20190708_SpeciesSharedPolymorphisms_QQPlot
Ben Fair
7/9/2019
Last updated: 2019-07-11
Checks: 6 1
Knit directory: Comparative_eQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.4.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190319) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/20190521_eQTL_CrossSpeciesEnrichment_cache/
Ignored: analysis_temp/.DS_Store
Ignored: code/.DS_Store
Ignored: code/snakemake_workflow/.DS_Store
Ignored: data/.DS_Store
Ignored: data/PastAnalysesDataToKeep/.DS_Store
Ignored: docs/.DS_Store
Ignored: docs/assets/.DS_Store
Untracked files:
Untracked: analysis/20190627_DiffContactsEgenes.Rmd
Untracked: analysis/20190708_DiffContactsEgenes_CisWindowControlled.Rmd
Untracked: analysis/20190708_SpeciesSharedPolymorphisms_InitialQQPlot.Rmd
Untracked: analysis/20190808_HumanLeadSnps.Rmd
Untracked: docs/figure/20190627_DiffContactsEgenes.Rmd/
Untracked: docs/figure/20190708_DiffContactsEgenes_CisWindowControlled.Rmd/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Lefler et al observed a number of species shared haplotpes (at least two shared snps) between chimp and human, which could be mainted through balancing selection. The targets of the selection were not always clear, and often the shared snps were non-coding, leading the authors to propose that they control gene expression. Some of the shared snps are in LD with eQTLs identified in human monocytes. Here I will check if I see these snps are eQTLs in my chimp heart samples. As an initial look at this, here I will plot a QQ-plot from my initial eQTL calling pipeline (250kb cis-window) for all snp-gene pairs for snps that were species shared polymorphisms, and a random sample of snp-gene pairs.
library(tidyverse)
shared.snps <- read.table('../output/SharedPolymorphisms.shared.chimpeqtls.txt', col.names = c('snp','gene','beta','stat','p','fdr','q'))
random.snps <- read.table('../output/SharedPolymorphisms.random.chimpeqtls.txt', col.names = c('snp','gene','beta','stat','p','fdr','q'))
ggplot(random.snps, aes(color="Random chimp variants", y=-log10(sort(p)), x=-log10(1:length(p)/length(p)))) +
geom_point() +
geom_point(data=shared.snps, aes(color="Variants shared with human")) +
xlab("-log10(Theoretical-Pvalues)") +
ylab("-log10(Observed-Pvalues)") +
geom_abline() +
theme_bw() +
theme(legend.position="bottom") +
theme(legend.title=element_blank())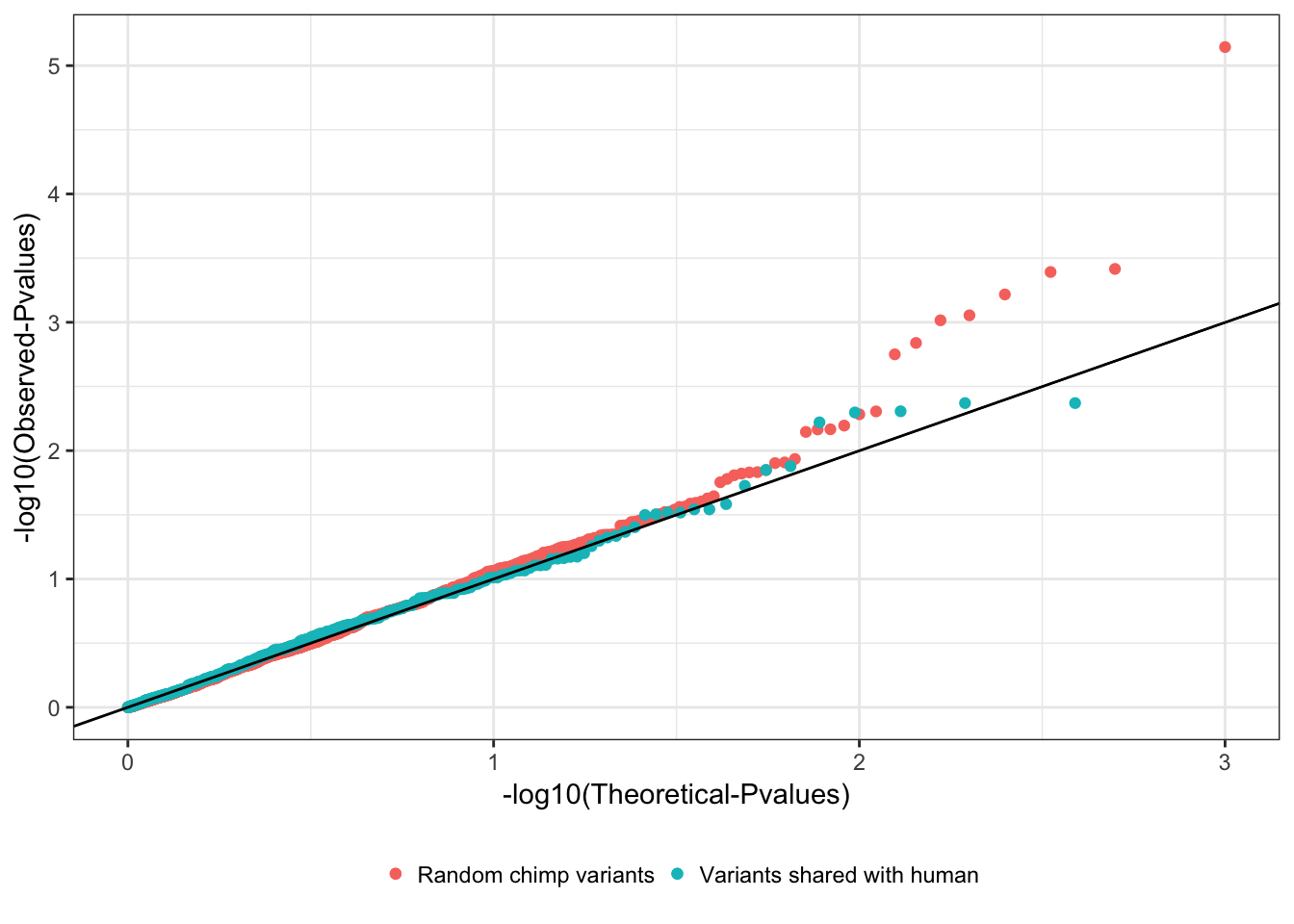
