Data integration and clustering of cardiomyocytes: fetal, young, adult
Belinda Phipson
10/17/2019
Last updated: 2019-10-21
Checks: 6 0
Knit directory: Porello-heart-snRNAseq/
This reproducible R Markdown analysis was created with workflowr (version 1.3.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190603) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: analysis/08-ClustEpicardial.Rmd
Untracked: analysis/PBapproachMarker-cardio.Rmd
Untracked: analysis/doublet-detection.Rmd
Untracked: broad_props.csv
Untracked: code/ReadDataObjects.R
Untracked: code/getTransformedProps.R
Untracked: data/adult-clust.txt
Untracked: data/dcm-clust.txt
Untracked: data/fetal-clust.txt
Untracked: data/gstlist-adult.Rdata
Untracked: data/gstlist-dcm-res03.Rdata
Untracked: data/gstlist-dcm.Rdata
Untracked: data/gstlist-fetal.Rdata
Untracked: data/gstlist-young.Rdata
Untracked: data/heart-markers-long.txt
Untracked: data/immune-markers-long.txt
Untracked: data/pseudobulk-pool.Rds
Untracked: data/pseudobulk.Rds
Untracked: data/targets_pools.txt
Untracked: data/young-clust.txt
Untracked: output/AllAdult-clustermarkers-v2.csv
Untracked: output/AllAdult-clustermarkers.csv
Untracked: output/AllFetal-clustermarkers.csv
Untracked: output/AllYoung-clustermarkers.csv
Untracked: output/Alldcm-clustermarkers.csv
Untracked: output/DEAnalysis/
Untracked: output/Figures/
Untracked: output/MarkerAnalysis/
Untracked: output/RDataObjects/
Untracked: output/cardio-numcells-clusters.csv
Untracked: output/cardio-numcells-clusters.txt
Untracked: output/fetal1-clustermarkers.csv
Untracked: output/fetal2-clustermarkers.csv
Untracked: output/fetal3-clustermarkers.csv
Untracked: output/heatmap-top10-adultmarkergenes.pdf
Untracked: output/young1-clustermarkers.csv
Unstaged changes:
Modified: analysis/01-QualityControl.Rmd
Modified: analysis/01a-DEpseudobulk.Rmd
Modified: analysis/02-ClusterFetal.Rmd
Modified: analysis/02c-ClusterFetal3.Rmd
Modified: analysis/03-ClusterYoung.Rmd
Modified: analysis/04-ClusterAdult.Rmd
Modified: analysis/07b-DECardio.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 6c4e378 | Belinda Phipson | 2019-10-21 | update cardiomyocyte clustering with filtered data |
| html | 67c1d63 | Belinda Phipson | 2019-10-18 | Build site. |
| Rmd | eaa7f38 | Belinda Phipson | 2019-10-18 | clustering of development samples |
Load libraries and functions
library(edgeR)
library(RColorBrewer)
library(org.Hs.eg.db)
library(limma)
library(Seurat)
library(monocle)
library(cowplot)
library(DelayedArray)
library(scran)
library(NMF)
library(workflowr)
library(ggplot2)
library(clustree)
library(dplyr)source("/misc/card2-single_cell_nuclei_rnaseq/Porello-heart-snRNAseq/code/normCounts.R")
source("/misc/card2-single_cell_nuclei_rnaseq/Porello-heart-snRNAseq/code/findModes.R")
source("/misc/card2-single_cell_nuclei_rnaseq/Porello-heart-snRNAseq/code/ggplotColors.R")targets <- read.delim("/misc/card2-single_cell_nuclei_rnaseq/Porello-heart-snRNAseq/data/targets.txt",header=TRUE, stringsAsFactors = FALSE)
targets$FileName2 <- paste(targets$FileName,"/",sep="")
targets$Group_ID2 <- gsub("LV_","",targets$Group_ID)
group <- c("Fetal_1","Fetal_2","Fetal_3",
"Young_1","Young_2","Young_3",
"Adult_1","Adult_2","Adult_3",
"Diseased_1","Diseased_2",
"Diseased_3","Diseased_4")
m <- match(group, targets$Group_ID2)
targets <- targets[m,]fetal.integrated <- readRDS(file="./output/RDataObjects/fetal-int.Rds")
load(file="./output/RDataObjects/fetalObjs.Rdata")
young.integrated <- readRDS(file="./output/RDataObjects/young-int.Rds")
load(file="./output/RDataObjects/youngObjs.Rdata")
adult.integrated <- readRDS(file="./output/RDataObjects/adult-int.Rds")
load(file="./output/RDataObjects/adultObjs.Rdata")Set default clustering resolution
# Default 0.3
Idents(fetal.integrated) <- fetal.integrated$integrated_snn_res.0.3
DimPlot(fetal.integrated, reduction = "tsne",label=TRUE,label.size = 6)+NoLegend()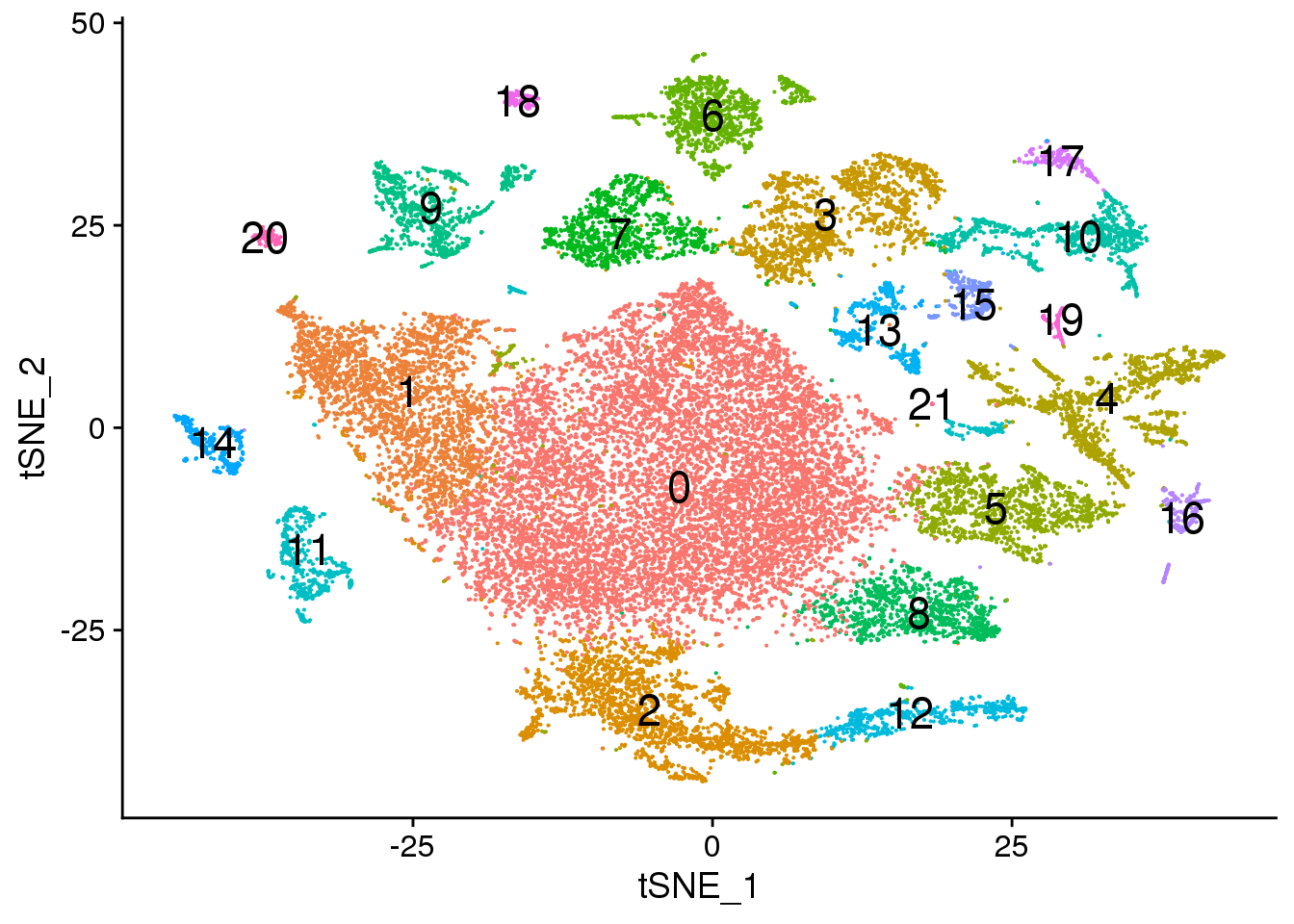
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
# Default 0.3
DimPlot(young.integrated, reduction = "tsne",label=TRUE,label.size = 6)+NoLegend()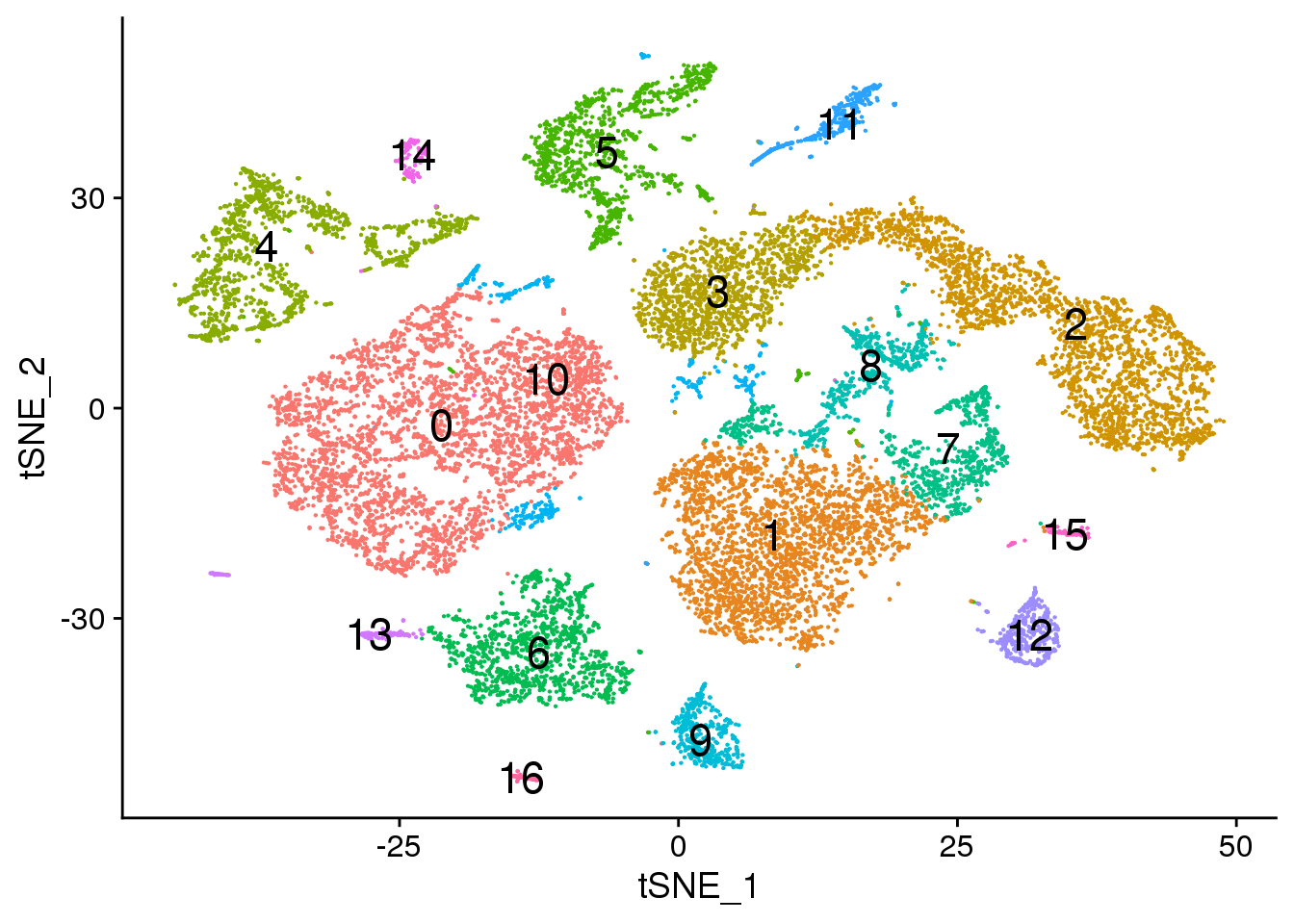
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
# Default 0.6
DimPlot(adult.integrated, reduction = "tsne",label=TRUE,label.size = 6)+NoLegend()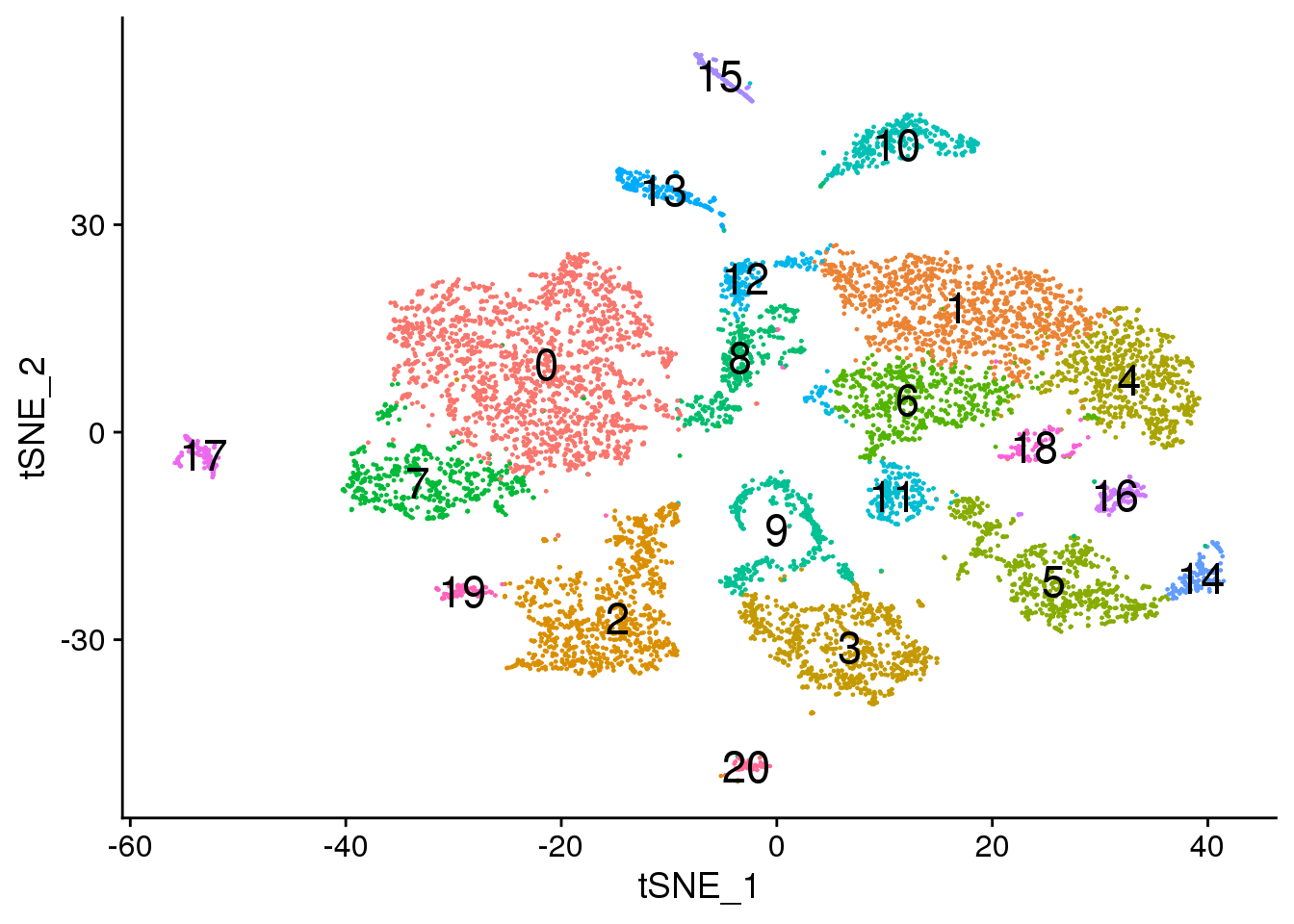
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
Merge all data together
heart <- merge(fetal.integrated, y = c(young.integrated, adult.integrated), project = "heart")
table(heart$orig.ident)
adult fetal young
9416 27760 16964 Get cardiomyocytes only
cardio <- subset(heart,subset = Broad_celltype == "Cardiomyocytes")Filter out crappy cells
Cardiomyocytes are fairly large cells and we wouldn’t expect them to only be expressing very few genes.
