ResponseToReviewer_Point6
Last updated: 2020-09-10
Checks: 6 1
Knit directory: Comparative_eQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.5.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190319) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: WorkingManuscript.zip
Ignored: WorkingManuscript/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis_temp/.DS_Store
Ignored: big_data/
Ignored: code/.DS_Store
Ignored: code/snakemake_workflow/.DS_Store
Ignored: code/snakemake_workflow/.Rhistory
Ignored: data/.DS_Store
Ignored: data/PastAnalysesDataToKeep/.DS_Store
Ignored: figures/
Ignored: output/.DS_Store
Unstaged changes:
Modified: analysis/20200907_Response_Point_06.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 5f95cbc | Benjmain Fair | 2020-09-10 | update site |
| html | 5f95cbc | Benjmain Fair | 2020-09-10 | update site |
Original reviewer point:
I would like to see more discussion about the inter-relatedness of the chimpanzees in the analysis of gene expression. Is that contributing to the power of the DE analysis, which has really high numbers of DE genes. That may certainly be due to the large samples size, but should be addressed. Related to that, the support that the gene-wise dispersion estimates are well correlated in humans and chimpanzees overall (Fig1C, and S4) seems qualitative. It looks like the chimpanzees might have less dispersion overall?
I will address the first point (about relatedness in DE analysis) empricially by reporfming DE analysis with subsamples of chimps that have some relatively high degree of inter-relatedness, compared to subsamples that do not have such relatedness.
First load necessary libraries for analysis…
library(tidyverse)
library(gplots)
library(readxl)
library(scales)Load relatedness matrix and RNA-seq sample batch info
#Relatedness matrix
SampleLabels <- read.table('../output/ForAssociationTesting.temp.fam', stringsAsFactors = F)$V2
GemmaMatrix <- as.matrix(read.table('../output/GRM.cXX.txt'))
colnames(GemmaMatrix) <- SampleLabels
row.names(GemmaMatrix) <- SampleLabels
#Metadata like RNA-seq batch
Metadata<-read_excel("../data/Metadata_SequencedChimps.xlsx")
