CheckTehranchiConcordance
Last updated: 2022-04-19
Checks: 6 1
Knit directory: ChromatinSplicingQTLs/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20191126) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 3c4d1c7. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.Rhistory
Ignored: code/.DS_Store
Ignored: code/.RData
Ignored: code/._.DS_Store
Ignored: code/._README.md
Ignored: code/._report.html
Ignored: code/.ipynb_checkpoints/
Ignored: code/.snakemake/
Ignored: code/Alignments/
Ignored: code/ENCODE/
Ignored: code/ExpressionAnalysis/
Ignored: code/FastqFastp/
Ignored: code/FastqFastpSE/
Ignored: code/FastqSE/
Ignored: code/Genotypes/
Ignored: code/IntronSlopes/
Ignored: code/Multiqc/
Ignored: code/Multiqc_chRNA/
Ignored: code/PeakCalling/
Ignored: code/Phenotypes/
Ignored: code/PlotGruberQTLs/
Ignored: code/ProCapAnalysis/
Ignored: code/QC/
Ignored: code/QTLs/
Ignored: code/ReferenceGenome/
Ignored: code/Rplots.pdf
Ignored: code/Session.vim
Ignored: code/SplicingAnalysis/
Ignored: code/TODO
Ignored: code/Tehranchi/
Ignored: code/bigwigs/
Ignored: code/bigwigs_FromNonWASPFilteredReads/
Ignored: code/config/.DS_Store
Ignored: code/config/._.DS_Store
Ignored: code/config/.ipynb_checkpoints/
Ignored: code/debug.ipynb
Ignored: code/debug_python.ipynb
Ignored: code/featureCounts/
Ignored: code/gwas_summary_stats/
Ignored: code/hyprcoloc/
Ignored: code/logs/
Ignored: code/notebooks/.ipynb_checkpoints/
Ignored: code/rules/.PlotQTLs.smk.swp
Ignored: code/rules/.ipynb_checkpoints/
Ignored: code/rules/OldRules/
Ignored: code/rules/notebooks/
Ignored: code/scratch/
Ignored: code/scripts/.ipynb_checkpoints/
Ignored: code/scripts/GTFtools_0.8.0/
Ignored: code/scripts/liftOverBedpe/liftOverBedpe.py
Ignored: code/snakemake.log
Ignored: code/snakemake.sbatch.log
Ignored: data/.DS_Store
Ignored: data/._.DS_Store
Ignored: data/._20220414203249_JASPAR2022_combined_matrices_25818_jaspar.txt
Ignored: data/._ColorsForPhenotypes.xlsx
Ignored: data/ColorsForPhenotypes.xls.sb-eaf20aec-pT1YzB/
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-10.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-11.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-2.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-3.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-4.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-5.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-6.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-7.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022-8.csv
Ignored: data/GWAS_catalog_summary_stats_sources/._list_gwas_summary_statistics_6_Apr_2022.csv
Untracked files:
Untracked: analysis/202204012_Cluster_TestGwasHarmonisation.Rmd
Untracked: analysis/20220415_Cluster_CheckTehranchiConcordance.Rmd
Untracked: code/envs/crossmap.yml
Untracked: code/scripts/GetGWASLeadVariantWindowsFromBed.py
Untracked: code/scripts/StandardizeGwasStats_FromGwasCatalog.R
Untracked: code/scripts/StandardizeGwasStats_FromGwasCatalogOR.R
Untracked: code/scripts/StandardizeGwasStats_MS.R
Untracked: code/scripts/TehranchiScanTFBS.py
Untracked: code/snakemake_profiles/slurm/__pycache__/
Untracked: data/20220414203249_JASPAR2022_combined_matrices_25818_jaspar.txt