par(mfrow=c(1,2))
plot(density(cardio$nFeature_RNA),main="Number of genes detected")
abline(v=500,col=2)
plot(density(cardio$nCount_RNA),main="Library size")
abline(v=2500,col=2)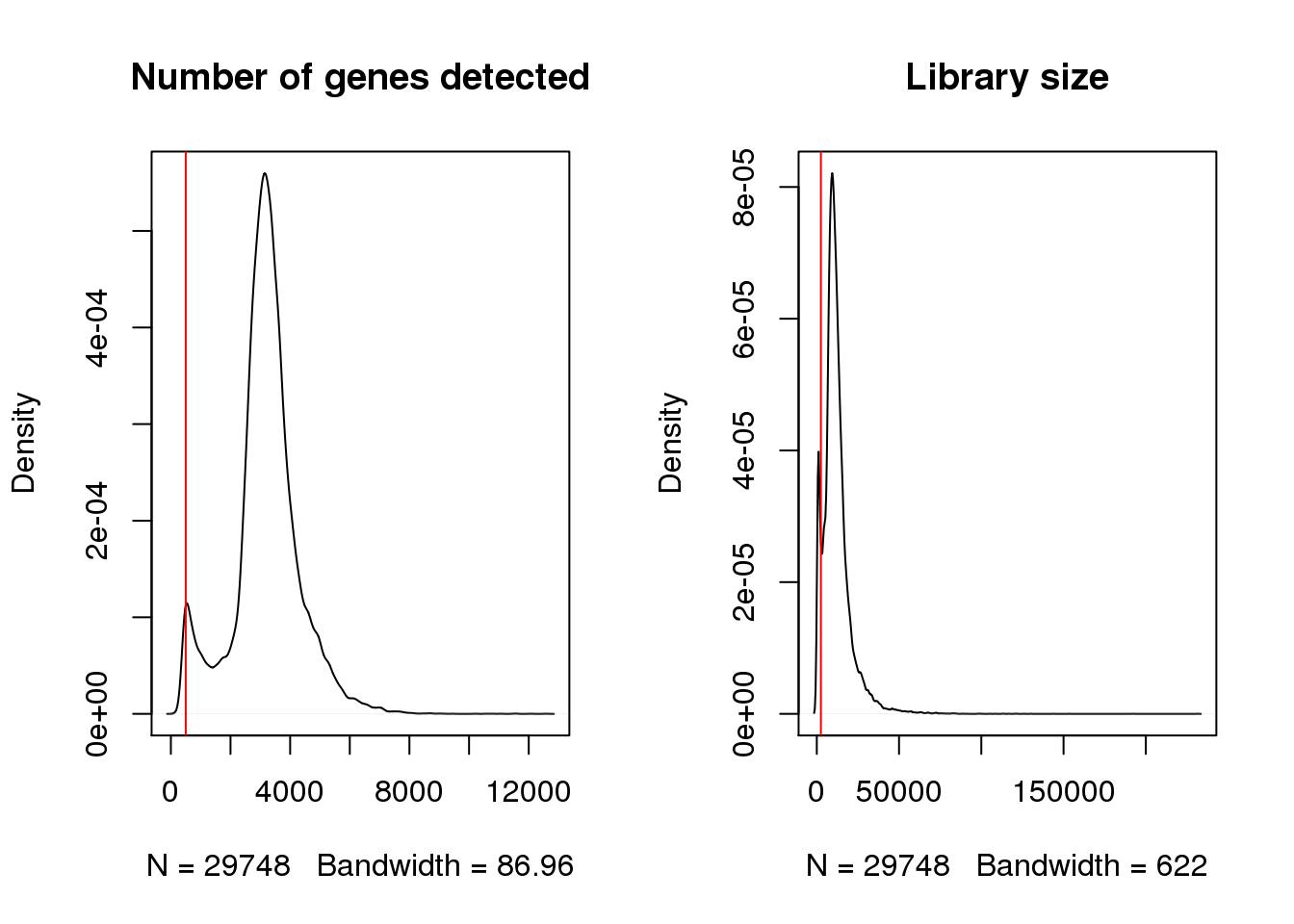
cardio <- subset(cardio, subset = nFeature_RNA > 500 & nCount_RNA > 2500)
dim(cardio)[1] 4256 27037table(cardio$biorep)
a1 a2 a3 f1 f2 f3 y1 y2 y3
1768 526 216 5435 8462 5114 1049 1962 2505 Run new integration with SCtransform normalisation
The new code does not work so I will take the same approach that I took integrating the samples within groups.
cardio.list <- SplitObject(cardio, split.by = "biorep")
min(sapply(cardio.list, ncol))[1] 216for (i in 1:length(cardio.list)) {
cardio.list[[i]] <- SCTransform(cardio.list[[i]], verbose = FALSE)
}cardio.anchors <- FindIntegrationAnchors(object.list = cardio.list, dims=1:30,anchor.features = 3000,k.filter=216)cardio.integrated <- IntegrateData(anchorset = cardio.anchors,dims=1:30)Perform clustering
DefaultAssay(object = cardio.integrated) <- "integrated"Perform scaling and PCA
cardio.integrated <- ScaleData(cardio.integrated, verbose = FALSE)
cardio.integrated <- RunPCA(cardio.integrated, npcs = 50, verbose = FALSE)
ElbowPlot(cardio.integrated,ndims=50)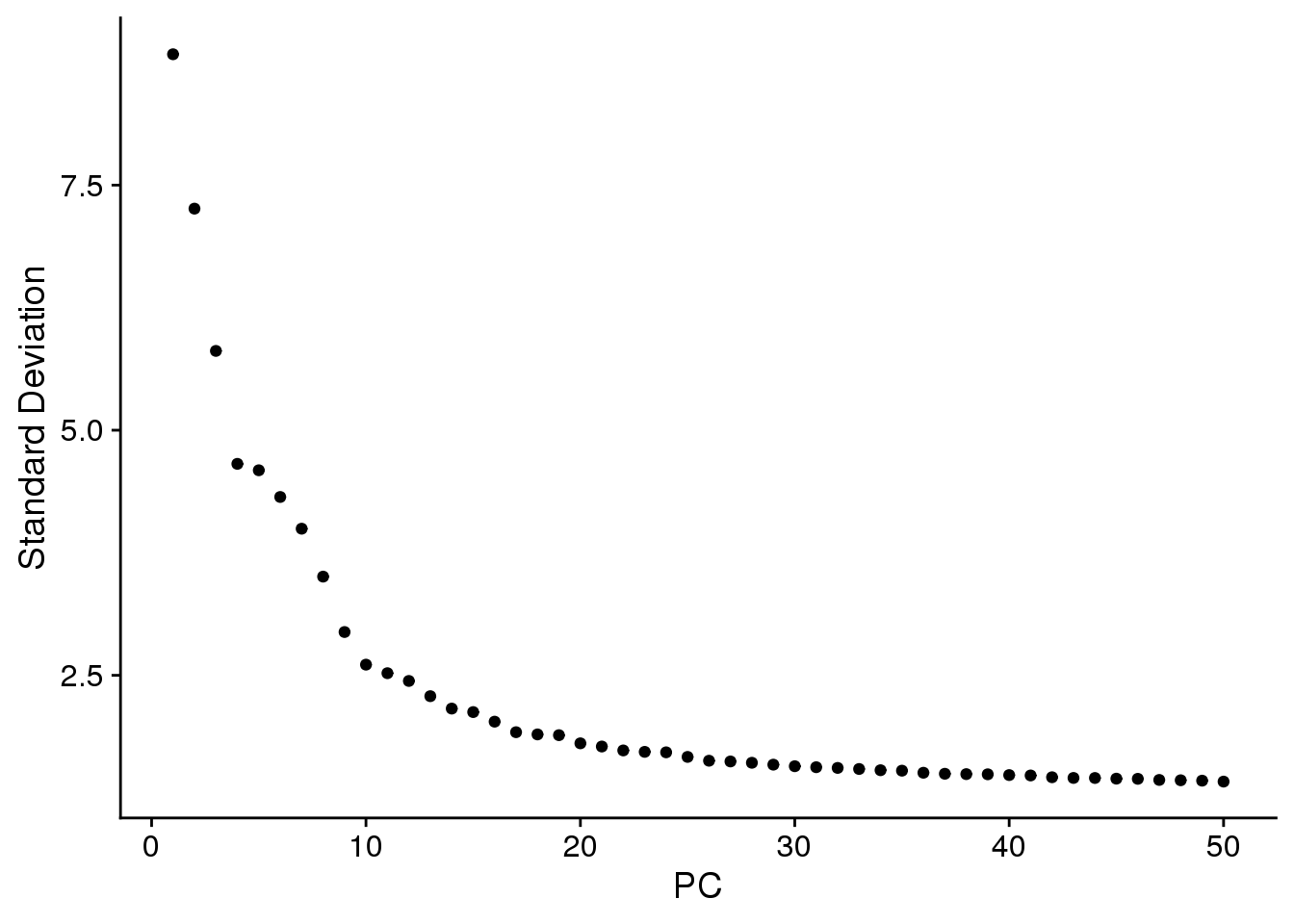
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
VizDimLoadings(cardio.integrated, dims = 1:4, reduction = "pca")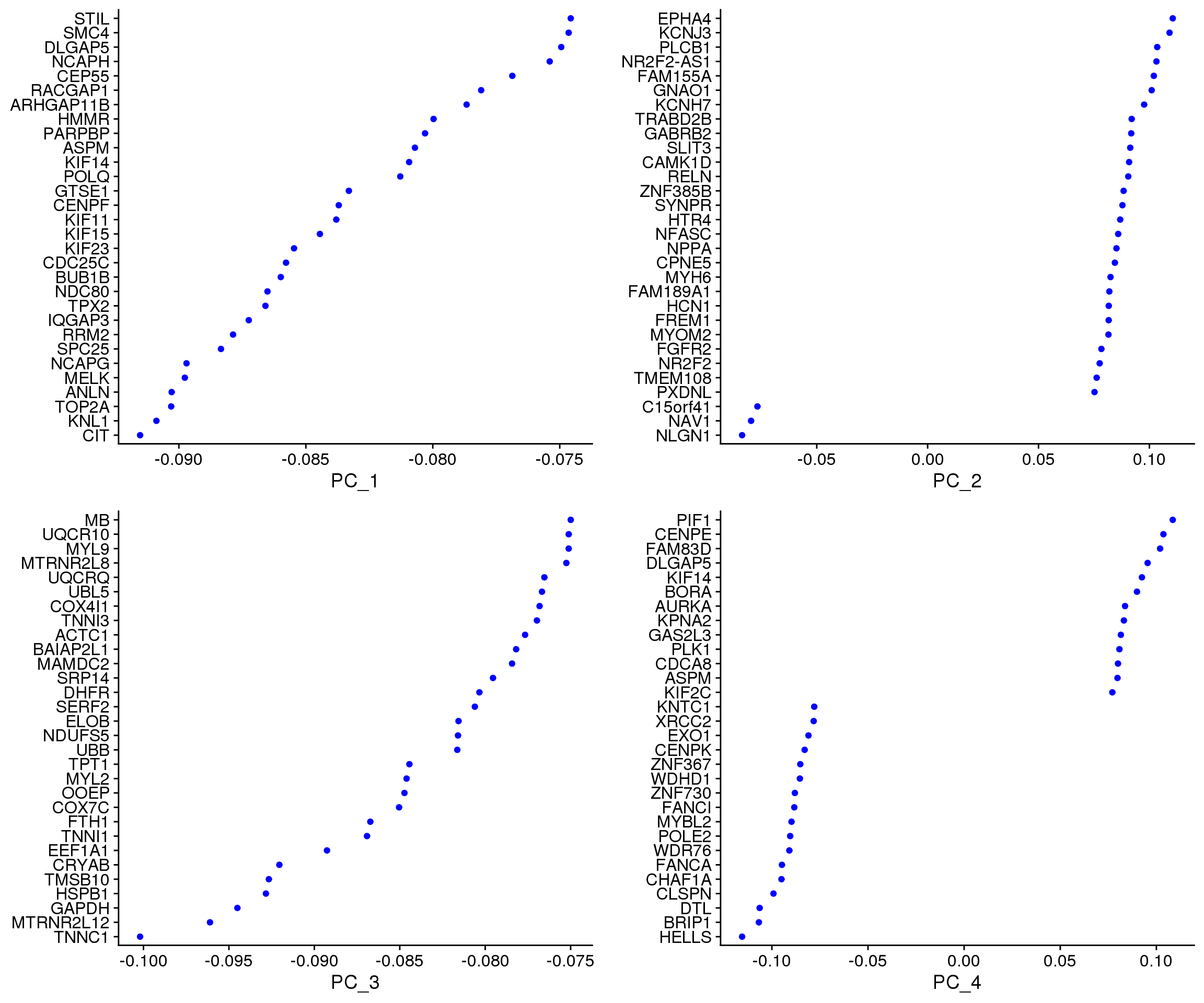
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "pca",group.by="orig.ident")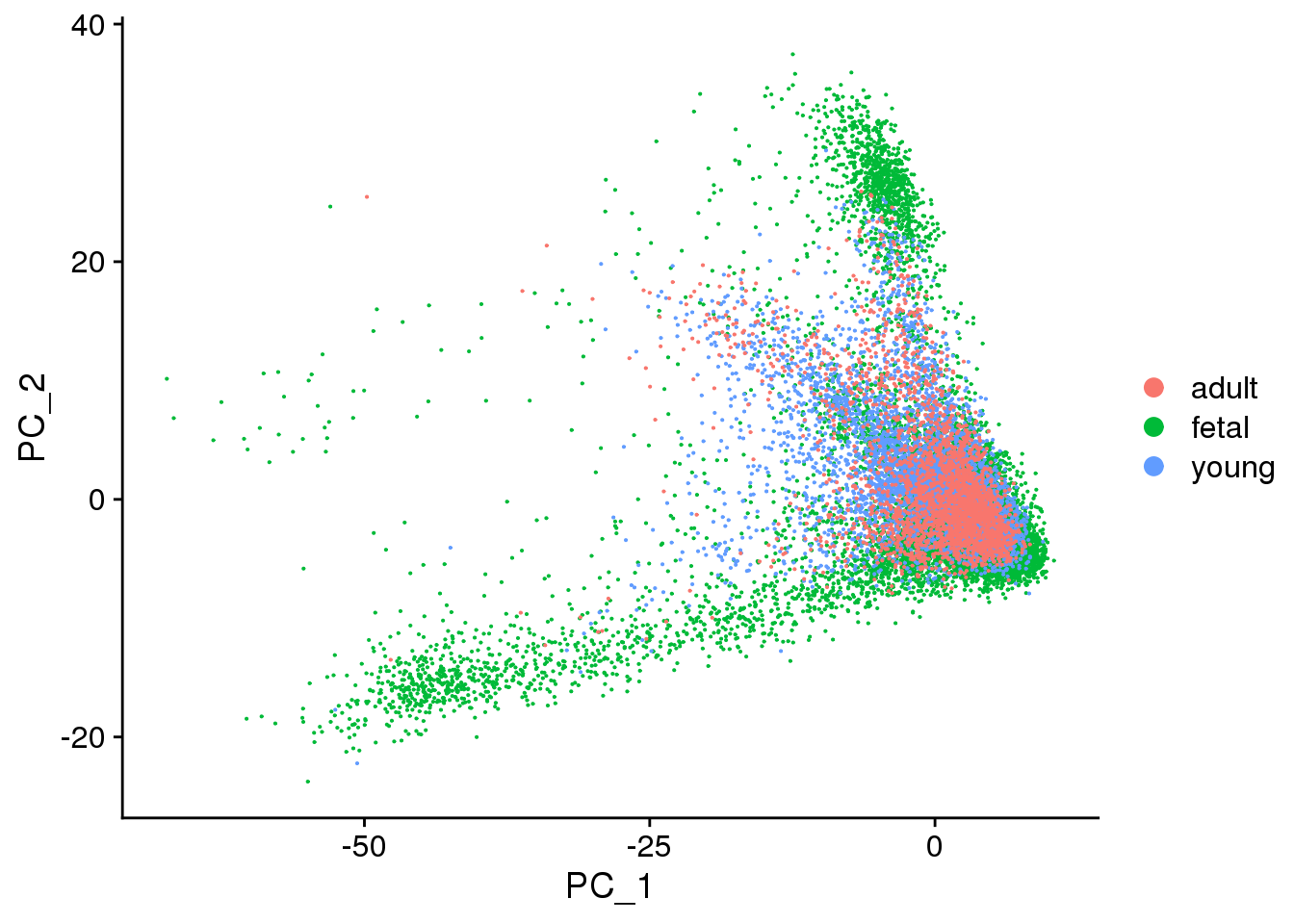
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "pca",group.by="biorep")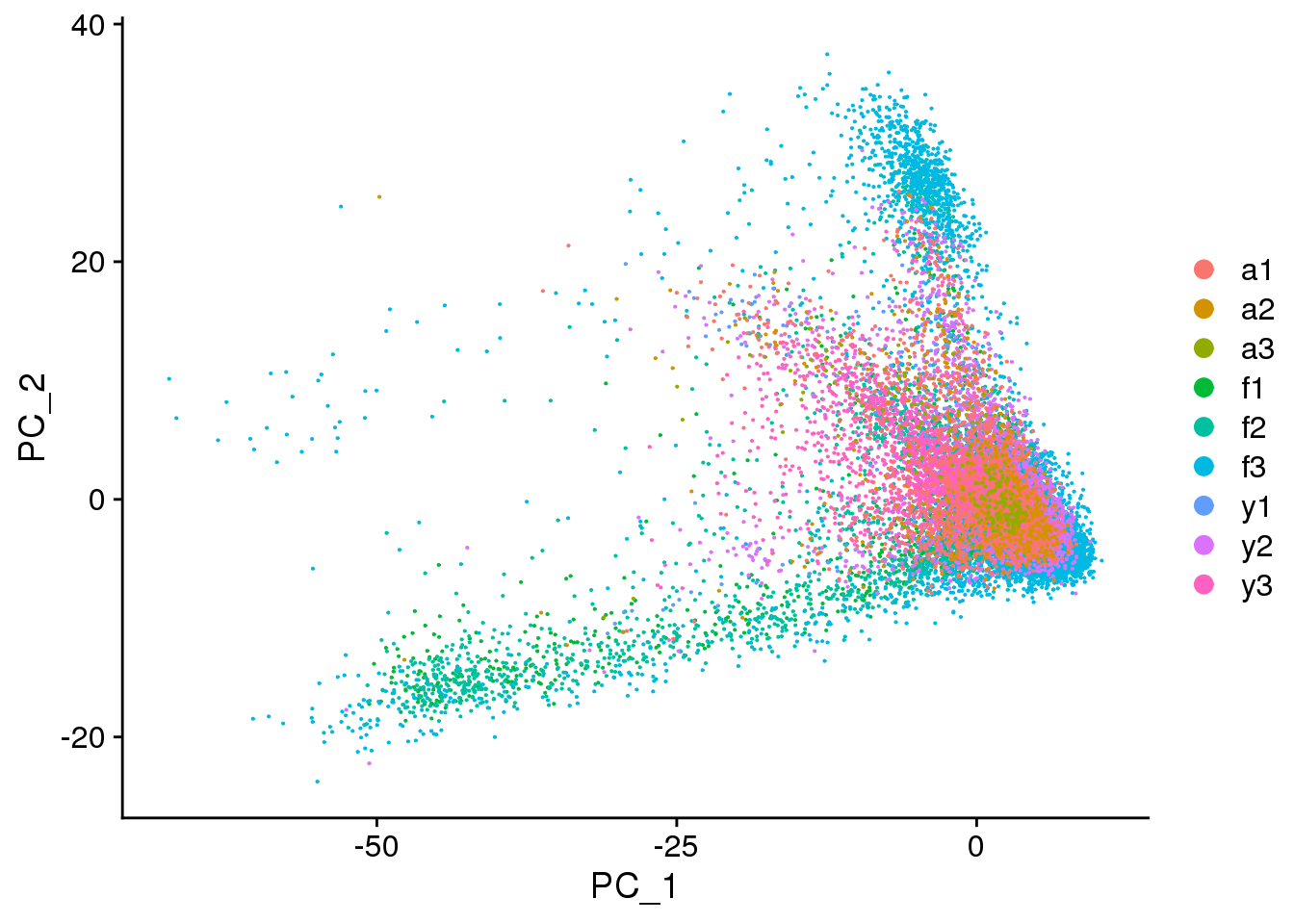
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "pca",group.by="sex")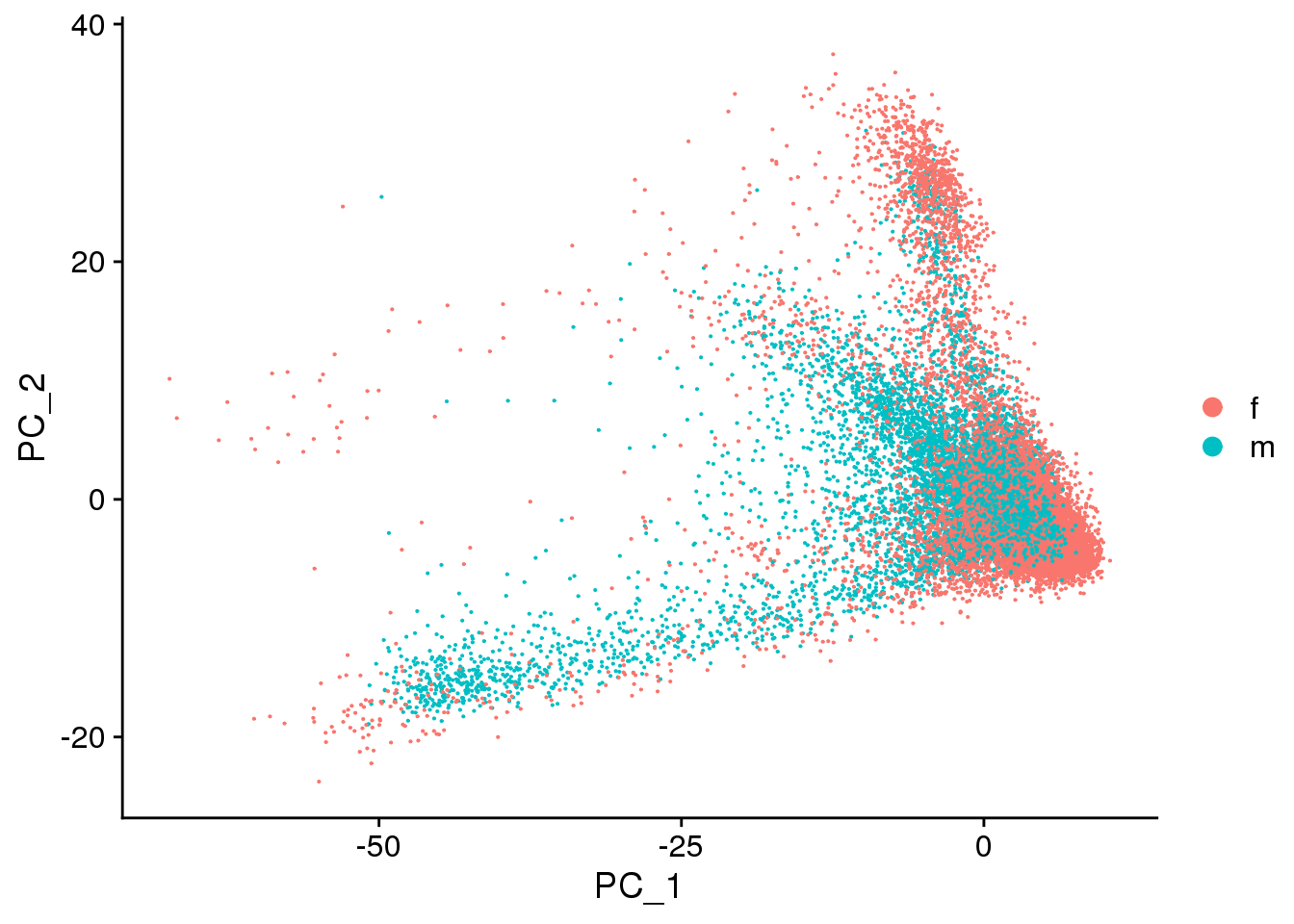
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "pca",group.by="batch")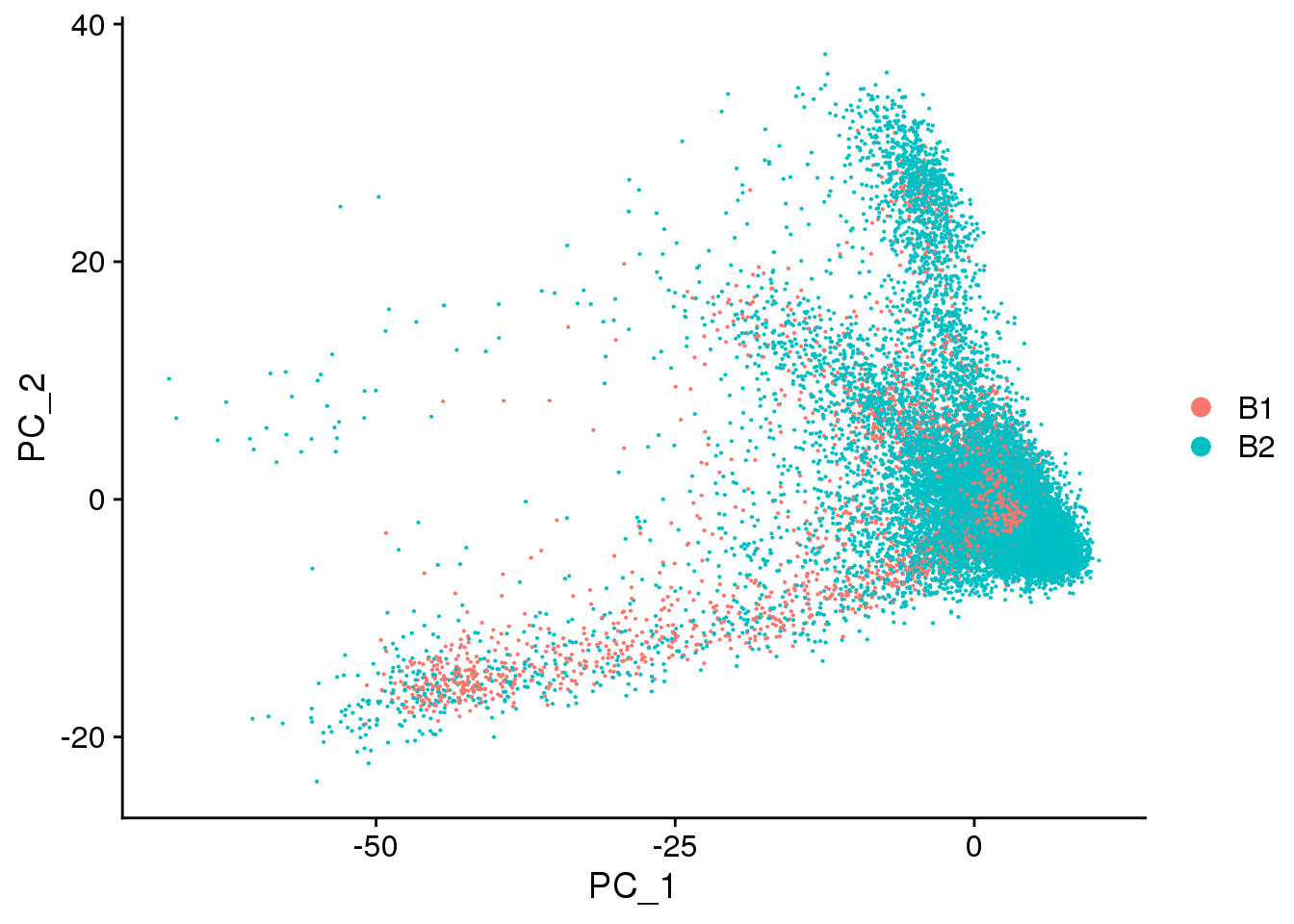
DimHeatmap(cardio.integrated, dims = 1:15, cells = 500, balanced = TRUE)
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimHeatmap(cardio.integrated, dims = 16:30, cells = 500, balanced = TRUE)
DimHeatmap(cardio.integrated, dims = 31:45, cells = 500, balanced = TRUE)