Metadata$RNA.Extract_date %>% unique() %>% length()[1] 5Metadata$RNA.Library.prep.batch %>% unique() %>% length()[1] 5Colors <- data.frame(Numbers=1:5, Colors=hue_pal()(5))
BatchColor <- data.frame(IndividualID=as.character(colnames(GemmaMatrix))) %>%
left_join(Metadata, by=c("IndividualID"="IndividualID (as listed in vcf)")) %>%
dplyr::select(IndividualID, RNA.Library.prep.batch) %>%
left_join(Colors, by=c("RNA.Library.prep.batch"="Numbers")) %>%
pull(Colors) %>% as.character()
diag(GemmaMatrix) <- NA #For plotting purposes, just don't show the diagonol so that the color scale is better for non-diagnol entries
heatmap.2(GemmaMatrix, trace="none", RowSideColors = BatchColor, ColSideColors = BatchColor)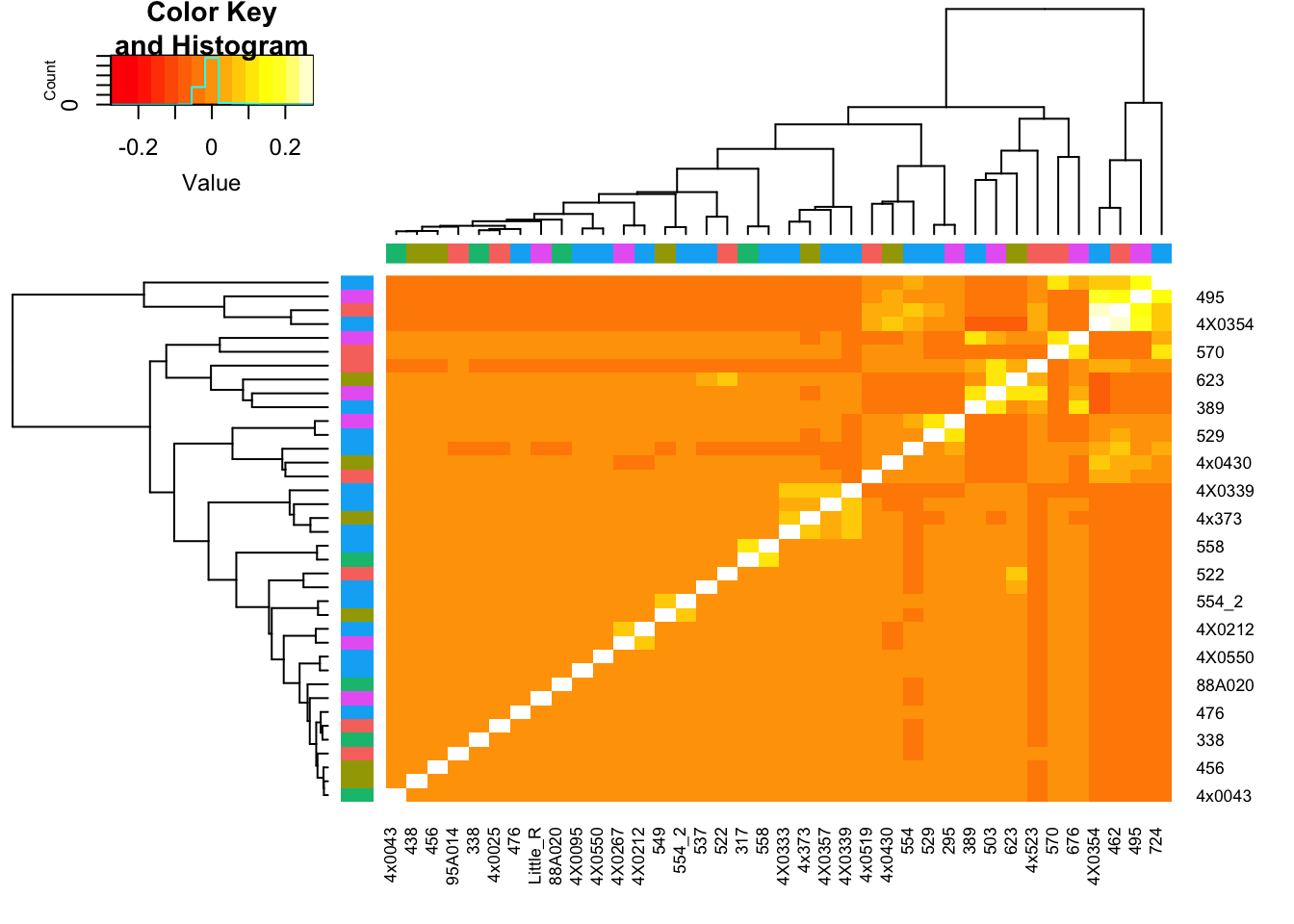
| Version | Author | Date |
|---|---|---|
| 5f95cbc | Benjmain Fair | 2020-09-10 |
I may pick out some of those somewhat related sample blocks for DE analysis… First, I should also try to match batches for this analysis.
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Catalina 10.15.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] scales_1.1.0 readxl_1.3.1 gplots_3.0.1.1 forcats_0.4.0
[5] stringr_1.4.0 dplyr_0.8.3 purrr_0.3.3 readr_1.3.1
[9] tidyr_1.0.0 tibble_2.1.3 ggplot2_3.2.1 tidyverse_1.3.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.5 lubridate_1.7.4 lattice_0.20-38 gtools_3.8.1
[5] assertthat_0.2.1 zeallot_0.1.0 rprojroot_1.3-2 digest_0.6.23
[9] R6_2.4.1 cellranger_1.1.0 backports_1.1.5 reprex_0.3.0
[13] evaluate_0.14 httr_1.4.1 pillar_1.4.2 rlang_0.4.1
[17] lazyeval_0.2.2 rstudioapi_0.10 gdata_2.18.0 whisker_0.4
[21] rmarkdown_1.18 munsell_0.5.0 broom_0.5.2 compiler_3.6.1
[25] httpuv_1.5.2 modelr_0.1.5 xfun_0.11 pkgconfig_2.0.3
[29] htmltools_0.4.0 tidyselect_0.2.5 workflowr_1.5.0 fansi_0.4.0
[33] crayon_1.3.4 dbplyr_1.4.2 withr_2.1.2 later_1.0.0
[37] bitops_1.0-6 grid_3.6.1 nlme_3.1-143 jsonlite_1.6
[41] gtable_0.3.0 lifecycle_0.1.0 DBI_1.0.0 git2r_0.26.1
[45] magrittr_1.5 KernSmooth_2.23-16 cli_2.0.0 stringi_1.4.3
[49] farver_2.0.1 fs_1.3.1 promises_1.1.0 xml2_1.2.2
[53] generics_0.0.2 vctrs_0.2.0 tools_3.6.1 glue_1.3.1
[57] hms_0.5.2 yaml_2.2.0 colorspace_1.4-1 caTools_1.17.1.3
[61] rvest_0.3.5 knitr_1.26 haven_2.2.0