Untracked: data/Tehranchi_PrimaryTFBinding-QTLs.bed.gz
Unstaged changes:
Modified: code/rules/DownloadAndPreprocess.smk
Modified: code/rules/GWAS_PrepForColoc.smk
Modified: code/rules/ProcessTehranchi.smk
Modified: code/rules/common.py
Modified: code/scripts/TehranchiTableS2_to_bed.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Intro
Tehranchi et al used pooled chip-seq assay on YRI LCLs to assess allele-specific binding QTLs for 5 transcription factors. They found that the QTL SNPs over TF motif hits were largely concordant with predicted TF-binding effects from position weight matrix. In our data, I want to check how TF binding may effect other processes, like splicing, for which NFKB for example was recently shown to affect. To do this, first I downloaded TableS2 from Tehranchi et al, lifted over the QTL variants to hg38, searched for TF motif hits (motifs downloaded from JASPAR) for both alleles, and caculated the change in PWM score. I did this all using a script in the snakemake, and in this notebook I will explore the results…
library(tidyverse)
library(qvalue)
dat <- read_tsv("../code/scratch/ScanTFBS.txt.gz")
head(dat)# A tibble: 6 x 9
chrom start stop TF P motif Delta MaxAnyValue
<chr> <dbl> <dbl> <chr> <dbl> <chr> <dbl> <dbl>
1 chr1 777808 777809 NF-kB 0.0210 MA01… 11.0 6.85
2 chr1 777808 777809 PU.1 0.00763 MA01… 11.0 6.85
3 chr1 777809 777810 PU.1 0.00763 MA01… 15.2 6.85
4 chr1 922659 922660 Pou2… 0.0333 MA01… -5.72 13.0
5 chr1 922659 922660 Pou2… 0.0333 MA01… -12.8 14.5
6 chr1 992360 992361 PU.1 0.0135 MA01… 12.1 4.01
# … with 1 more variable: hg38refIsHighbind <lgl>dat$motif %>% unique() [1] "MA0105.4.NFKB1" "MA0137.2.STAT1" "MA0137.3.STAT1"
[4] "MA0778.1.NFKB2" "MA0080.2.SPI1" "MA0105.3.NFKB1"
[7] "MA0491.2.JUND" "MA0105.2.NFKB1" "MA0492.1.JUND"
[10] "MA0785.1.POU2F1"dat$TF %>% unique()[1] "NF-kB" "PU.1" "Pou2f1" "JunD" "H3K4me3" "Stat1" Each row is a Tehranchi QTL with a TF PWM motif hit. Note that for each QTL, I searched across 10 different JASPAR motifs, since some of the different TF’s that were probed had more than one motif that I thought would be worthwhile checking. One of the first things I should do is condense SNPs for the best hit for the motifs for a single TF, and also select only the direct binding hits (where the QTL TF matches the motif TF)
MotifToTF <- c("MA0105.4.NFKB1"="NF-kB",
"MA0137.2.STAT1"="Stat1",
"MA0137.3.STAT1"="Stat1",
"MA0778.1.NFKB2"="NF-kB",
"MA0080.2.SPI1"="PU.1",
"MA0105.3.NFKB1"="NF-kB",
"MA0491.2.JUND"="JunD",
"MA0785.1.POU2F1"="Pou2f1"
)
dat.filtered <- dat %>%
mutate(motif = recode(motif, !!!MotifToTF)) %>%
filter(TF == motif) %>%
group_by(chrom, start, TF) %>%
arrange(desc(MaxAnyValue)) %>%
distinct(chrom, start, TF, .keep_all = T) %>%
ungroup()The P values here are what is reported in TableS2 from Tehranchi. I think they are nominal P-values, and filtered for nominal Pvalue < 0.05.
dat.filtered$P %>% hist()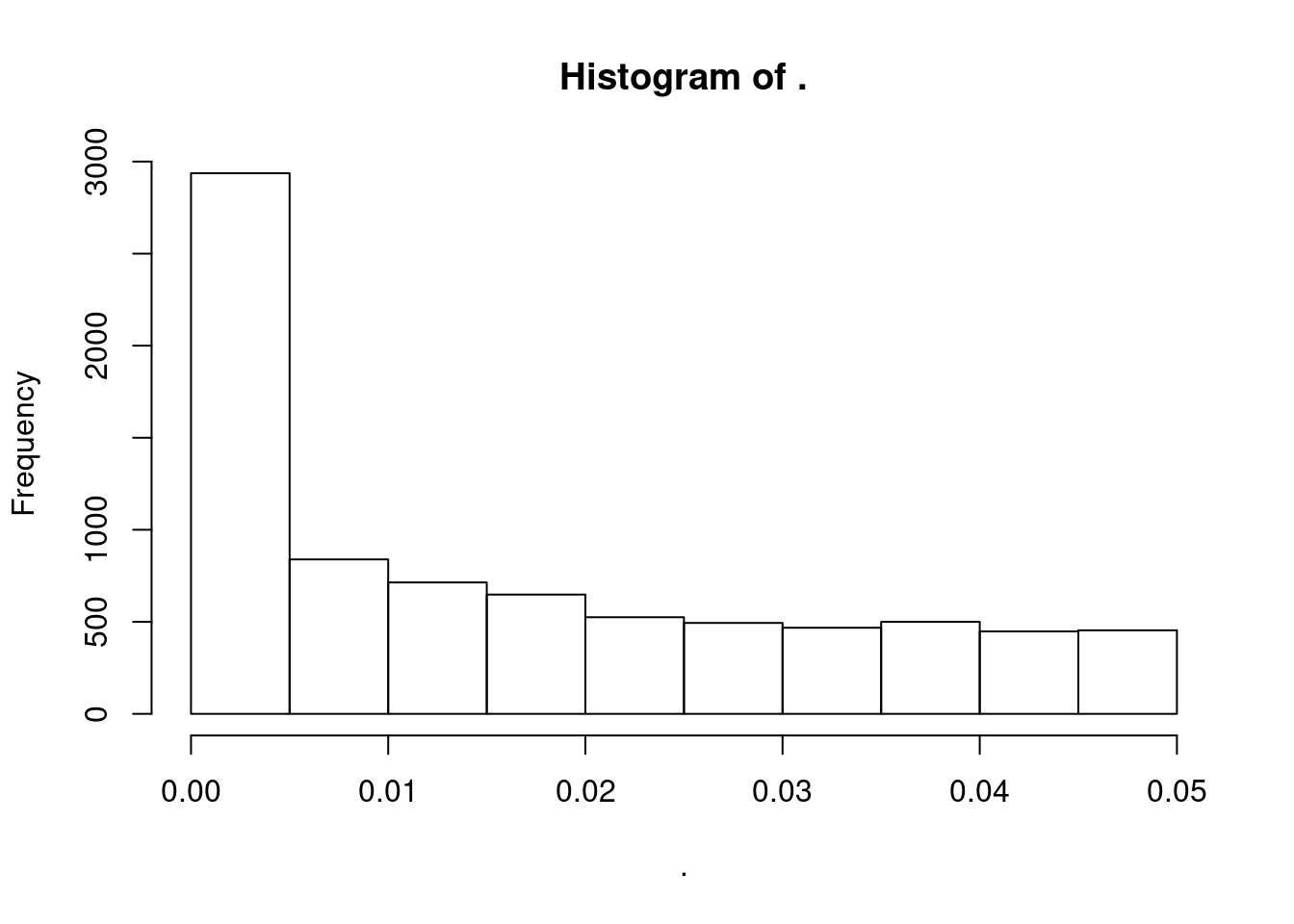
Based on the histogram, most of these QTLs are probably false positives, most of the Pvalue distribution mass is in a flat region. If we had the whole distribution of P-values it would be trivial to estimate FDR and filter out most of the false discoveries.