Perform nearest neighbours clustering
cardio.integrated <- FindNeighbors(cardio.integrated, dims = 1:20)
cardio.integrated <- FindClusters(cardio.integrated, resolution = 0.1)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9410
Number of communities: 7
Elapsed time: 10 secondstable(Idents(cardio.integrated))
0 1 2 3 4 5 6
18330 2849 2101 1280 1121 1016 340 par(mar=c(5,4,2,2))
barplot(table(Idents(cardio.integrated)),ylab="Number of cells",xlab="Clusters")
title("Number of cells in each cluster")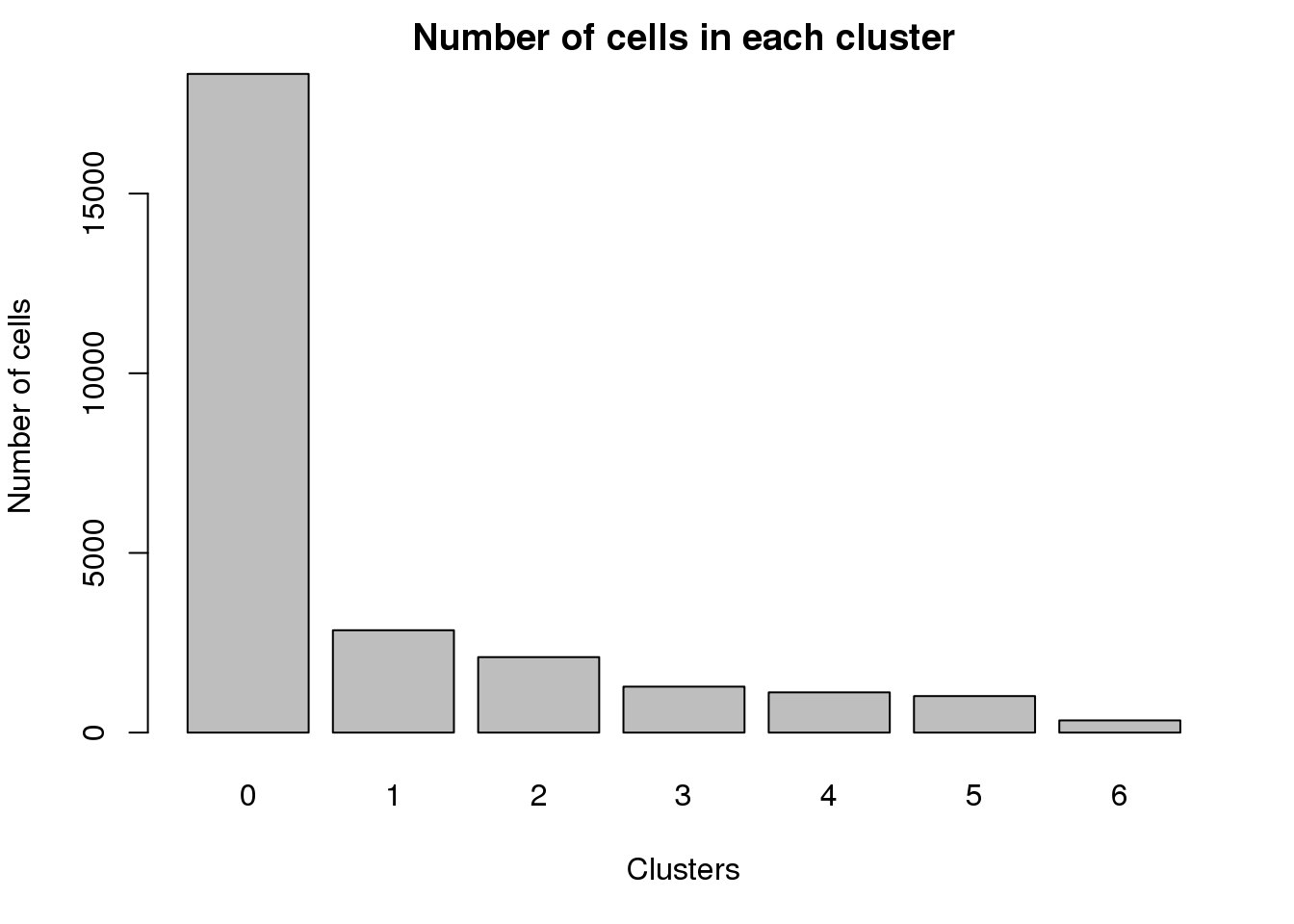
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
Visualisation with TSNE
set.seed(10)
cardio.integrated <- RunTSNE(cardio.integrated, reduction = "pca", dims = 1:20)DimPlot(cardio.integrated, reduction = "tsne",label=TRUE,label.size = 6,pt.size = 0.5)+NoLegend()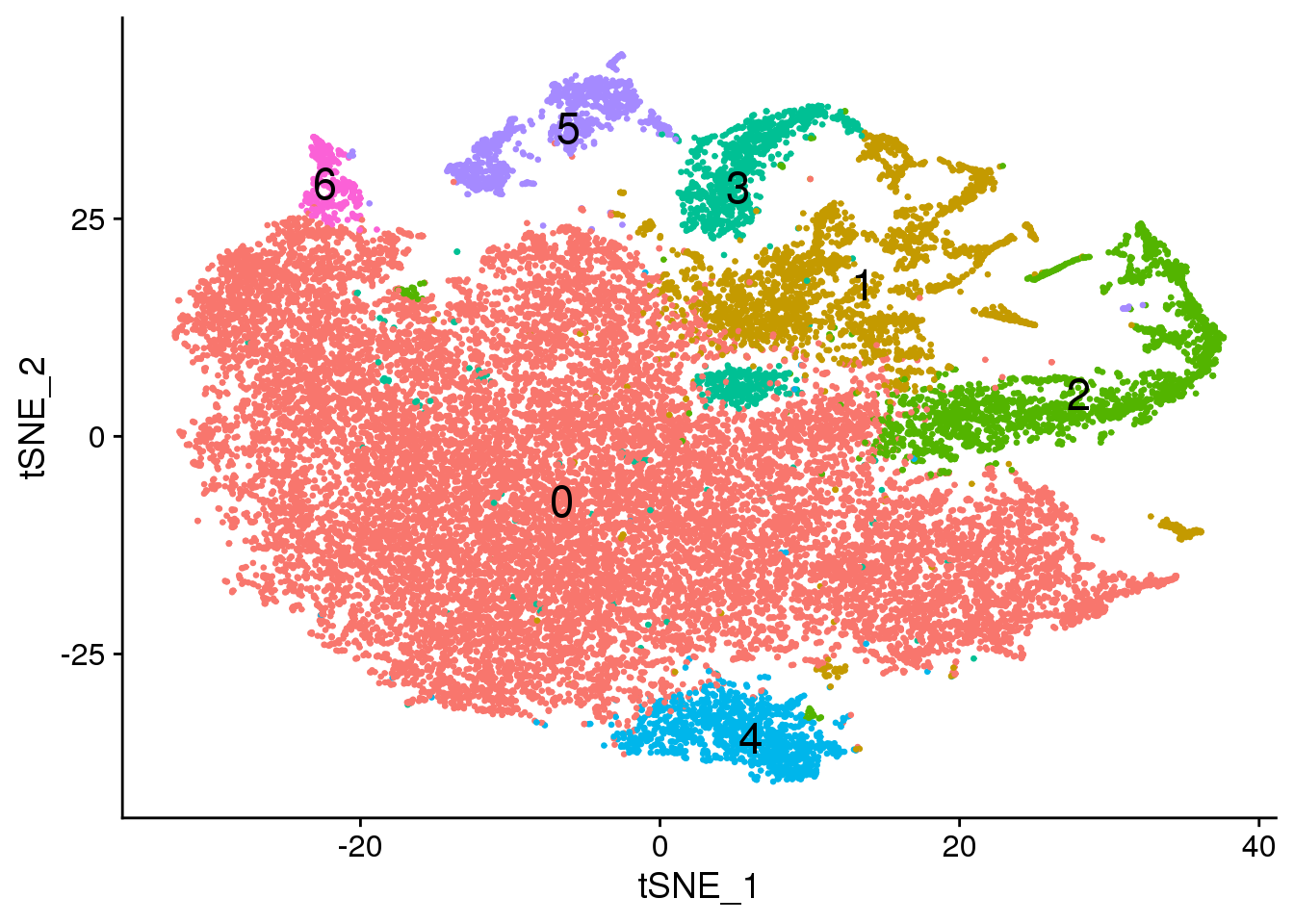
pdf(file="./output/Figures/NormalDev/tsne-cardioALL-res01.pdf",width=10,height=8,onefile = FALSE)
DimPlot(cardio.integrated, reduction = "tsne",label=TRUE,label.size = 6,pt.size = 0.5)+NoLegend()
dev.off()png
2 DimPlot(cardio.integrated, reduction = "tsne", group.by = "orig.ident")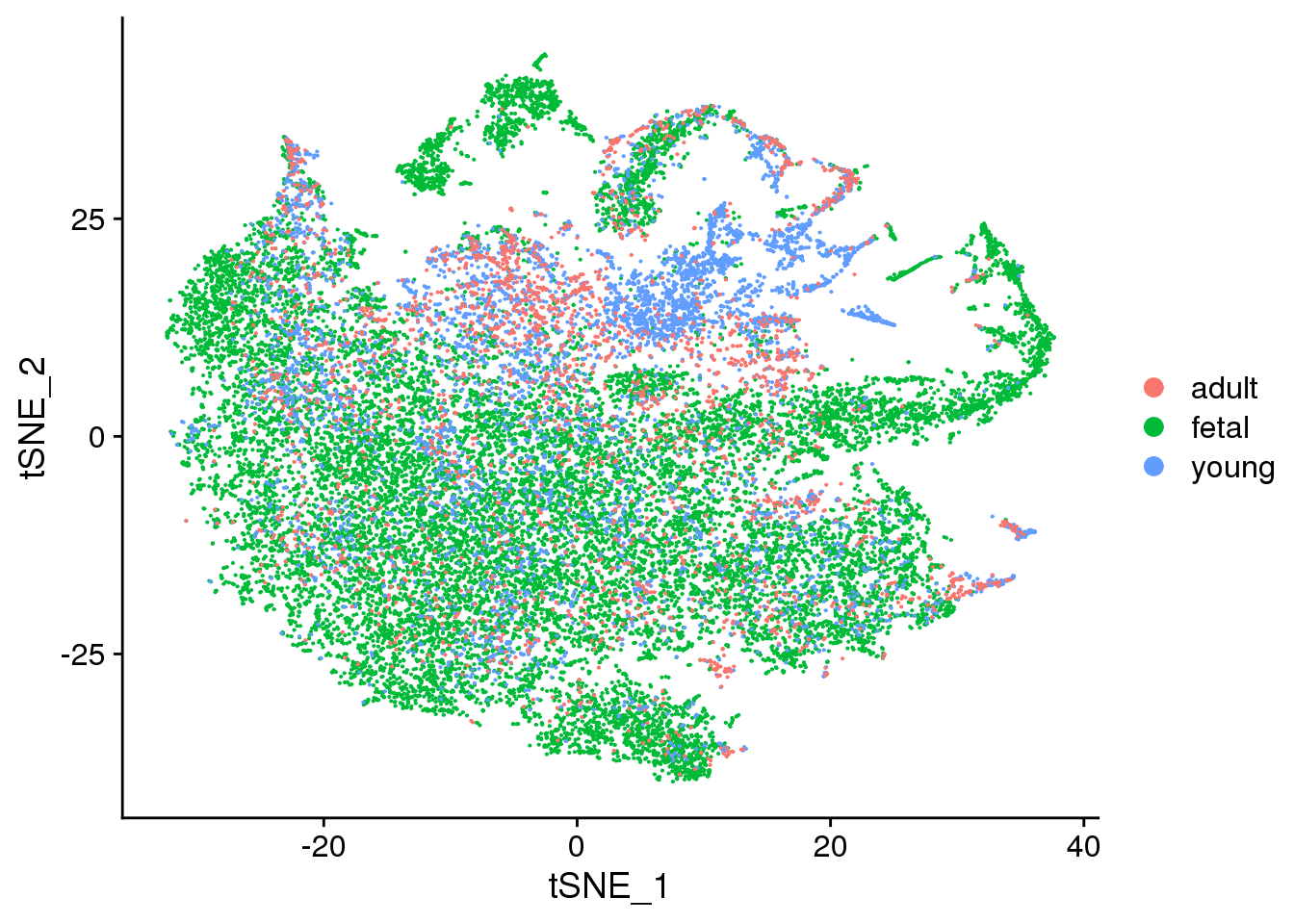
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "tsne", split.by = "orig.ident")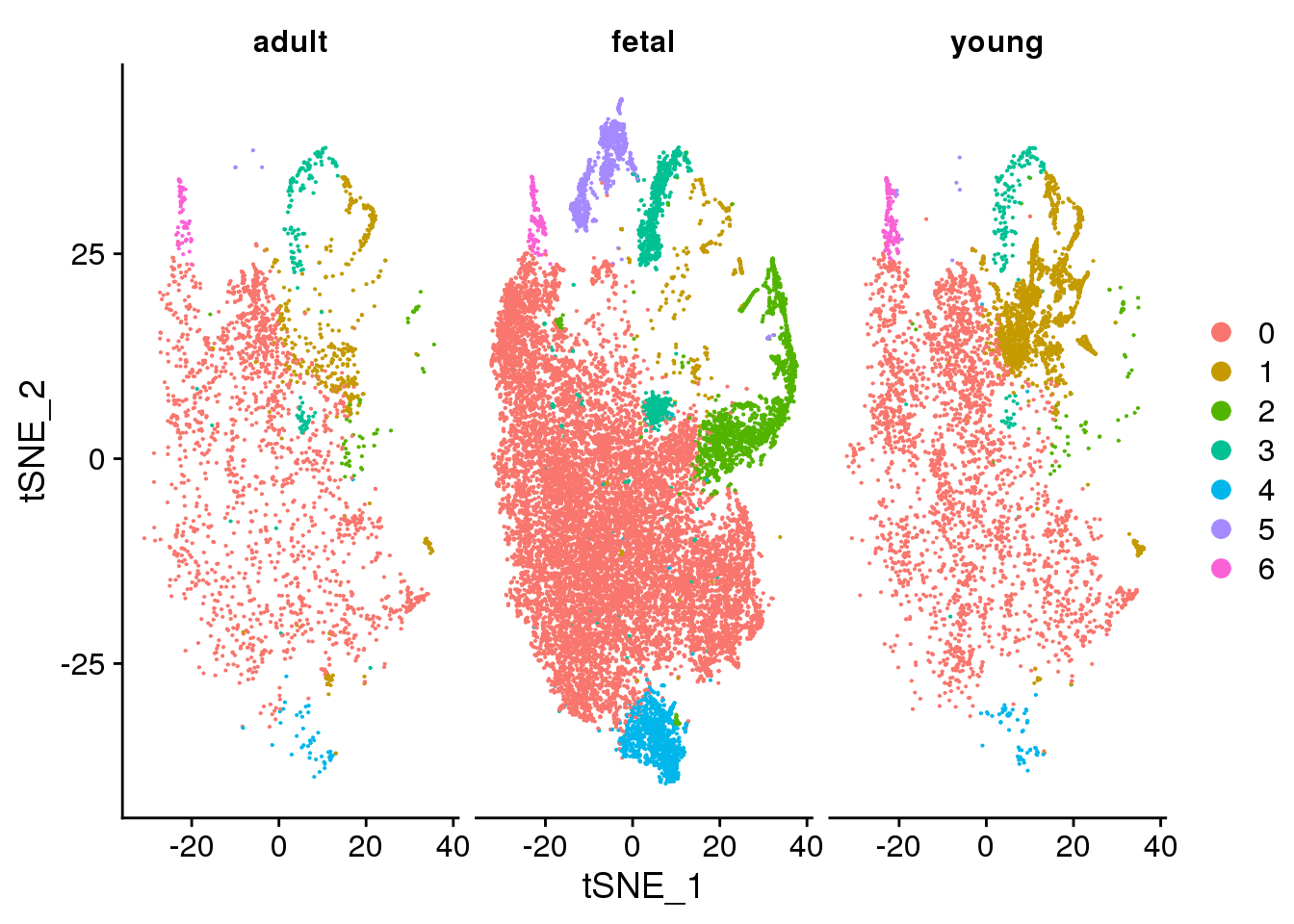
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "tsne", group.by = "biorep")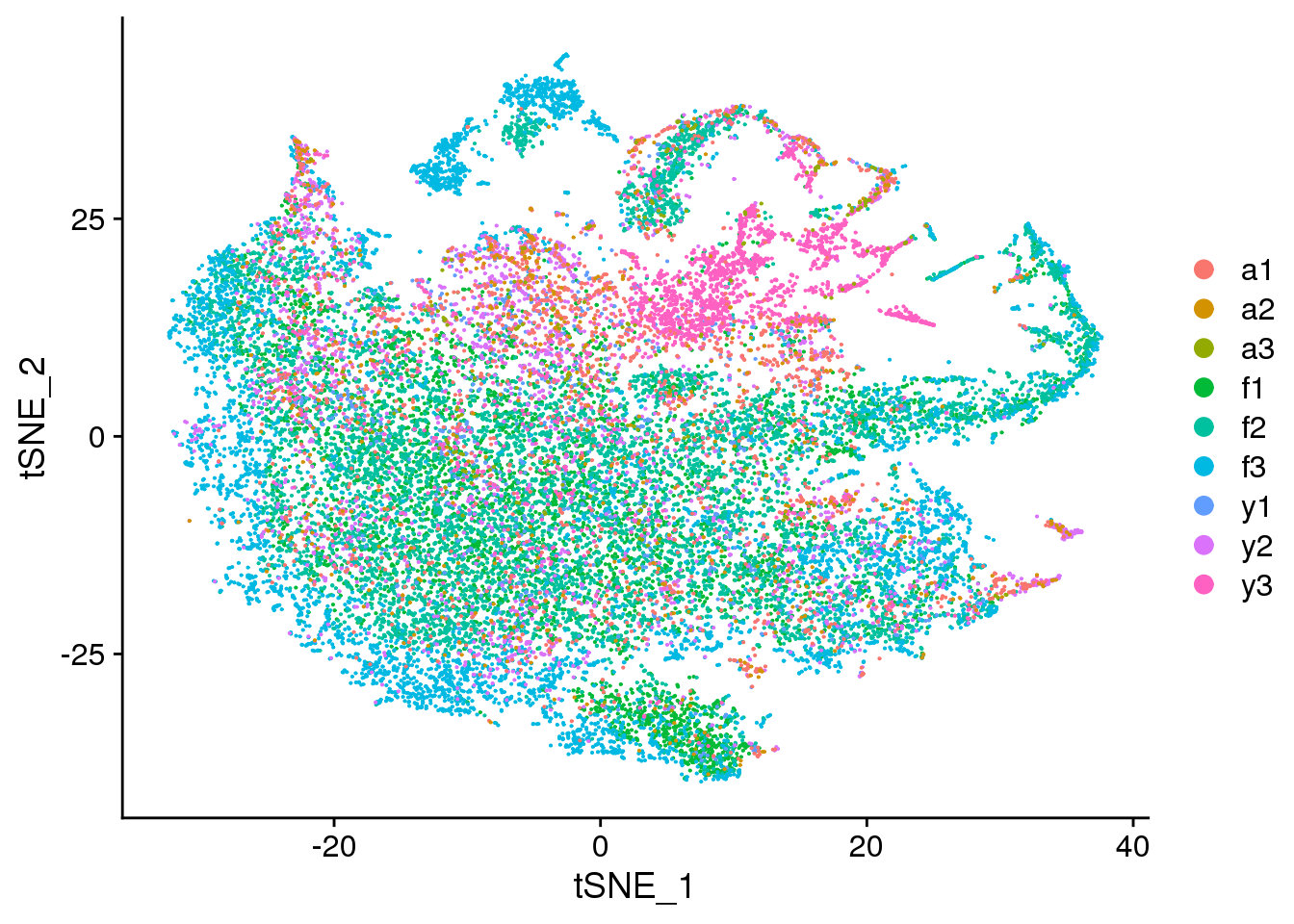
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "tsne", group.by = "sex")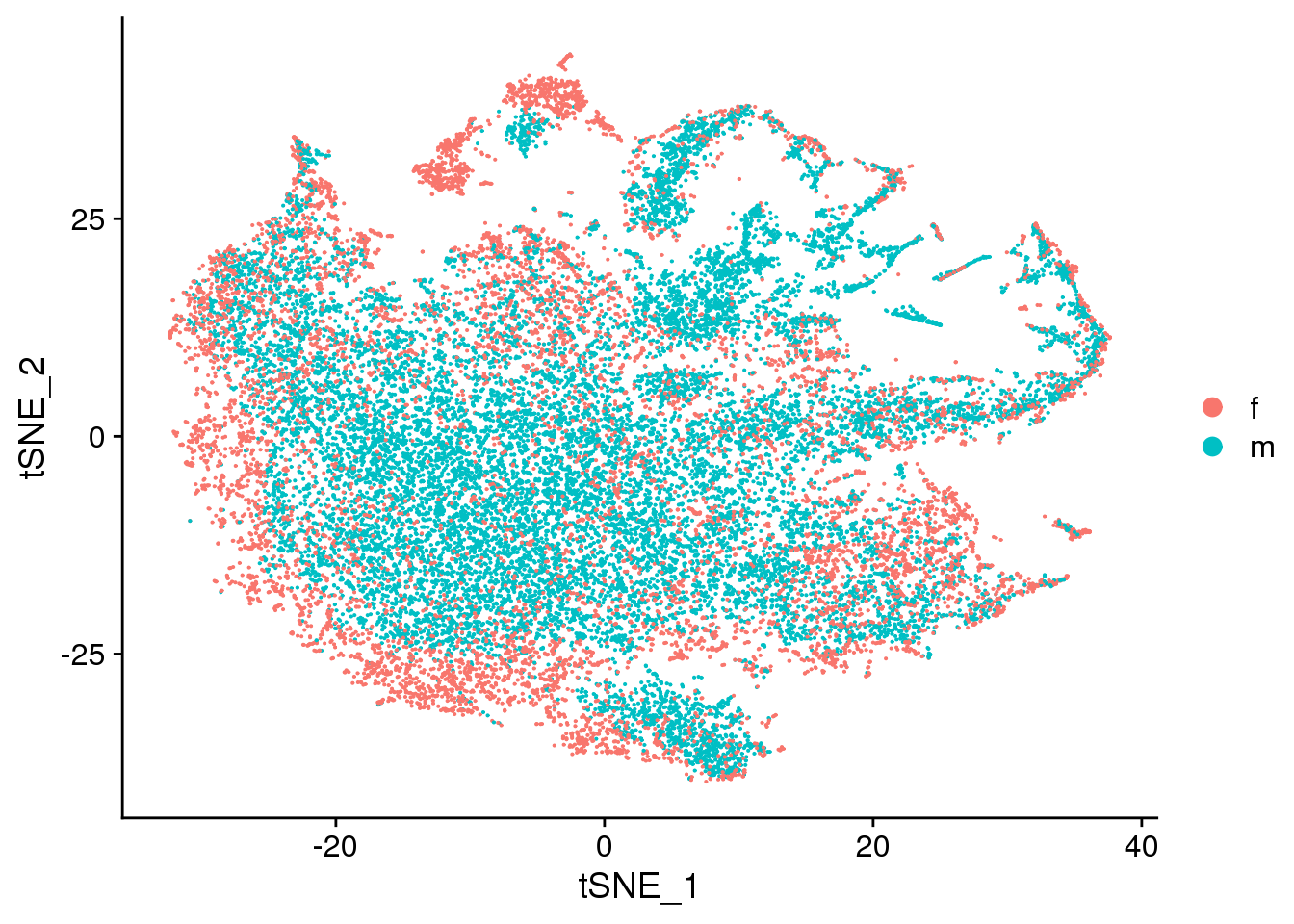
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "tsne", split.by = "sex")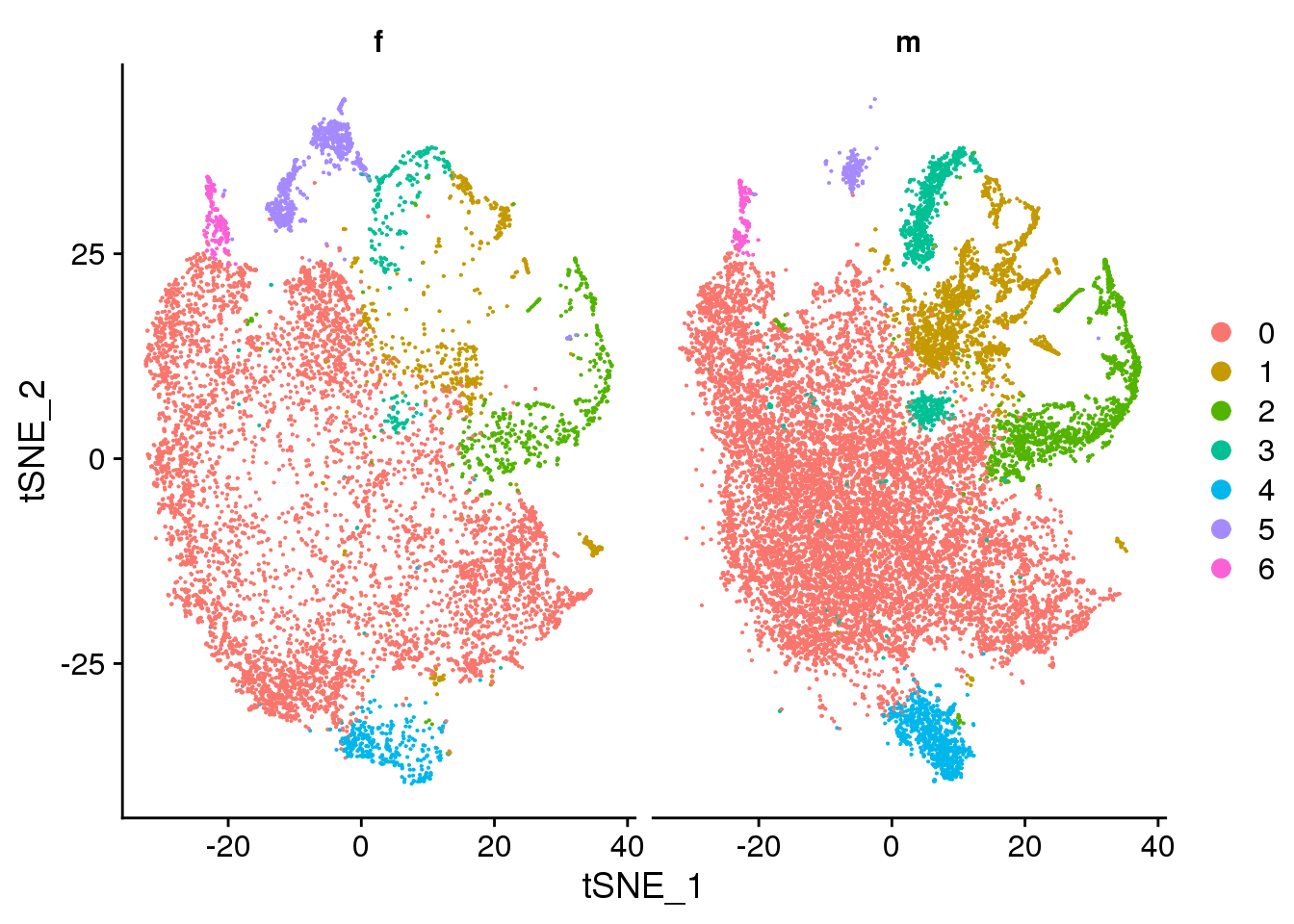
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
DimPlot(cardio.integrated, reduction = "tsne", group.by = "batch")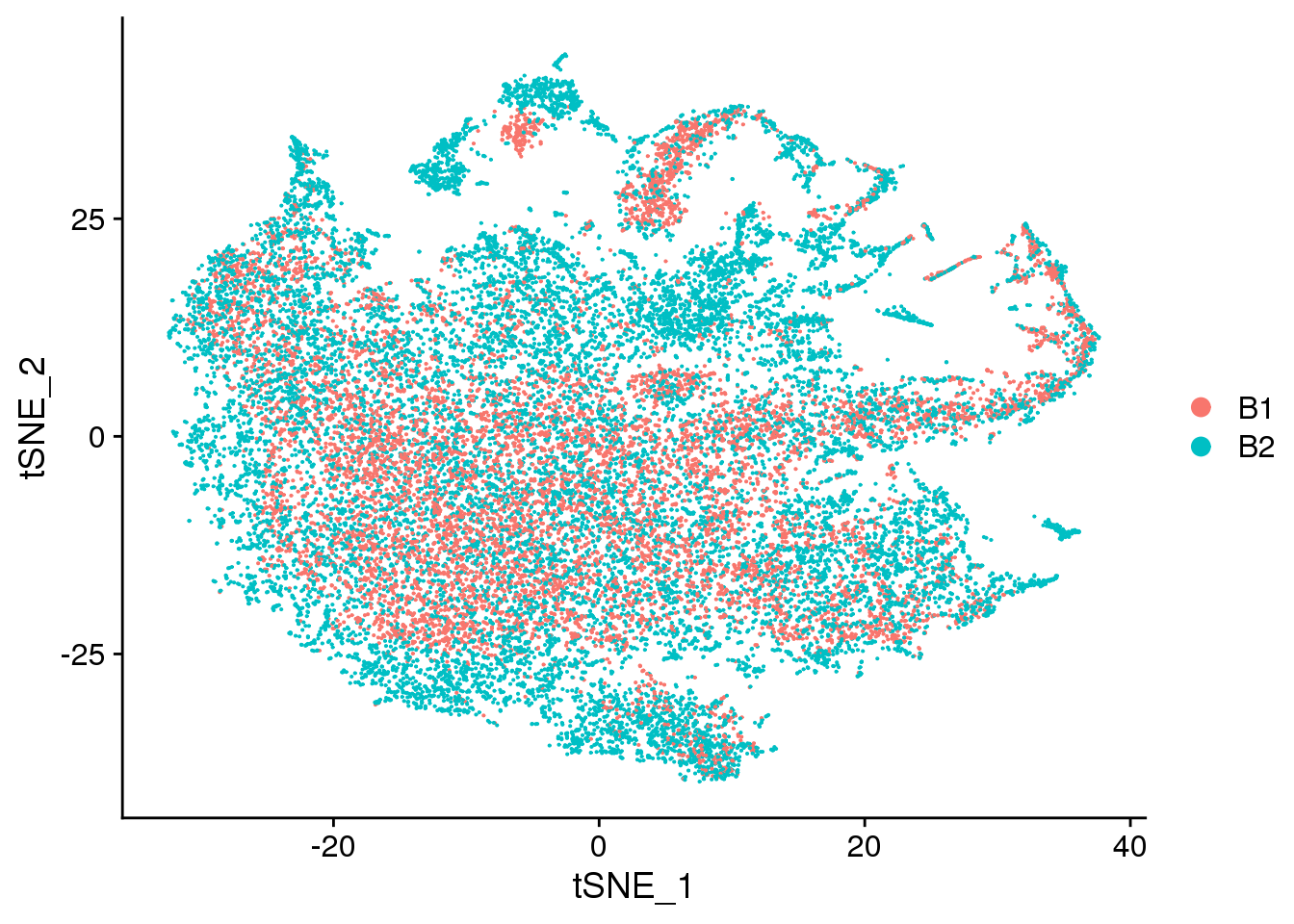
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
par(mfrow=c(1,1))
par(mar=c(4,4,2,2))
tab <- table(Idents(cardio.integrated),cardio.integrated$biorep)
barplot(t(tab/rowSums(tab)),beside=TRUE,col=ggplotColors(9),legend=TRUE)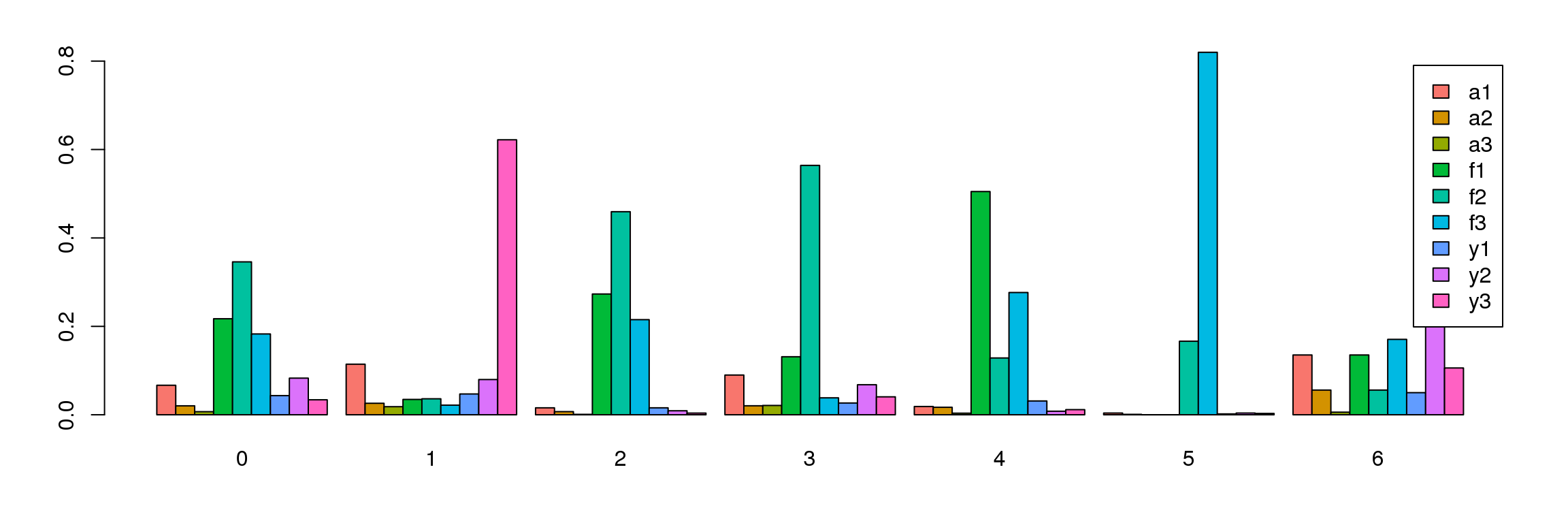
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