wilcox.test(random.snps$p, shared.snps$p, alternative='greater')
Wilcoxon rank sum test with continuity correction
data: random.snps$p and shared.snps$p
W = 198880, p-value = 0.2572
alternative hypothesis: true location shift is greater than 0A few things to note that I could do more carefully to assess if the shared polymorphisms are acting as eQTLs:
- a lot of the snps are not within 250kb of the target gene that they may be acting through. For example, the snps around FREM/GYPE locus are probably acting throuh GYPE, as this, like HLA, is involved in pathogen defense and under odd selection constraints. This is what Lefler proposed. However, the I did not test that snp-gene pair.
- I should make some locus zoom plots in human and chimp for heart, since maybe these snps don’t even act as eqtls in human heart.
- IGFBP7 (ENSPTRP00000027670) might be a particularly interesting example of a putative balanced eQTL from Lefler et al to make a locuszoom plot for both species, as in GTEx it is a left-ventricle heart specific eQTL. However I see now this snp was not called in my chimp data. Maybe a locuszoom plot will still see tags. Why this snp was genotyped could also point to a genotype error… For example, it could be a duplicated region in the genome that was thrown out of my snp-calling pipeline but not Lefler et al’s.
- May be worthwhile making the same plot from human GTEx data for multiple tissues, since my chimp data is relatively underpowered.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.4.0 stringr_1.4.0 dplyr_0.8.1 purrr_0.3.2
[5] readr_1.3.1 tidyr_0.8.3 tibble_2.1.3 ggplot2_3.1.1
[9] tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.1 cellranger_1.1.0 plyr_1.8.4 pillar_1.4.1
[5] compiler_3.5.1 git2r_0.25.2 workflowr_1.4.0 tools_3.5.1
[9] digest_0.6.19 lubridate_1.7.4 jsonlite_1.6 evaluate_0.14
[13] nlme_3.1-140 gtable_0.3.0 lattice_0.20-38 pkgconfig_2.0.2
[17] rlang_0.3.4 cli_1.1.0 rstudioapi_0.10 yaml_2.2.0
[21] haven_2.1.0 xfun_0.7 withr_2.1.2 xml2_1.2.0
[25] httr_1.4.0 knitr_1.23 hms_0.4.2 generics_0.0.2
[29] fs_1.3.1 rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5
[33] glue_1.3.1 R6_2.4.0 readxl_1.3.1 rmarkdown_1.13
[37] modelr_0.1.4 magrittr_1.5 backports_1.1.4 scales_1.0.0
[41] htmltools_0.3.6 rvest_0.3.4 assertthat_0.2.1 colorspace_1.4-1
[45] labeling_0.3 stringi_1.4.3 lazyeval_0.2.2 munsell_0.5.0
[49] broom_0.5.2 crayon_1.3.4