For now, let’s plot the concordance of deltaPWM (ref-alt) versus if the high binding allele was the ref by plotting histograms of the deltaPWM, colored by if TableS2 reports the the reference (lifted to hg38) is the high binding allele. So I would expect most of the true QTLs with a positive deltaPWM to also be where reference is the high binding allele.
dat.filtered %>%
filter(!Delta==0) %>%
ggplot(aes(x=Delta, fill=hg38refIsHighbind)) +
geom_histogram() +
facet_wrap(~TF, scales = "free") +
theme_classic()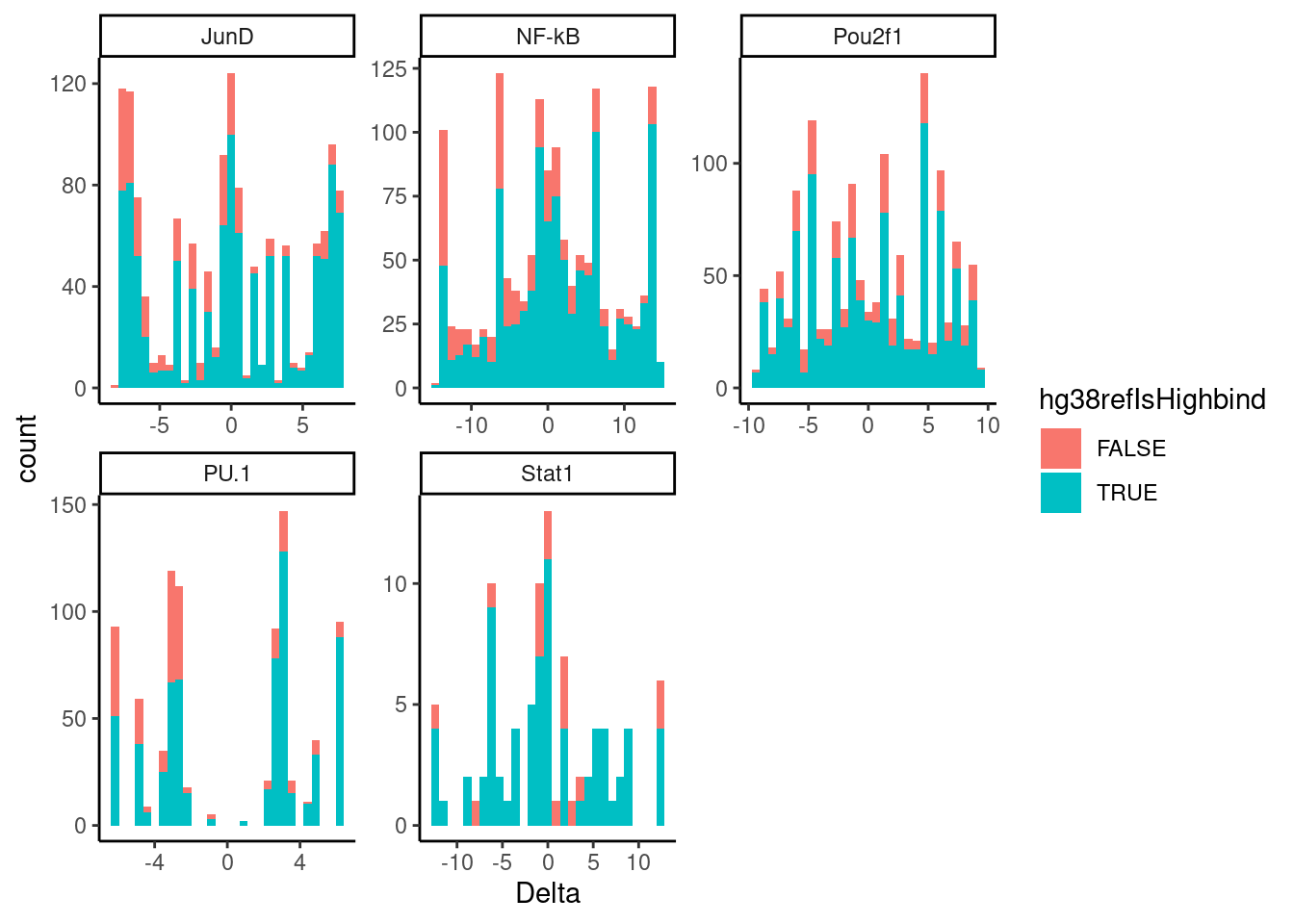
Not much concordance… But what happens if i use a more stringent Pvalue filter to filter out some of the false discoveries…
dat.filtered %>%
filter(!Delta==0) %>%
filter(P<0.001) %>%
ggplot(aes(x=Delta, fill=hg38refIsHighbind)) +
geom_histogram() +
facet_wrap(~TF, scales = "free") +
theme_classic()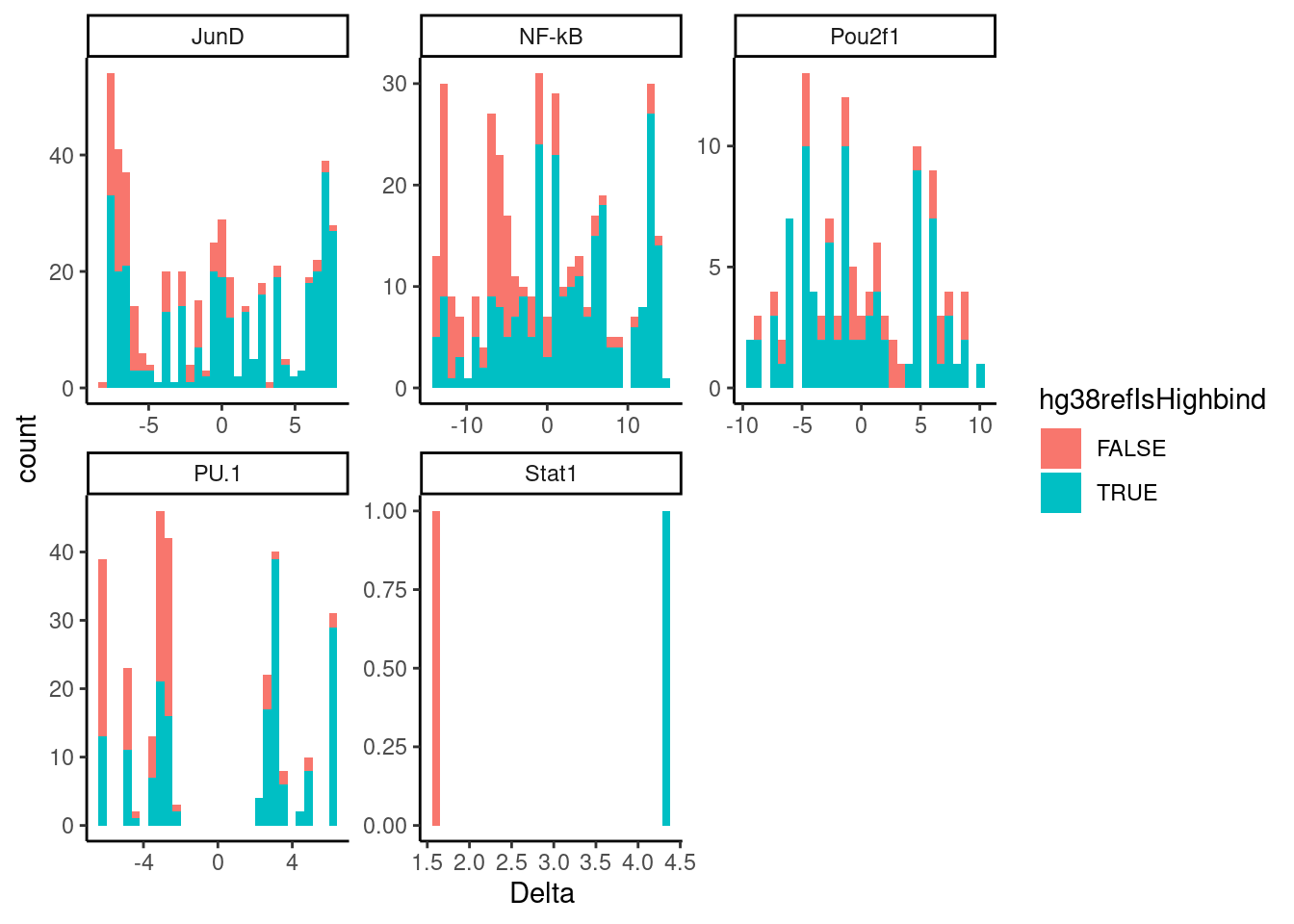