par(mfrow=c(1,1))
par(mar=c(4,4,2,2))
tab <- table(Idents(cardio.integrated),cardio.integrated$orig.ident)
barplot(t(tab/rowSums(tab)),beside=TRUE,col=ggplotColors(3))
legend("topleft",legend=colnames(tab),fill=ggplotColors(3))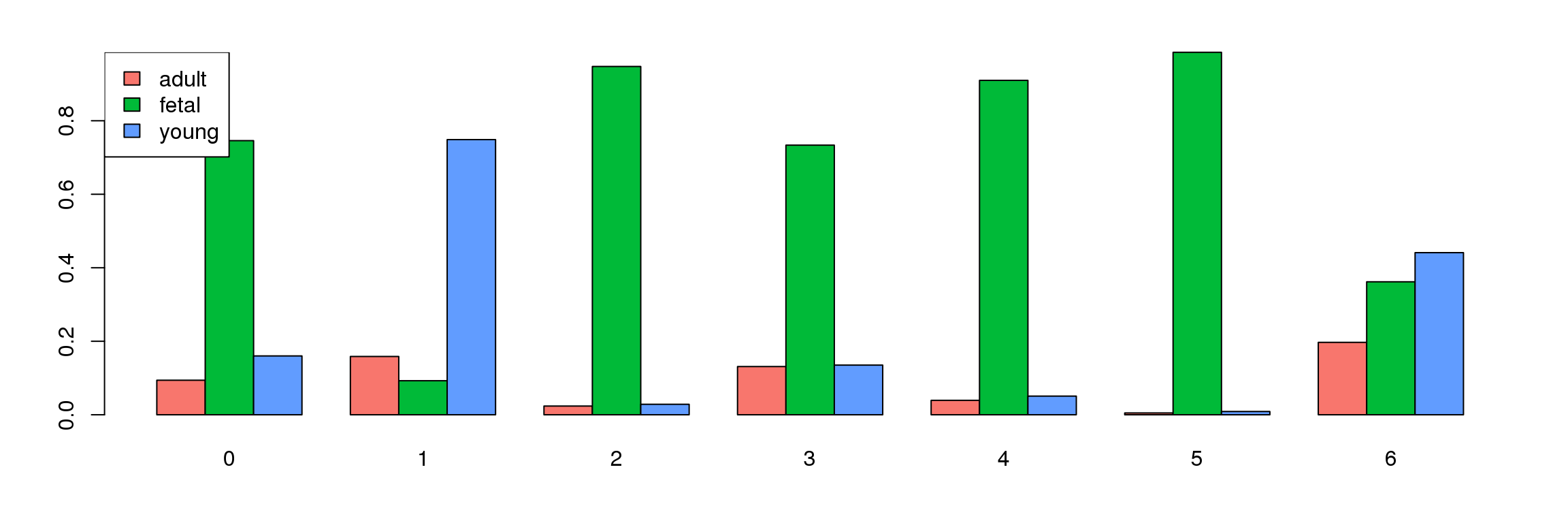
Visualisation with clustree
clusres <- c(0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9,1,1.1,1.2)for(i in 1:length(clusres)){
cardio.integrated <- FindClusters(cardio.integrated,
resolution = clusres[i])
}Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9410
Number of communities: 7
Elapsed time: 10 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9183
Number of communities: 11
Elapsed time: 9 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9023
Number of communities: 13
Elapsed time: 8 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8873
Number of communities: 16
Elapsed time: 10 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8735
Number of communities: 17
Elapsed time: 8 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8601
Number of communities: 18
Elapsed time: 9 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8486
Number of communities: 19
Elapsed time: 8 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8401
Number of communities: 18
Elapsed time: 9 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8299
Number of communities: 20
Elapsed time: 8 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8232
Number of communities: 23
Elapsed time: 9 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8173
Number of communities: 23
Elapsed time: 9 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 27037
Number of edges: 943610
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8105
Number of communities: 23
Elapsed time: 7 secondspct.male <- function(x) {mean(x=="m")}
pct.female <- function(x) {mean(x=="f")}
pct.fetal <- function(x) {mean(x=="fetal")}
pct.young <- function(x) {mean(x=="young")}
pct.adult <- function(x) {mean(x=="adult")}clustree(cardio.integrated, prefix = "integrated_snn_res.")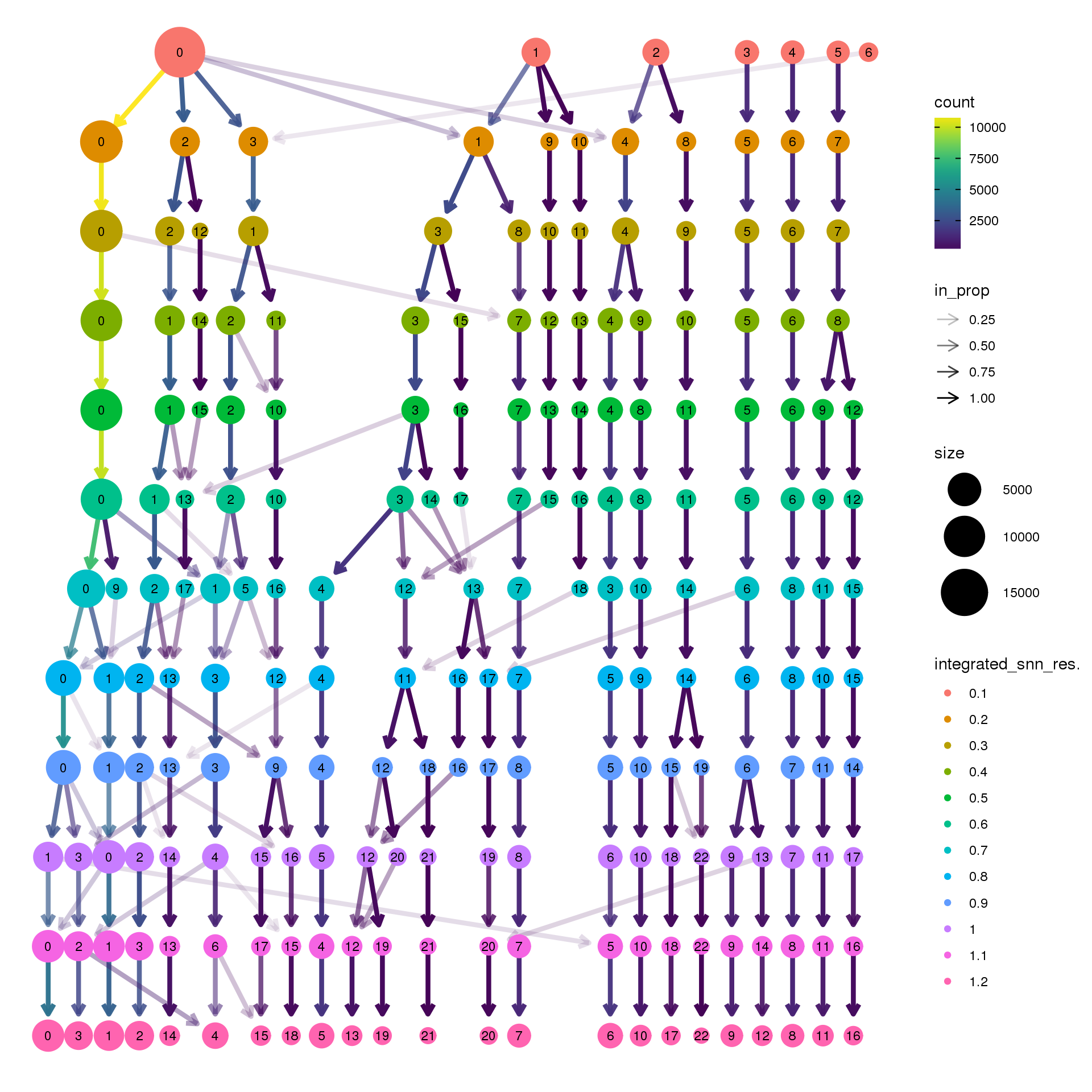
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
clustree(cardio.integrated, prefix = "integrated_snn_res.",
