 globals_0.16.3
[69] lifecycle_1.0.4 miniUI_0.1.1.1
[71] filelock_1.0.3 rsvd_1.0.5
[73] rprojroot_2.1.1 polyclip_1.10-6
[75] lmtest_0.9-40 graph_1.72.0
[77] Matrix_1.6-5 Rhdf5lib_1.16.0
[79] zoo_1.8-12 beeswarm_0.4.0
[81] whisker_0.4 ggridges_0.5.6
[83] processx_3.8.0 rjson_0.2.21
[85] png_0.1-8 viridisLite_0.4.2
[87] bitops_1.0-9 getPass_0.2-2
[89] R.oo_1.24.0 KernSmooth_2.23-20
[91] spam_2.10-0 rhdf5filters_1.6.0
[93] Biostrings_2.62.0 blob_1.2.4
[95] DelayedMatrixStats_1.16.0 parallelly_1.37.1
[97] spatstat.random_3.2-3 beachmat_2.10.0
[99] scales_1.3.0 memoise_2.0.1
[101] magrittr_2.0.3 plyr_1.8.9
[103] ica_1.0-3 zlibbioc_1.48.2
[105] compiler_4.1.2 BiocIO_1.8.0
[107] dqrng_0.3.2 RColorBrewer_1.1-3
[109] fitdistrplus_1.1-11 Rsamtools_2.10.0
[111] snakecase_0.11.0 cli_3.6.2
[113] XVector_0.34.0 listenv_0.9.1
[115] pbapply_1.7-2 ps_1.7.2
[117] MASS_7.3-55 tidyselect_1.2.1
[119] stringi_1.8.3 highr_0.10
[121] yaml_2.3.8 BiocSingular_1.10.0
[123] locfit_1.5-9.4 ggrepel_0.9.1
[125] grid_4.1.2 sass_0.4.9
[127] tools_4.1.2 future.apply_1.11.2
[129] parallel_4.1.2 rstudioapi_0.13
[131] git2r_0.31.0 gridExtra_2.3
[133] farver_2.1.1 Rtsne_0.17
[135] digest_0.6.35 BiocManager_1.30.25
[137] shiny_1.8.1.1 Rcpp_1.0.12
[139] BiocVersion_3.18.1 later_1.3.2
[141] RcppAnnoy_0.0.22 httr_1.4.7
[143] colorspace_2.1-0 XML_3.99-0.16.1
[145] fs_1.6.4 tensor_1.5.1
[147] reticulate_1.36.1.9000 splines_4.1.2
[149] RBGL_1.70.0 uwot_0.1.14
[151] statmod_1.5.0 spatstat.utils_3.1-3
[153] sp_2.1-3 plotly_4.10.4.9000
[155] xtable_1.8-4 jsonlite_1.8.8
[157] R6_2.6.1 pillar_1.9.0
[159] htmltools_0.5.8.1 mime_0.12
[161] DT_0.20 glue_1.7.0
[163] fastmap_1.1.1 BiocParallel_1.28.3
[165] BiocNeighbors_1.12.0 interactiveDisplayBase_1.32.0
[167] codetools_0.2-20 utf8_1.2.4
[169] lattice_0.22-7 bslib_0.3.1
[171] spatstat.sparse_3.0-3 tibble_3.2.1
[173] curl_5.2.1 ggbeeswarm_0.6.0
[175] leiden_0.4.3.1 survival_3.2-13
[177] limma_3.62.2 rmarkdown_2.26
[179] munsell_0.5.1 rhdf5_2.38.1
[181] GenomeInfoDbData_1.2.11 HDF5Array_1.22.1
[183] reshape2_1.4.4 gtable_0.3.5