Ok this is a bit more believable. Maybe I should try estimating a FDR and filtering on an appropriate FDR threshold. First let’s plot the Pvalue distribition (at least up to 0.05) for each TF
dat.filtered %>%
ggplot(aes(x=P)) +
geom_histogram() +
facet_wrap(~TF, scales = "free") +
theme_classic()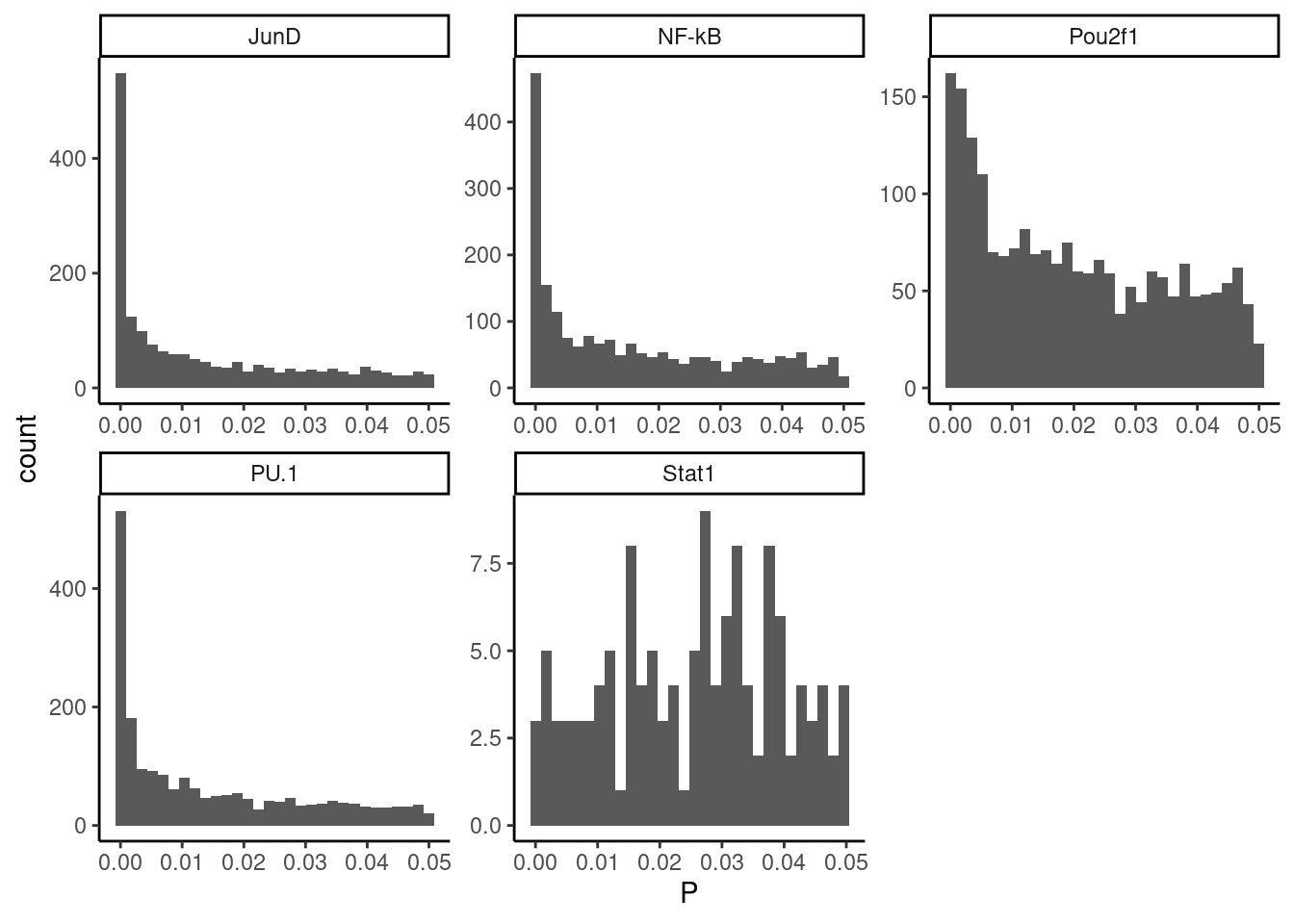
Ok I think I should just drop STAT1 from analysis, and for the others I will try to apply Storey’s q value FDR estimation approach somehow. How about we count the number of observations from 0.04 to 0.05 (mostly flat region, representing 1% of the interval from 0-1), then create 95X as many observations sampled randomly from uniform (from 0.05 to 1) to fill in the rest of the Pvalue distribution with these mock data. Then we can use qvalue package to estimate FDR.
MockPValsToFillIn <- dat.filtered %>%
filter(!TF == "Stat1") %>%
filter(P<0.05 & P>0.04) %>%