node_colour = "sex", node_colour_aggr = "pct.female",assay="RNA")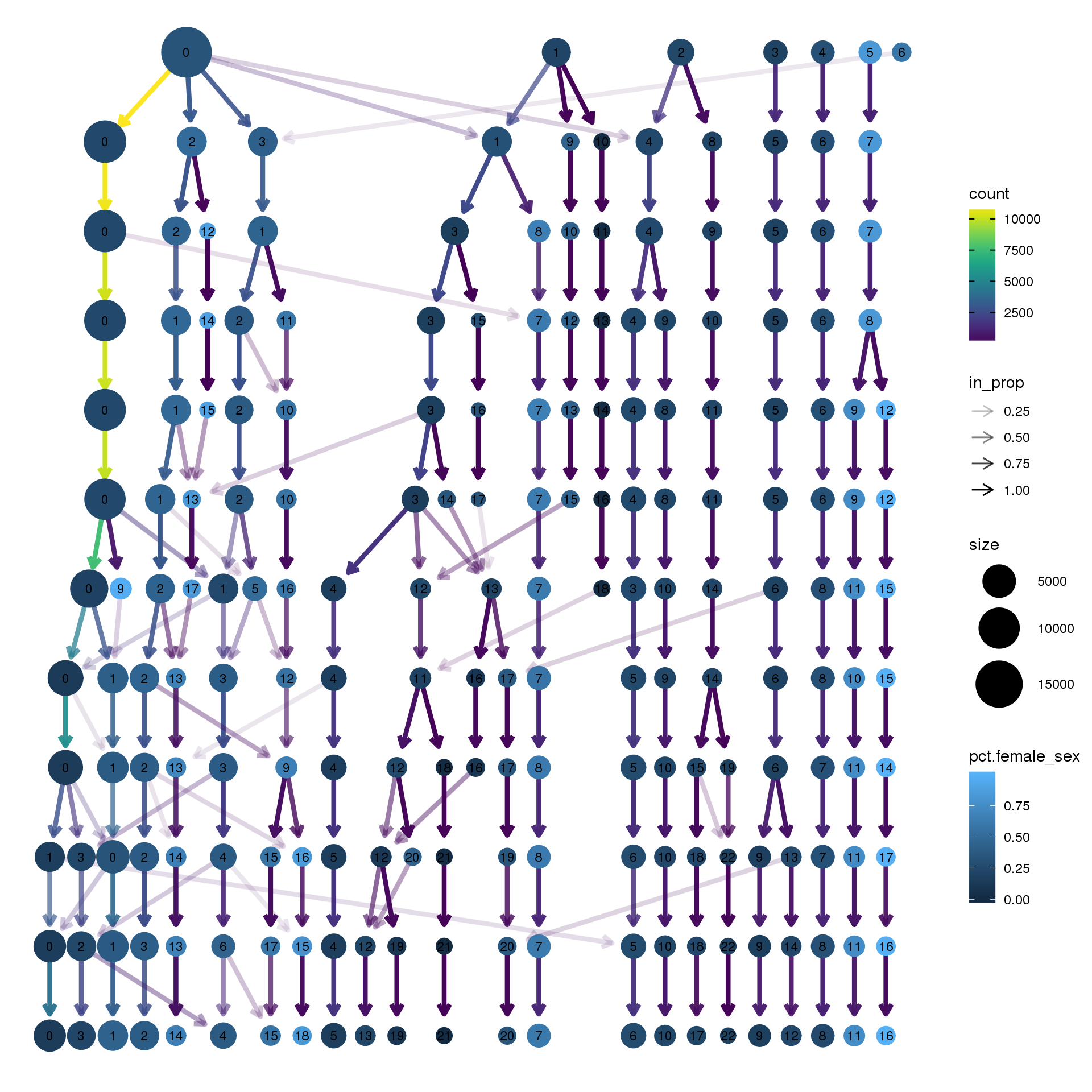
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
clustree(cardio.integrated, prefix = "integrated_snn_res.",
node_colour = "sex", node_colour_aggr = "pct.male",assay="RNA")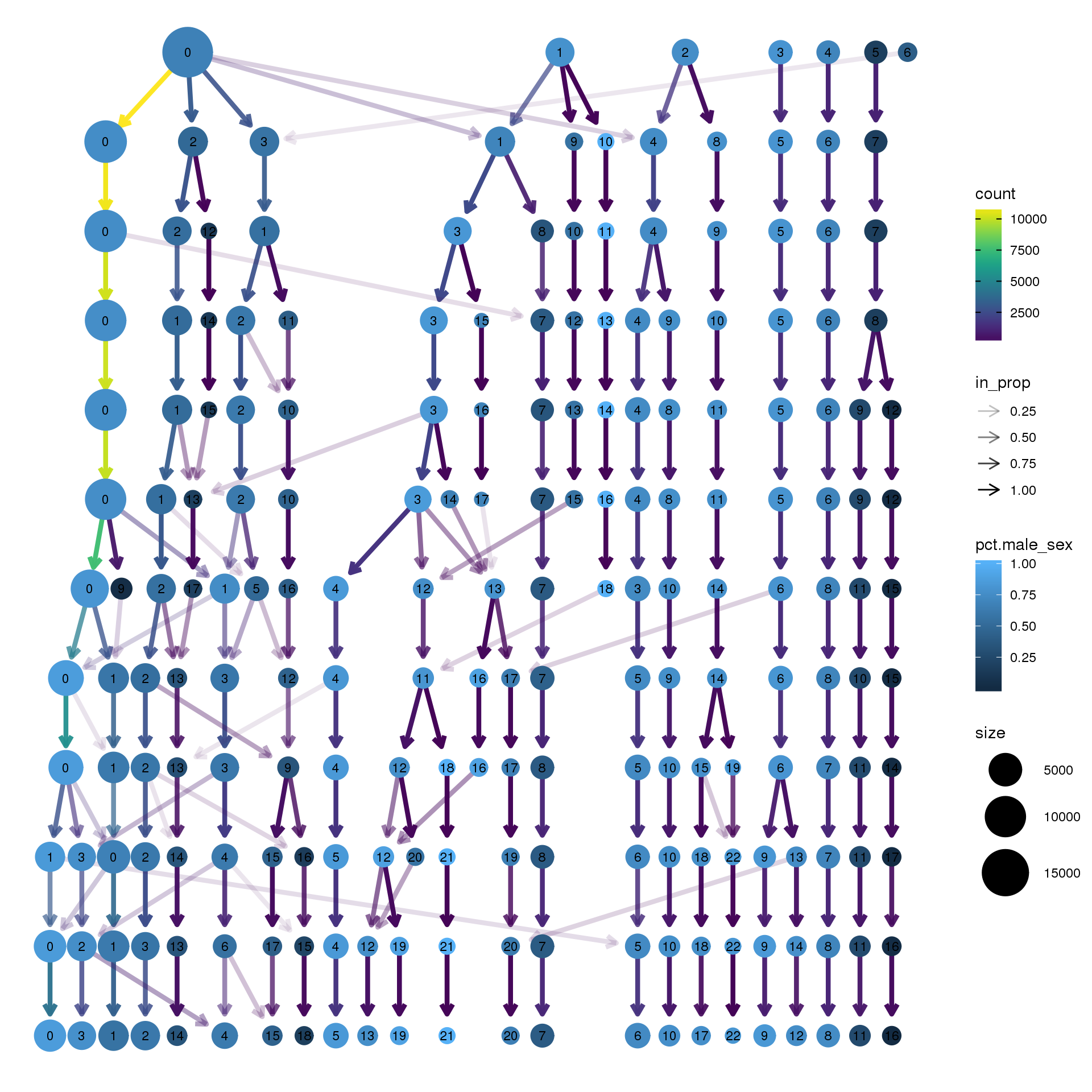
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
clustree(cardio.integrated, prefix = "integrated_snn_res.",
node_colour = "orig.ident", node_colour_aggr = "pct.fetal",assay="RNA")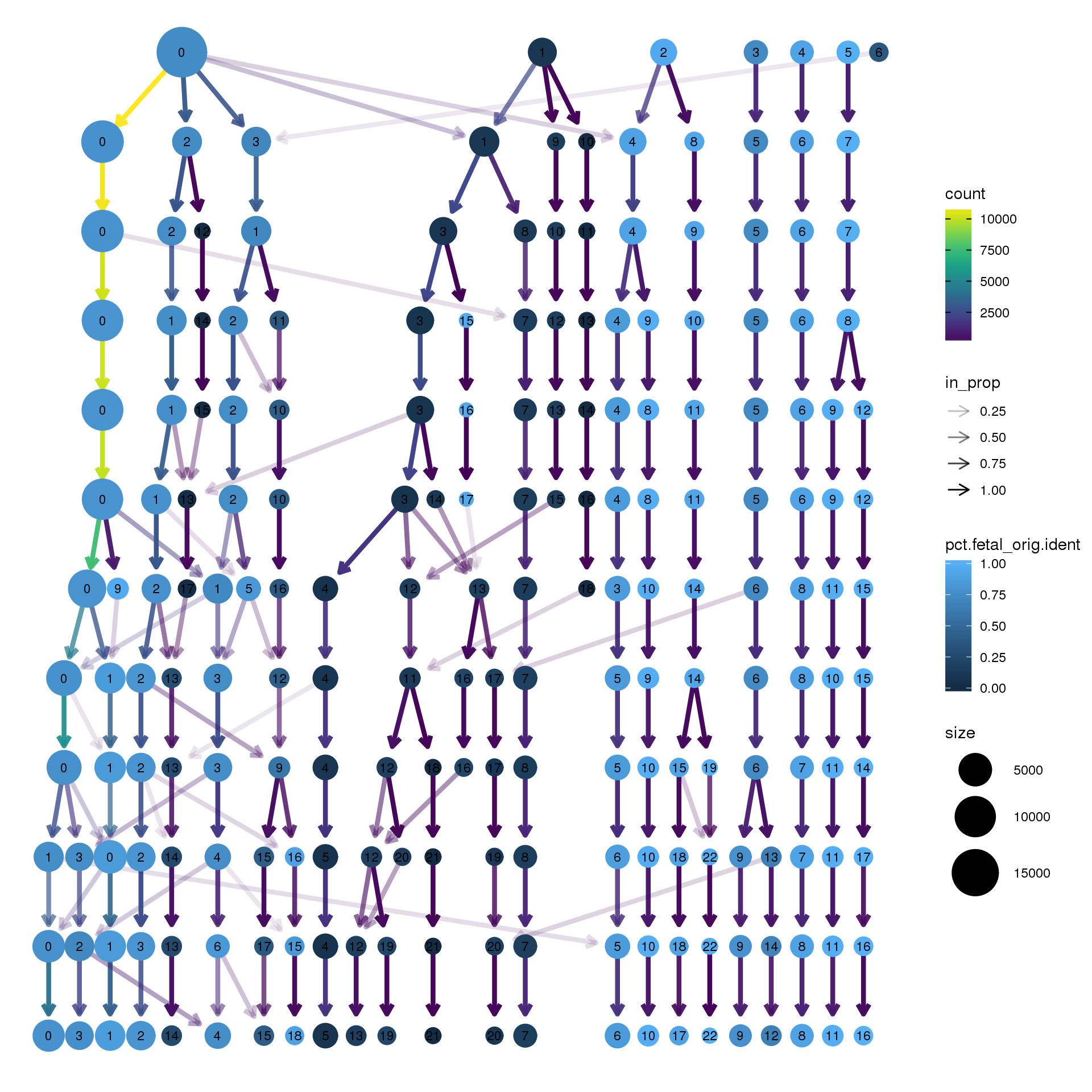
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
clustree(cardio.integrated, prefix = "integrated_snn_res.",
node_colour = "orig.ident", node_colour_aggr = "pct.young",assay="RNA")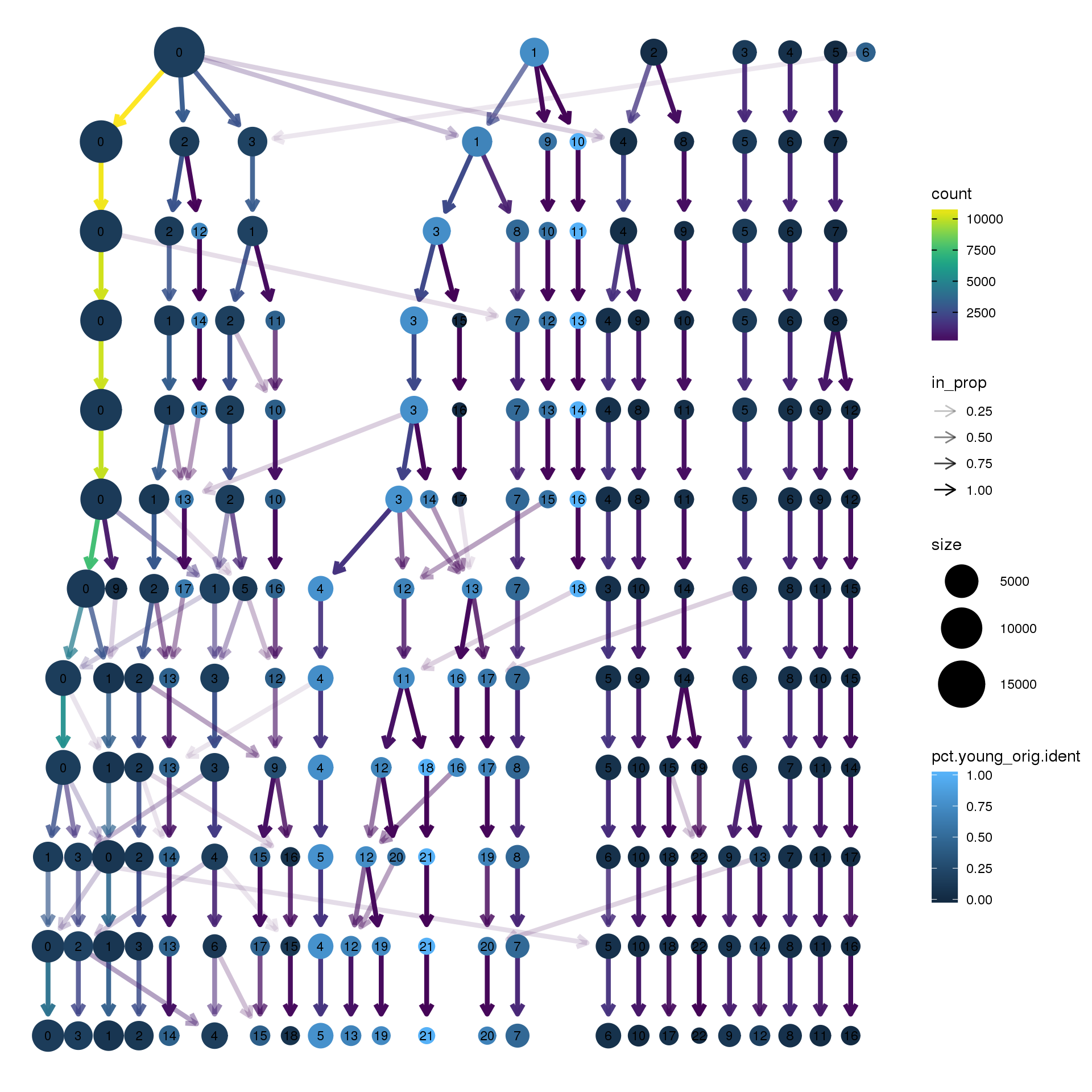
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
clustree(cardio.integrated, prefix = "integrated_snn_res.",
node_colour = "orig.ident", node_colour_aggr = "pct.adult",assay="RNA")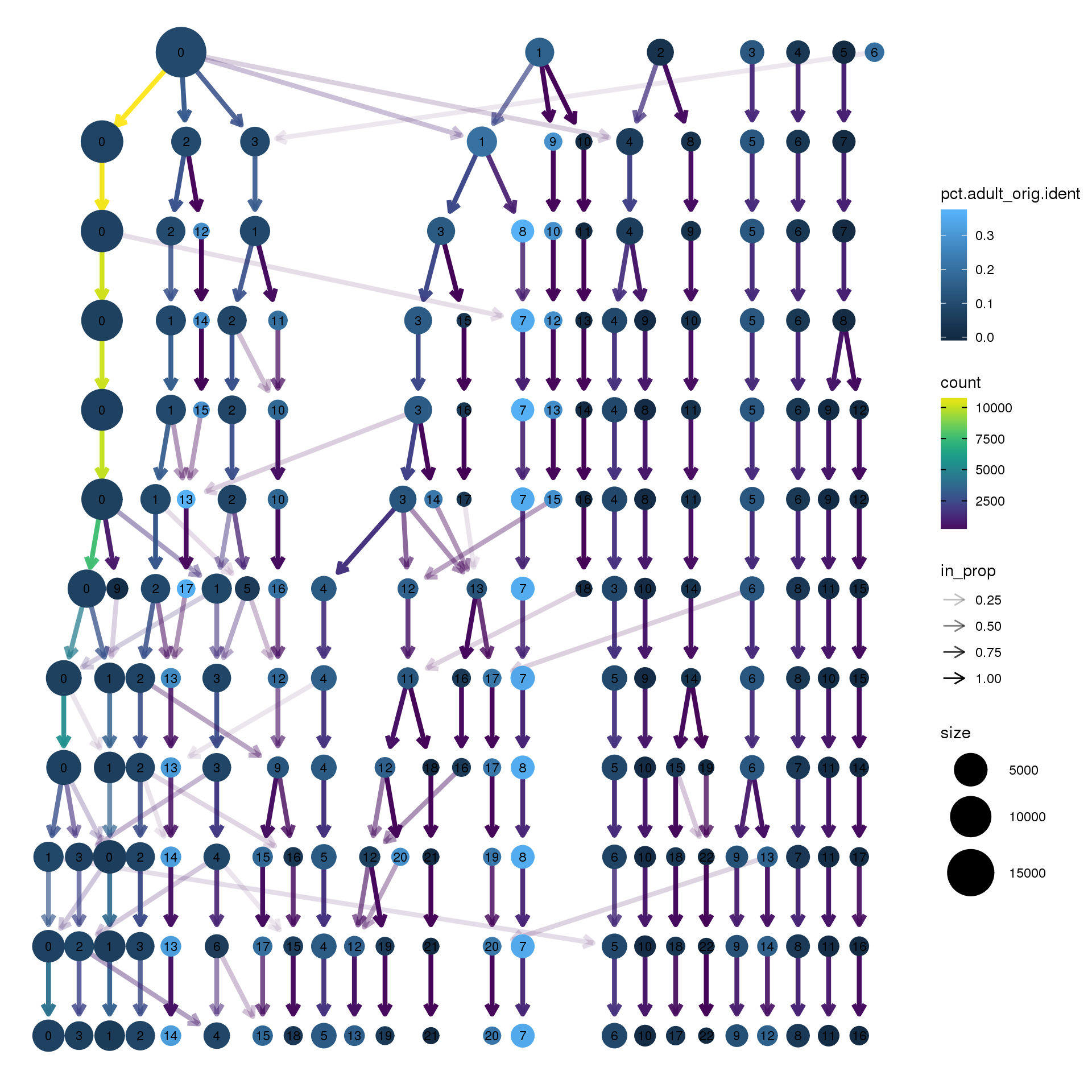
Save Seurat object
DefaultAssay(cardio.integrated) <- "RNA"
Idents(cardio.integrated) <- cardio.integrated$integrated_snn_res.0.1saveRDS(cardio.integrated,file="./output/RDataObjects/cardio-int-FYA-filtered.Rds")
#cardio.integrated <- readRDS(file="./output/RDataObjects/cardio-int-FYA.Rds")
# Load unfiltered counts matrix for every sample (object all)
load("./output/RDataObjects/all-counts.Rdata")columns(org.Hs.eg.db) [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT"
[5] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE"
[9] "EVIDENCEALL" "GENENAME" "GO" "GOALL"
[13] "IPI" "MAP" "OMIM" "ONTOLOGY"
[17] "ONTOLOGYALL" "PATH" "PFAM" "PMID"
[21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG"
[25] "UNIGENE" "UNIPROT" ann <- AnnotationDbi:::select(org.Hs.eg.db,keys=rownames(all),columns=c("SYMBOL","ENTREZID","ENSEMBL","GENENAME","CHR"),keytype = "SYMBOL")
m <- match(rownames(all),ann$SYMBOL)
ann <- ann[m,]
table(ann$SYMBOL==rownames(all))
TRUE
33939 mito <- grep("mitochondrial",ann$GENENAME)
length(mito)[1] 226ribo <- grep("ribosomal",ann$GENENAME)
length(ribo)[1] 198missingEZID <- which(is.na(ann$ENTREZID))
