Establish Expression cutoff with 3’ Seq
Briana Mittleman
1/16/2020
Last updated: 2020-04-03
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/TrialFiltersMeta.txt.sb-9845453e-R58Y0Q/
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: ApplyDoubleFilt.Rmd
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/HandC.TvN
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/annotationBias.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: analysis/pol2.Rmd
Untracked: code/._AlignmentScores.sh
Untracked: code/._BothFCMM.sh
Untracked: code/._BothFCMMPrim.sh
Untracked: code/._BothFCnewOInclusive.sh
Untracked: code/._ChimpStarMM2.sh
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._ClosestorthoEx.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._Filter255MM.sh
Untracked: code/._FilterPrimSec.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._MismatchNumbers.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._SAF2Bed.py
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._StarMM2.sh
Untracked: code/._TestFC.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2Bedbothstrand.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chimpMultiCov.sh
Untracked: code/._chimpMultiCov255.sh
Untracked: code/._chimpMultiCovInclusive.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopGeneral.ph
Untracked: code/._extractPhylopGeneral.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._filterPrimaryread.py
Untracked: code/._filterSecondaryread.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixFCheadforExp.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._humanMultiCov.sh
Untracked: code/._humanMultiCov255.sh
Untracked: code/._humanMultiCov_inclusive.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapMMandOrthoexon.sh
Untracked: code/._overlapPASandOrthoexon.sh
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantLiftedPASPrimary.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/AlignmentScores.err
Untracked: code/AlignmentScores.out
Untracked: code/AlignmentScores.sh
Untracked: code/BothFCMM.err
Untracked: code/BothFCMM.out
Untracked: code/BothFCMM.sh
Untracked: code/BothFCMMPrim.err
Untracked: code/BothFCMMPrim.out
Untracked: code/BothFCMMPrim.sh
Untracked: code/BothFCnewOInclusive.sh
Untracked: code/BothFCnewOInclusive.sh.err
Untracked: code/BothFCnewOInclusive.sh.out
Untracked: code/ChimpStarMM2.err
Untracked: code/ChimpStarMM2.out
Untracked: code/ChimpStarMM2.sh
Untracked: code/ClassifyLeafviz.sh
Untracked: code/ClosestorthoEx.err
Untracked: code/ClosestorthoEx.out
Untracked: code/ClosestorthoEx.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/Filter255.err
Untracked: code/Filter255.out
Untracked: code/Filter255MM.sh
Untracked: code/FilterPrimSec.err
Untracked: code/FilterPrimSec.out
Untracked: code/FilterPrimSec.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/IntersectMMandOrtho.err
Untracked: code/IntersectMMandOrtho.out
Untracked: code/IntersectPASandOrtho.err
Untracked: code/IntersectPASandOrtho.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/MismatchNumbers.err
Untracked: code/MismatchNumbers.out
Untracked: code/MismatchNumbers.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/SAF2Bed.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/StarMM2.err
Untracked: code/StarMM2.out
Untracked: code/StarMM2.sh
Untracked: code/TestFC.err
Untracked: code/TestFC.out
Untracked: code/TestFC.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2Bedbothstrand.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chimpMultiCov.err
Untracked: code/chimpMultiCov.out
Untracked: code/chimpMultiCov.sh
Untracked: code/chimpMultiCov255.sh
Untracked: code/chimpMultiCovInclusive.err
Untracked: code/chimpMultiCovInclusive.out
Untracked: code/chimpMultiCovInclusive.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopGeneral.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterPrimaryread.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSecondaryread.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFCheadforExp.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getAlloverlap.py
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/humanMultiCov.err
Untracked: code/humanMultiCov.out
Untracked: code/humanMultiCov.sh
Untracked: code/humanMultiCov255.err
Untracked: code/humanMultiCov255.out
Untracked: code/humanMultiCov255.sh
Untracked: code/humanMultiCovInclusive.err
Untracked: code/humanMultiCovInclusive.out
Untracked: code/humanMultiCov_inclusive.sh
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapMMandOrthoexon.sh
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapPASandOrthoexon.sh
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quantLiftedPASPrimary.err
Untracked: code/quantLiftedPASPrimary.out
Untracked: code/quantLiftedPASPrimary.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runChimpDiffIsoDF.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runHumanDiffIsoDF.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_dF.err
Untracked: code/run_Humanleafcutter_dF.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Chimp_tvN_DF.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN_DF.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/test.txt
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._TrialFiltersMeta.txt
Untracked: data/._TrialFiltersMeta.txt.sb-9845453e-R58Y0Q
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/BaseComp/
Untracked: data/CleanLiftedPeaks_FC_primary/
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OrthoExonBed/
Untracked: data/OverlapBenchmark/
Untracked: data/OverlappingPAS/
Untracked: data/PAS/
Untracked: data/PAS_SAF/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/TestAnnoBiasOE/
Untracked: data/TestMM2/
Untracked: data/TestMM2_AS/
Untracked: data/TestMM2_PrimaryRead/
Untracked: data/TestMM2_SeondaryRead/
Untracked: data/TestMM2_mismatch/
Untracked: data/TestMM2_quality/
Untracked: data/TestWithinMergePAS/
Untracked: data/Test_FC_methods/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalHvC/
Untracked: data/TrialFiltersMeta.txt
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/bioGRID/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/multimap/
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/testQuant/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/DiffUsedIntronic.Rmd
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/ExploredAPA_DF.Rmd
Modified: analysis/MMExpreiment.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/SpliceSiteStrength.Rmd
Modified: analysis/TotalVNuclearBothSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/changeMisprimcut.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/dAPAandapaQTL_DF.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/speciesSpecific.Rmd
Modified: analysis/upsetter_DF.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | f161386 | brimittleman | 2020-01-18 | Build site. |
| Rmd | 289ba9b | brimittleman | 2020-01-18 | add expression cutoff code |
| html | 5eef3eb | brimittleman | 2020-01-16 | Build site. |
| Rmd | 5c24c0c | brimittleman | 2020-01-16 | add cutoff code files |
In this analysis I will use feature counts to count all the 3’ seq reads in each gene. I will then use a similar pipeline to the RNA seq to establish a cutoff for normalized expression. This will be used as a data filter on the pas for the humans and chimps.