select(TF) %>%
uncount(95) %>%
mutate(P = runif(nrow(.), 0.05, 1))
#Plot histogram with mock pvalues filled in
dat.filtered %>%
filter(!TF == "Stat1") %>%
bind_rows(MockPValsToFillIn) %>%
ggplot(aes(x=P)) +
geom_histogram() +
facet_wrap(~TF, scales = "free") +
theme_classic()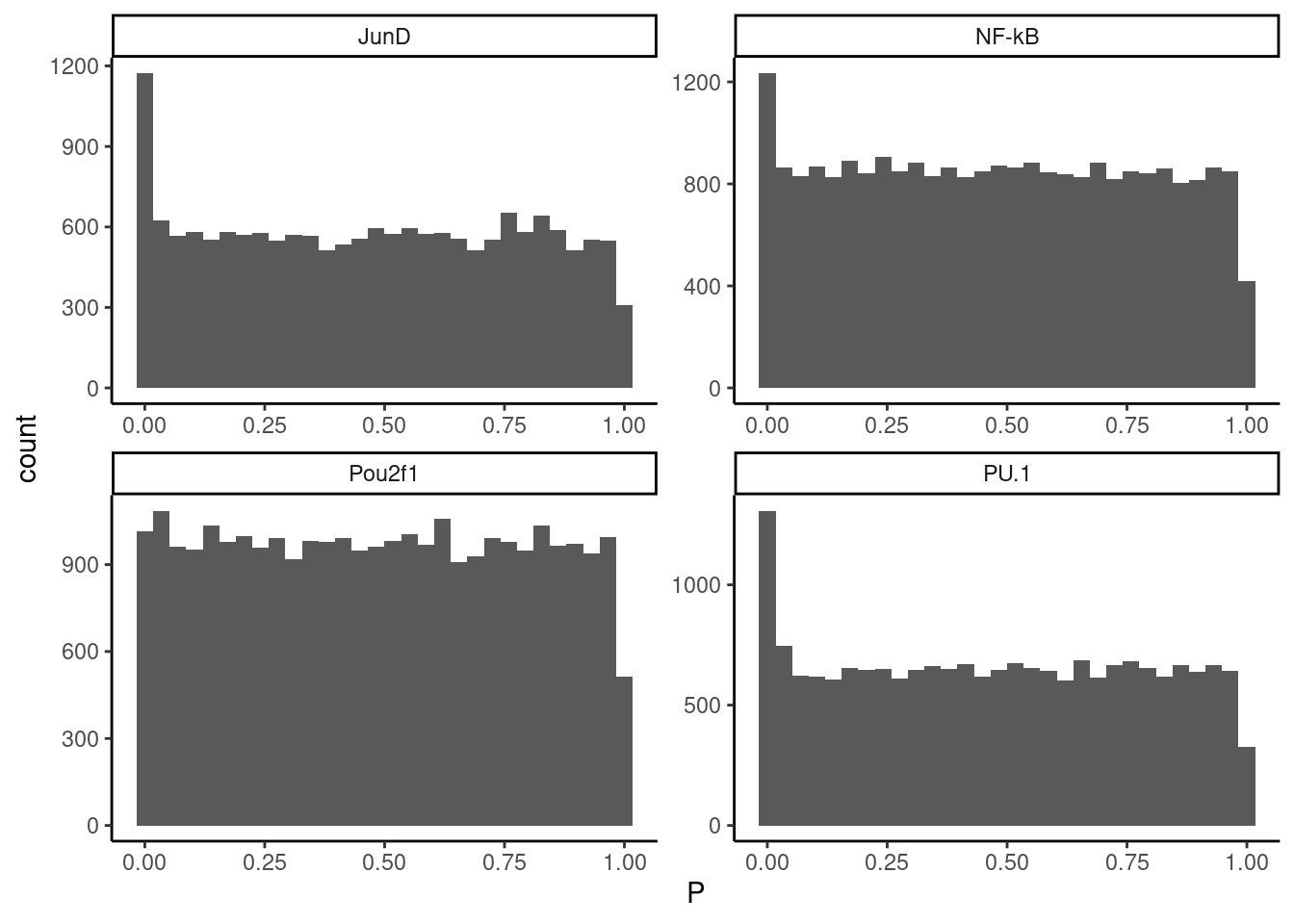
dat.filted.with.FDR <- dat.filtered %>%
filter(!TF == "Stat1") %>%
bind_rows(MockPValsToFillIn) %>%
group_by(TF) %>%
mutate(q = qvalue(P)$qvalues) %>%
ungroup()
# Plot qvalues per TF. Based on the histogram, Pou2f1 should have basically no QTLs that pass a stringent (10%) FDR threshold
dat.filted.with.FDR %>%
ggplot(aes(x=q)) +
geom_histogram() +
facet_wrap(~TF, scales = "free") +
theme_classic()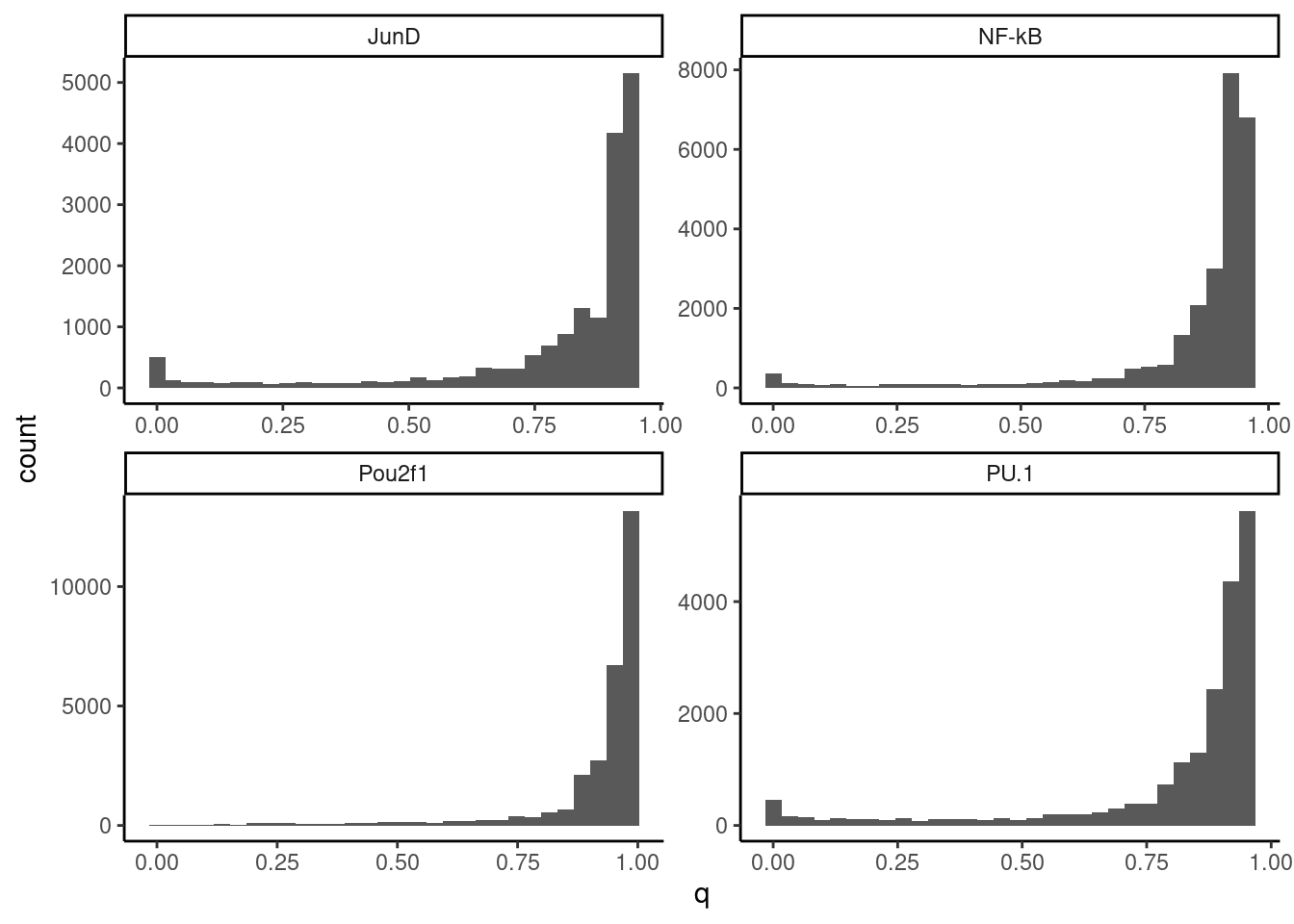
Ok great. I think that worked fine. Now let’s replot the concordance of deltaPWM versus observed high binding allele for FDR10% QTLs