length(missingEZID)[1] 10530Find Markers
#adultmarkers <- FindAllMarkers(adult.integrated, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
# Limma-trend for DE
m <- match(colnames(cardio.integrated),colnames(all))
all.counts <- all[,m]chuck <- unique(c(mito,ribo,missingEZID))
length(chuck)[1] 10875all.counts.keep <- all.counts[-chuck,]
ann.keep <- ann[-chuck,]
table(ann.keep$SYMBOL==rownames(all.counts.keep))
TRUE
23064 numzero.genes <- rowSums(all.counts.keep==0)
#avg.exp <- rowMeans(cpm.DGEList(y.kid,log=TRUE))
#plot(avg.exp,numzero.genes,xlab="Average log-normalised-counts",ylab="Number zeroes per gene")
table(numzero.genes > (ncol(all.counts.keep)-20))
FALSE TRUE
18407 4657 keep.genes <- numzero.genes < (ncol(all.counts.keep)-20)
table(keep.genes)keep.genes
FALSE TRUE
4698 18366 all.keep <- all.counts.keep[keep.genes,]
dim(all.keep)[1] 18366 27037ann.keep <- ann.keep[keep.genes,]y.cardio <- DGEList(all.keep)
logcounts <- normCounts(y.cardio,log=TRUE,prior.count=0.5)
#logcounts.n <- normalizeBetweenArrays(logcounts, method = "cyclicloess")
maxclust <- length(levels(Idents(cardio.integrated)))-1
grp <- paste("c",Idents(cardio.integrated),sep = "")
grp <- factor(grp,levels = paste("c",0:maxclust,sep=""))
design <- model.matrix(~0+grp+cardio.integrated$biorep)
colnames(design)[1:(maxclust+1)] <- levels(grp)
mycont <- matrix(0,ncol=length(levels(grp)),nrow=length(levels(grp)))
colnames(mycont)<-levels(grp)
diag(mycont)<-1
mycont[upper.tri(mycont)]<- -1/(length(levels(factor(grp)))-1)
mycont[lower.tri(mycont)]<- -1/(length(levels(factor(grp)))-1)
# Fill out remaining rows with 0s
zero.rows <- matrix(0,ncol=length(levels(grp)),nrow=(ncol(design)-length(levels(Idents(cardio.integrated)))))
test <- rbind(mycont,zero.rows)
fit <- lmFit(logcounts,design)
fit.cont <- contrasts.fit(fit,contrasts=test)
fit.cont <- eBayes(fit.cont,trend=TRUE,robust=TRUE)
fit.cont$genes <- ann.keep
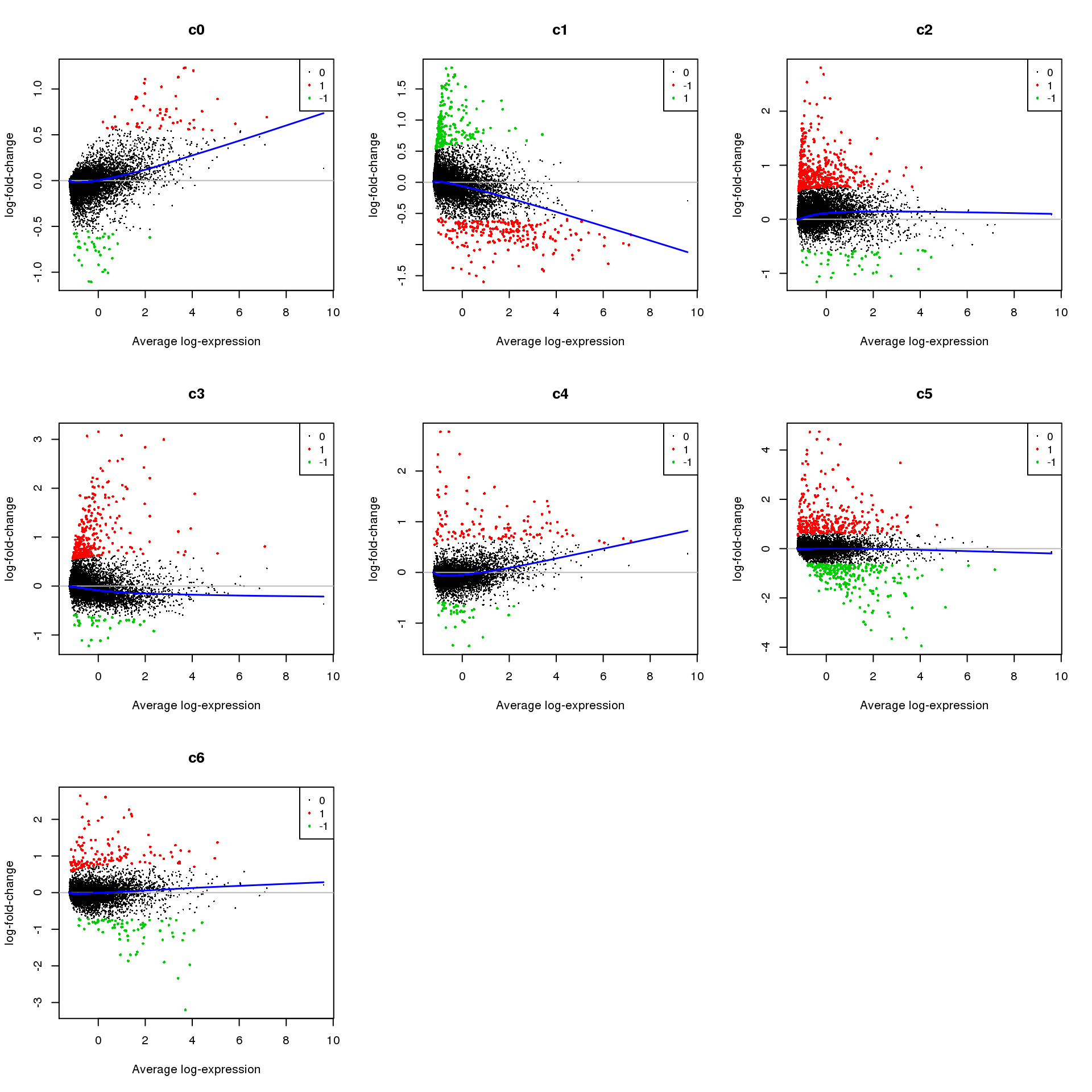
summary(decideTests(fit.cont)) c0 c1 c2 c3 c4 c5 c6
Down 7235 4032 2697 5758 6760 3880 2650
NotSig 8667 8790 9107 9337 10182 11538 14120
Up 2464 5544 6562 3271 1424 2948 1596treat <- treat(fit.cont,lfc=0.5)
dt <- decideTests(treat)
summary(dt) c0 c1 c2 c3 c4 c5 c6
Down 39 231 48 37 42 253 71
NotSig 18274 17920 17850 18045 18208 17808 18163
Up 53 215 468 284 116 305 132par(mfrow=c(3,3))
for(i in 1:ncol(mycont)){
plotMD(treat,coef=i,status = dt[,i],hl.cex=0.5)
abline(h=0,col=colours()[c(226)])
lines(lowess(treat$Amean,treat$coefficients[,i]),lwd=1.5,col=4)
}
Write out marker genes for each cluster
contnames <- colnames(mycont)
for(i in 1:length(contnames)){
topsig <- topTreat(treat,coef=i,n=Inf)
write.csv(topsig,file=paste("./output/MarkerAnalysis/Cardiomyocytes/Development/Filtered/Cluster-",contnames[i],".csv",sep=""))
}fdr <- apply(treat$p.value, 2, function(x) p.adjust(x, method="BH"))
output <- data.frame(treat$genes,LogFC=treat$coefficients,AveExp=treat$Amean,tstat=treat$t, pvalue=treat$p.value, fdr=fdr)
write.csv(output,file="./output/MarkerAnalysis/Cardiomyocytes/Development/Filtered/MarkerAnalysis.csv")Perform gene set testing on C2 and GO sets
contnames <- colnames(mycont)
load("./output/RDataObjects/human_c2_v5p2.rdata")
load("./output/RDataObjects/human_c5_v5p2.rdata")
c2.id <- ids2indices(Hs.c2,treat$genes$ENTREZID)
c5.id <- ids2indices(Hs.c5,treat$genes$ENTREZID)
reactome.id <-c2.id[grep("REACTOME",names(c2.id))]
c2.c0 <- cameraPR(treat$t[,1],c2.id)
reactome.c0 <- cameraPR(treat$t[,1],reactome.id)
go.c0 <- cameraPR(treat$t[,1],c5.id)
for(i in 1:length(contnames)){
write.csv(cameraPR(treat$t[,i],c2.id),file=paste("./output/MarkerAnalysis/Cardiomyocytes/Development/Filtered/c2-",contnames[i],".csv",sep=""))
write.csv(cameraPR(treat$t[,i],reactome.id),file=paste("./output/MarkerAnalysis/Cardiomyocytes/Development/Filtered/reactome-",contnames[i],".csv",sep=""))
write.csv(cameraPR(treat$t[,i],c5.id),file=paste("./output/MarkerAnalysis/Cardiomyocytes/Development/Filtered/go-",contnames[i],".csv",sep=""))
}Check quality of clusters
The quality of the clusters look good.
par(mfrow=c(1,1))
numgenes <- colSums(all.keep!=0)
boxplot(numgenes~grp)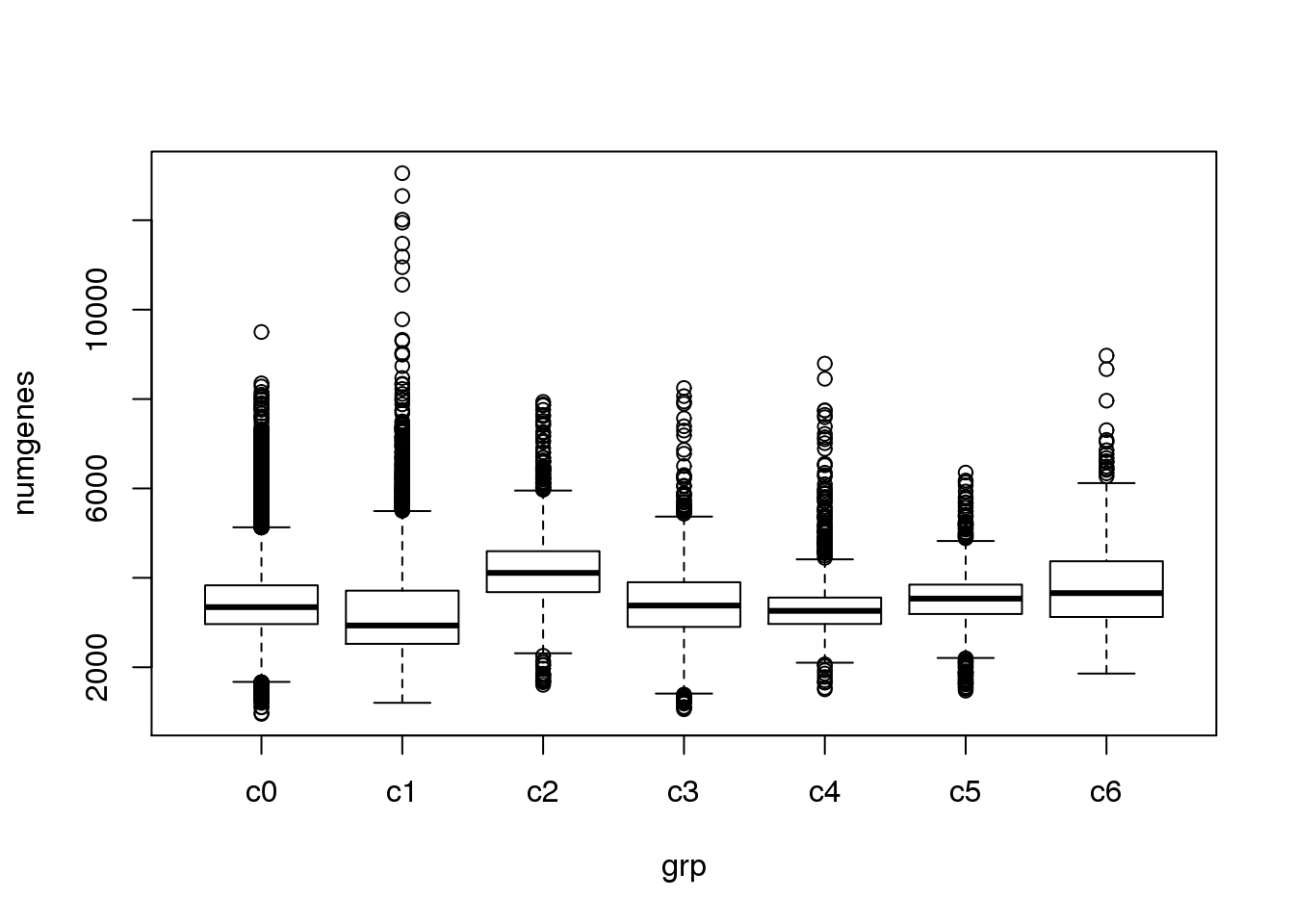
Heatmap of marker genes
sam <- factor(cardio.integrated$biorep,levels=c("f1","f2","f3","y1","y2","y3","a1","a2","a3"))
newgrp <- paste(grp,sam,sep=".")