dat.filted.with.FDR %>%
filter(q < 0.1) %>%
filter(!Delta==0) %>%
ggplot(aes(x=Delta, fill=hg38refIsHighbind)) +
geom_histogram() +
facet_wrap(~TF, scales = "free") +
theme_classic()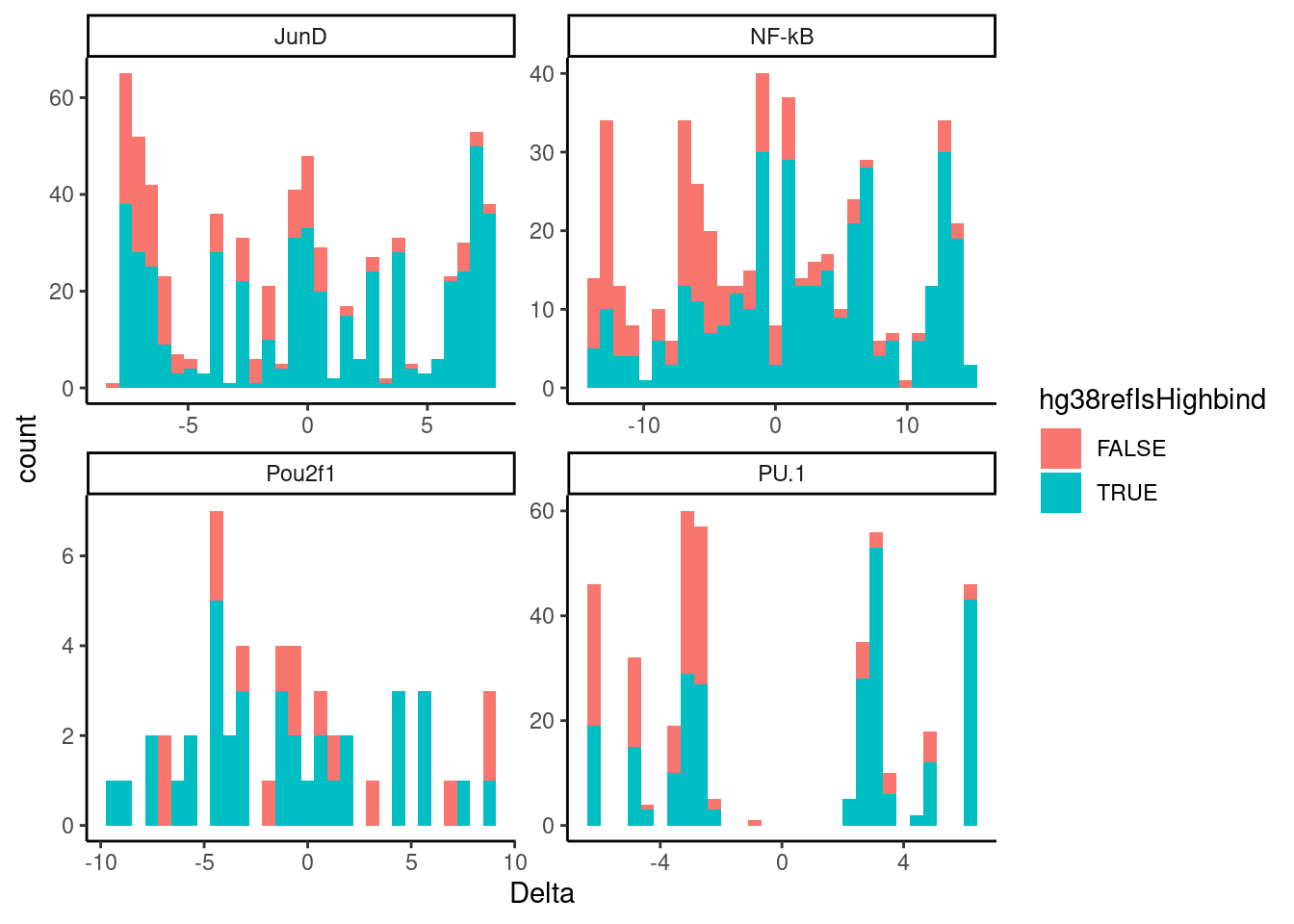
Ok that looks better. Let’s replot a different way, for simplicity:
dat.filted.with.FDR %>%
filter(q < 0.1) %>%
filter(!Delta==0) %>%
mutate(IsRefenceAlleleHigherPWM = Delta>0) %>%
count(IsRefenceAlleleHigherPWM, hg38refIsHighbind, TF) %>%
ggplot(aes(x=IsRefenceAlleleHigherPWM, y=n, fill=hg38refIsHighbind)) +
geom_col() +
facet_wrap(~TF, scales = "free") +
labs(x="Is RefAllele higher predicted binding (PWM)", y="Number TF-binding QTLs", fill="Is RefAllele higher observed binding") +
theme_classic()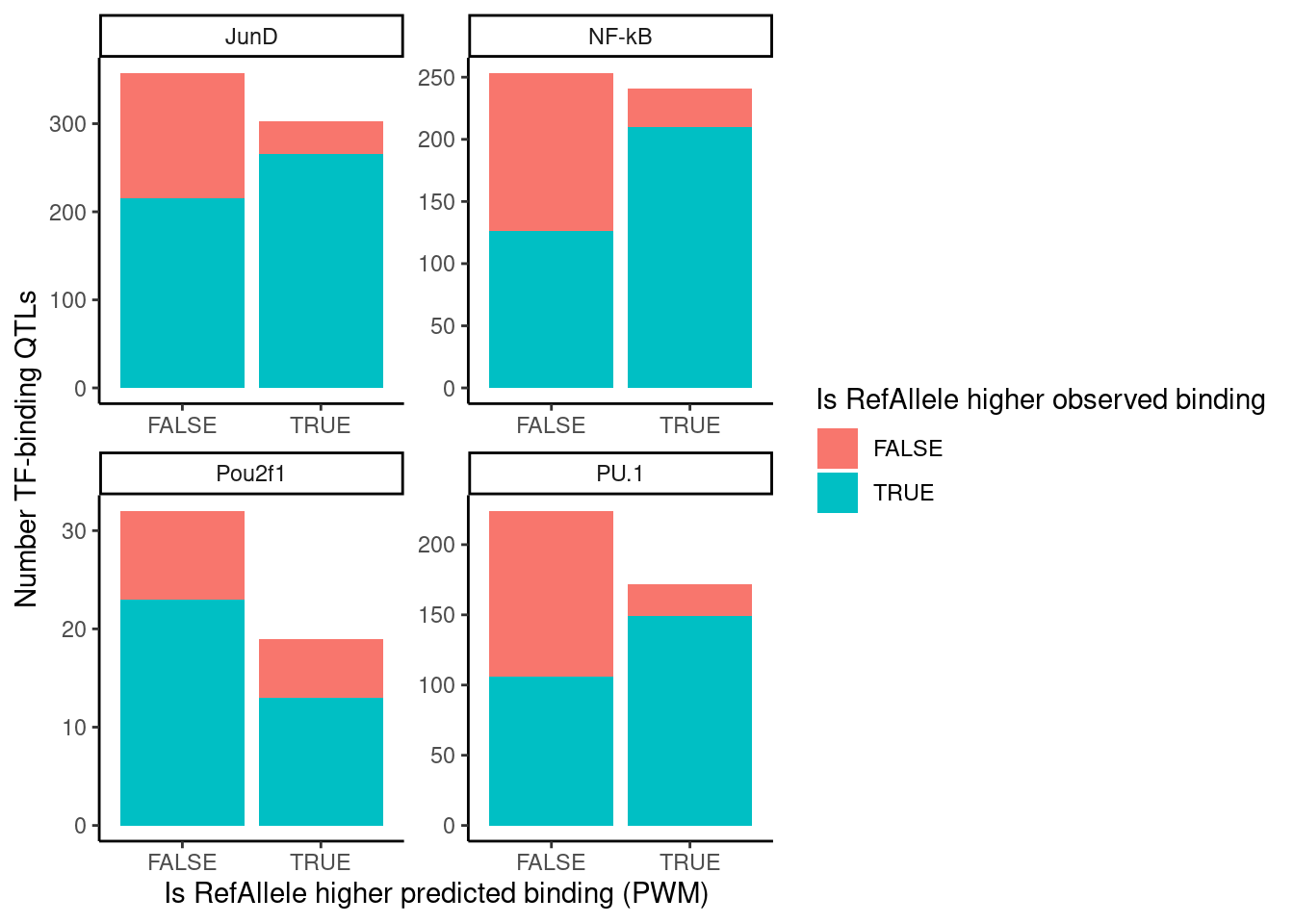