newgrp <- factor(newgrp,levels=paste(rep(levels(grp),each=9),levels(sam),sep="."))
o <-order(newgrp)
clust <- rep(levels(grp),each=9)
samps <- rep(levels(sam),length(levels(grp)))Summarise expression across cells
sumexpr <- matrix(NA,nrow=nrow(logcounts),ncol=length(levels(newgrp)))
rownames(sumexpr) <- rownames(logcounts)
colnames(sumexpr) <- levels(newgrp)
for(i in 1:nrow(sumexpr)){
sumexpr[i,] <- tapply(logcounts[i,],newgrp,mean)
}sig.genes <- gene.label <- vector("list", length(levels(grp)))
for(i in 1:length(sig.genes)){
top <- topTreat(treat,coef=i,n=Inf)
sig.genes[[i]] <- rownames(top)[top$logFC>0][1:10]
gene.label[[i]] <- paste(rownames(top)[top$logFC>0][1:10],levels(grp)[i],sep="-")
}
csig <- unlist(sig.genes)
genes <- unlist(gene.label)
myColors <- list(Clust=NA,Sample=NA)
myColors$Clust<-sample(ggplotColors(length(levels(grp))),length(levels(grp)))
names(myColors$Clust)<-levels(grp)
myColors$Sample <- sample(ggplotColors(length(levels(sam))),length(levels(sam)))
names(myColors$Sample) <- levels(sam)
pdf(file="./output/Figures/NormalDev/cardio-heatmap-siggenes-summarised-FYA-filtered.pdf",width=20,height=20,onefile = FALSE)
aheatmap(sumexpr[csig,],Rowv = NA,Colv = NA, labRow = genes,
annCol=list(Clust=clust,Sample=samps),
annColors=myColors,
fontsize=16,color="-RdYlBu",
scale="none")
dev.off()png
2 aheatmap(sumexpr[csig,],Rowv = NA,Colv = NA, labRow = genes,
annCol=list(Clust=clust,Sample=samps),
annColors=myColors,
fontsize=16,color="-RdYlBu",
scale="none")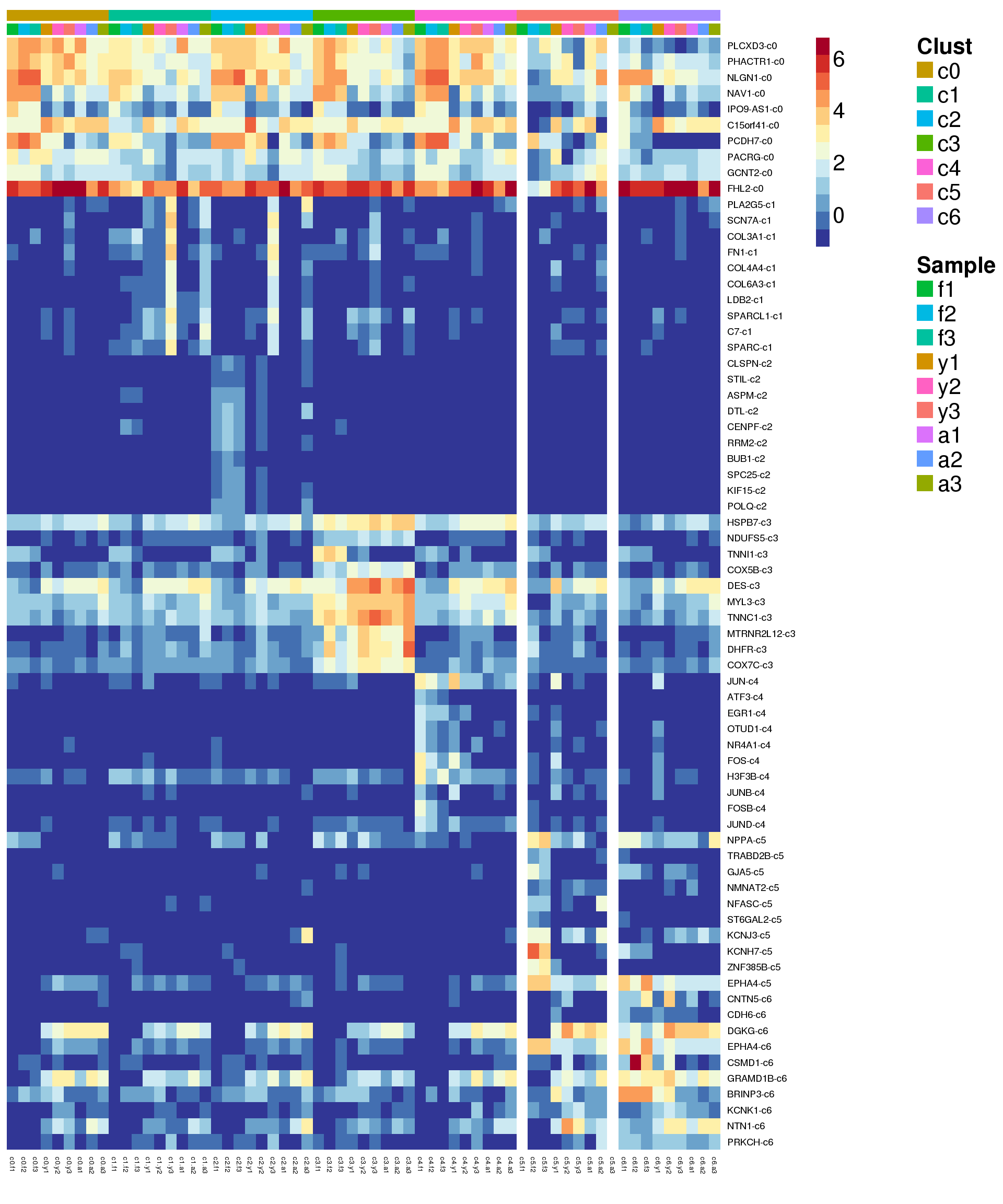
| Version | Author | Date |
|---|---|---|
| 67c1d63 | Belinda Phipson | 2019-10-18 |
Heatmap of pre-identified heart genes
hm <- read.delim("./data/heart-markers-long.txt",stringsAsFactors = FALSE)
hgene <- toupper(hm$Gene)
hgene <- unique(hgene)
m <- match(hgene,rownames(sumexpr))
m <- m[!is.na(m)]
mycelltypes <- hm$Celltype[match(rownames(sumexpr)[m],toupper(hm$Gene))]
mycelltypes <- factor(mycelltypes)
mygenes <- rownames(sumexpr)[m]
mygenelab <- paste(mygenes,mycelltypes,sep="_")
pdf(file="./output/Figures/NormalDev/cardio-heatmap-hmarkers-summarised-FYA-filtered.pdf",width=20,height=15,onefile = FALSE)
aheatmap(sumexpr[m,],Rowv = NA,Colv = NA, labRow = mygenelab,
annCol=list(Clust=clust,Sample=samps),
# annRow=list(Celltypes=mycelltypes),
annColors=myColors,
fontsize=14,color="-RdYlBu")
dev.off()png
2 aheatmap(sumexpr[m,],Rowv = NA,Colv = NA, labRow = mygenelab,
annCol=list(Clust=clust,Sample=samps),
# annRow=list(Celltypes=mycelltypes),
annColors=myColors,
fontsize=14,color="-RdYlBu")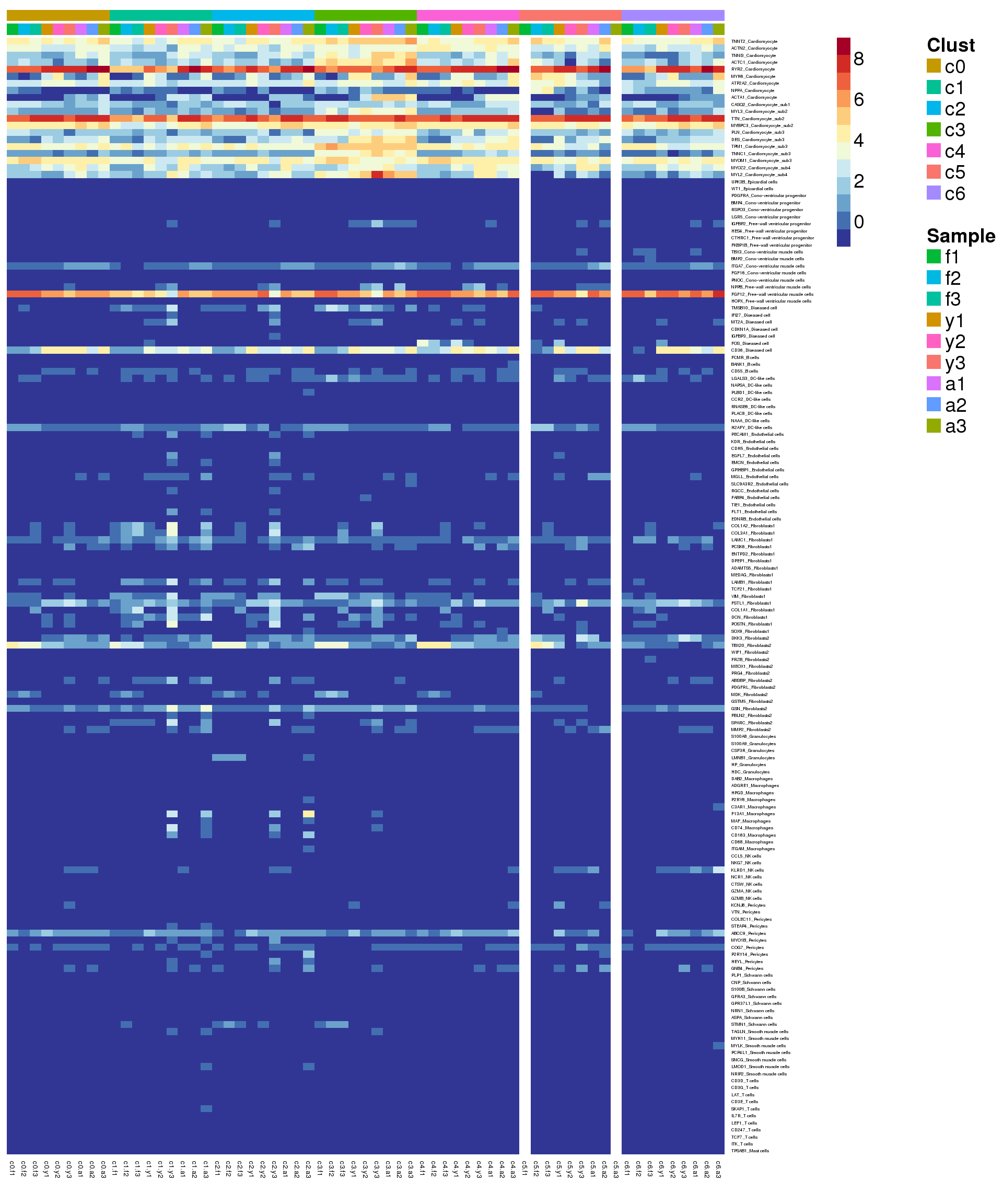
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS release 6.7 (Final)
Matrix products: default
BLAS: /usr/local/installed/R/3.6.0/lib64/R/lib/libRblas.so
LAPACK: /usr/local/installed/R/3.6.0/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] splines parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] dplyr_0.8.3 clustree_0.4.0
[3] ggraph_1.0.2 workflowr_1.3.0
[5] NMF_0.21.0 bigmemory_4.5.33
[7] cluster_2.1.0 rngtools_1.4
[9] pkgmaker_0.27 registry_0.5-1
[11] scran_1.12.0 SingleCellExperiment_1.6.0
[13] SummarizedExperiment_1.14.1 GenomicRanges_1.36.0
[15] GenomeInfoDb_1.20.0 DelayedArray_0.10.0
[17] BiocParallel_1.18.1 matrixStats_0.55.0
[19] cowplot_1.0.0 monocle_2.12.0
[21] DDRTree_0.1.5 irlba_2.3.3
[23] VGAM_1.1-1 ggplot2_3.2.1
[25] Matrix_1.2-17 Seurat_3.0.3.9019
[27] org.Hs.eg.db_3.8.2 AnnotationDbi_1.46.1
[29] IRanges_2.18.1 S4Vectors_0.22.0
[31] Biobase_2.44.0 BiocGenerics_0.30.0
[33] RColorBrewer_1.1-2 edgeR_3.26.3
[35] limma_3.40.2
loaded via a namespace (and not attached):
[1] reticulate_1.13 R.utils_2.9.0
[3] tidyselect_0.2.5 RSQLite_2.1.2
[5] htmlwidgets_1.5 grid_3.6.0
[7] combinat_0.0-8 docopt_0.6.1
[9] Rtsne_0.15 munsell_0.5.0
[11] codetools_0.2-16 ica_1.0-2
[13] statmod_1.4.30 future_1.14.0
[15] withr_2.1.2 colorspace_1.4-1
[17] fastICA_1.2-2 knitr_1.25
[19] ROCR_1.0-7 gbRd_0.4-11
[21] listenv_0.7.0 labeling_0.3
[23] Rdpack_0.11-0 git2r_0.26.1
[25] slam_0.1-45 GenomeInfoDbData_1.2.1
[27] polyclip_1.10-0 farver_1.1.0
[29] bit64_0.9-7 pheatmap_1.0.12
[31] rprojroot_1.3-2 vctrs_0.2.0
[33] xfun_0.10 R6_2.4.0
[35] doParallel_1.0.15 ggbeeswarm_0.6.0
[37] rsvd_1.0.2 locfit_1.5-9.1
[39] bitops_1.0-6 assertthat_0.2.1
[41] SDMTools_1.1-221.1 scales_1.0.0
[43] beeswarm_0.2.3 gtable_0.3.0
[45] npsurv_0.4-0 globals_0.12.4
[47] tidygraph_1.1.2 rlang_0.4.0
[49] zeallot_0.1.0 lazyeval_0.2.2
[51] checkmate_1.9.4 yaml_2.2.0
[53] reshape2_1.4.3 backports_1.1.5
[55] tools_3.6.0 gridBase_0.4-7
[57] gplots_3.0.1.1 dynamicTreeCut_1.63-1
[59] ggridges_0.5.1 Rcpp_1.0.2
[61] plyr_1.8.4 zlibbioc_1.30.0
[63] purrr_0.3.2 RCurl_1.95-4.12
[65] densityClust_0.3 pbapply_1.4-1
[67] viridis_0.5.1 zoo_1.8-6
[69] ggrepel_0.8.1 fs_1.3.1
[71] magrittr_1.5 data.table_1.12.4
[73] lmtest_0.9-37 RANN_2.6.1
[75] whisker_0.3-2 fitdistrplus_1.0-14
[77] lsei_1.2-0 evaluate_0.14
[79] xtable_1.8-4 sparsesvd_0.1-4
[81] gridExtra_2.3 HSMMSingleCell_1.4.0
[83] compiler_3.6.0 scater_1.12.2
[85] tibble_2.1.3 KernSmooth_2.23-15
[87] crayon_1.3.4 R.oo_1.22.0
[89] htmltools_0.4.0 tidyr_0.8.3
[91] DBI_1.0.0 tweenr_1.0.1
[93] MASS_7.3-51.4 R.methodsS3_1.7.1
[95] gdata_2.18.0 metap_1.1
[97] igraph_1.2.4.1 pkgconfig_2.0.3
[99] bigmemory.sri_0.1.3 plotly_4.9.0
[101] foreach_1.4.7 vipor_0.4.5
[103] dqrng_0.2.1 XVector_0.24.0
[105] bibtex_0.4.2 stringr_1.4.0
[107] digest_0.6.21 sctransform_0.2.0
[109] RcppAnnoy_0.0.12 tsne_0.1-3
[111] rmarkdown_1.14 DelayedMatrixStats_1.6.0
[113] gtools_3.8.1 nlme_3.1-141
[115] jsonlite_1.6 BiocNeighbors_1.2.0
[117] viridisLite_0.3.0 pillar_1.4.2
[119] lattice_0.20-38 httr_1.4.1
[121] survival_2.44-1.1 glue_1.3.1
[123] qlcMatrix_0.9.7 FNN_1.1.3
[125] png_0.1-7 iterators_1.0.12
[127] bit_1.1-14 ggforce_0.3.0
[129] stringi_1.4.3 blob_1.2.0
[131] BiocSingular_1.0.0 caTools_1.17.1.2
[133] memoise_1.1.0 future.apply_1.3.0
[135] ape_5.3