Most of the Pou2f1 might still be false positives, and there weren’t that many QTLs for that TF to begin with… Let’s just drop that from futher analysis. and in general there may still be some bias towards reference allele, perhaps due to mapping biases or something of the sort… But I think I am still picking up on a little signal in that when the referenceAllele has a higher PWM, it is more likely the higher binding allele. Let’s write out these TF QTLs to a new file, and consider quantifying splicing in-around these TF sites.
dat.filted.with.FDR %>%
filter(!TF == "Pou2f1") %>%
filter(q < 0.1) %>%
filter(!Delta==0) %>%
select(chrom, start, stop, DeltaPWM=Delta, P, TF) %>%
arrange(chrom, start, stop) %>%
write_tsv("../data/Tehranchi_PrimaryTFBinding-QTLs.bed.gz")
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] qvalue_2.16.0 forcats_0.4.0 stringr_1.4.0 dplyr_0.8.3
[5] purrr_0.3.4 readr_1.3.1 tidyr_1.1.0 tibble_2.1.3
[9] ggplot2_3.3.5 tidyverse_1.3.0
loaded via a namespace (and not attached):
[1] tidyselect_1.1.0 xfun_0.8 reshape2_1.4.3 splines_3.6.1
[5] haven_2.3.1 lattice_0.20-38 colorspace_1.4-1 vctrs_0.3.1
[9] generics_0.0.2 htmltools_0.3.6 yaml_2.2.0 utf8_1.1.4
[13] rlang_0.4.10 later_0.8.0 pillar_1.4.2 withr_2.4.1
[17] glue_1.3.1 DBI_1.1.0 dbplyr_1.4.2 modelr_0.1.8
[21] readxl_1.3.1 plyr_1.8.4 lifecycle_0.1.0 munsell_0.5.0
[25] gtable_0.3.0 workflowr_1.6.2 cellranger_1.1.0 rvest_0.3.5
[29] evaluate_0.14 labeling_0.3 knitr_1.23 httpuv_1.5.1
[33] fansi_0.4.0 broom_0.5.2 Rcpp_1.0.5 promises_1.0.1
[37] backports_1.1.4 scales_1.1.0 jsonlite_1.6 farver_2.1.0
[41] fs_1.3.1 hms_0.5.3 digest_0.6.20 stringi_1.4.3
[45] grid_3.6.1 rprojroot_2.0.2 cli_2.2.0 tools_3.6.1
[49] magrittr_1.5 crayon_1.3.4 pkgconfig_2.0.2 xml2_1.3.2
[53] reprex_0.3.0 lubridate_1.7.4 rstudioapi_0.10 assertthat_0.2.1
[57] rmarkdown_1.13 httr_1.4.1 R6_2.4.0 nlme_3.1-140
[61] git2r_0.26.1 compiler_3.6.1