library(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary("gplots")
Attaching package: 'gplots'The following object is masked from 'package:stats':
lowesslibrary("R.utils")Loading required package: R.ooLoading required package: R.methodsS3R.methodsS3 v1.7.1 (2016-02-15) successfully loaded. See ?R.methodsS3 for help.R.oo v1.22.0 (2018-04-21) successfully loaded. See ?R.oo for help.
Attaching package: 'R.oo'The following objects are masked from 'package:methods':
getClasses, getMethodsThe following objects are masked from 'package:base':
attach, detach, gc, load, saveR.utils v2.7.0 successfully loaded. See ?R.utils for help.
Attaching package: 'R.utils'The following object is masked from 'package:utils':
timestampThe following objects are masked from 'package:base':
cat, commandArgs, getOption, inherits, isOpen, parse, warningslibrary(tidyverse)── Attaching packages ─────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ tidyr::extract() masks R.utils::extract()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library("edgeR")Loading required package: limmalibrary("limma")
library("scales")
Attaching package: 'scales'The following object is masked from 'package:purrr':
discardThe following object is masked from 'package:readr':
col_factorlibrary("RColorBrewer")I will sum over all PAS for a psuedo gene count. I will use the full set of PAS before cutting to 5%.
Human nuclear only
humanPAS=read.table("../Human/data/CleanLiftedPeaks_FC/ALLPAS_postLift_LocParsed_Human_fixed.fc", header=T, stringsAsFactors = F) %>%
separate(Geneid, into=c("disc","PAS","chrom", "start","end","strand","geneid"), sep=":") %>%
separate(geneid,into=c("gene","loc"),sep="_") %>%
dplyr::select(gene,contains("_N")) %>%
gather(key="ind", value="count", -gene) %>%
group_by(ind, gene) %>%
summarize(GeneCount=sum(count)) %>%
spread(ind, GeneCount)Warning: Expected 2 pieces. Additional pieces discarded in 4 rows [48528,
48529, 48530, 92432].chimpPAS=read.table("../Chimp/data/CleanLiftedPeaks_FC/ALLPAS_postLift_LocParsed_Chimp_fixed.fc", header=T, stringsAsFactors = F) %>%
separate(Geneid, into=c("disc","PAS","chrom", "start","end","strand","geneid"), sep=":") %>%
separate(geneid,into=c("gene","loc"),sep="_") %>%
dplyr::select(gene,contains("_N")) %>%
gather(key="ind", value="count", -gene) %>%
group_by(ind, gene) %>%
summarize(GeneCount=sum(count)) %>%
spread(ind, GeneCount)Warning: Expected 2 pieces. Additional pieces discarded in 4 rows [48528,
48529, 48530, 92432].Join these together:
metadata=read.table("../data/metadata_HCpanel.txt",header = T) %>% mutate(id2=ifelse(grepl("pt", ID), ID, paste("X", ID, sep=""))) %>% filter(Fraction=="Nuclear")
order=c(metadata$id2[1:10], "pt30_N", "pt91_N")
BothbyGene= chimpPAS %>% inner_join(humanPAS,by="gene") %>% dplyr::select(gene,order)
#count matrix:
Genematrix=as.matrix(BothbyGene %>% column_to_rownames(var="gene"))colors <- colorRampPalette(c(brewer.pal(9, "Blues")[1],brewer.pal(9, "Blues")[9]))(100)
pal <- c(brewer.pal(9, "Set1"), brewer.pal(8, "Set2"), brewer.pal(12, "Set3"))
labels <- paste(metadata$Species,metadata$Line, sep=" ")# Clustering (original code from Julien Roux)
cors <- cor(Genematrix, method="spearman", use="pairwise.complete.obs")
heatmap.2( cors, scale="none", col = colors, margins = c(12, 12), trace='none', denscol="white", labCol=labels, ColSideColors=pal[as.integer(as.factor(metadata$Species))], RowSideColors=pal[as.integer(as.factor(metadata$Collection))+9], cexCol = 0.2 + 1/log10(15), cexRow = 0.2 + 1/log10(15))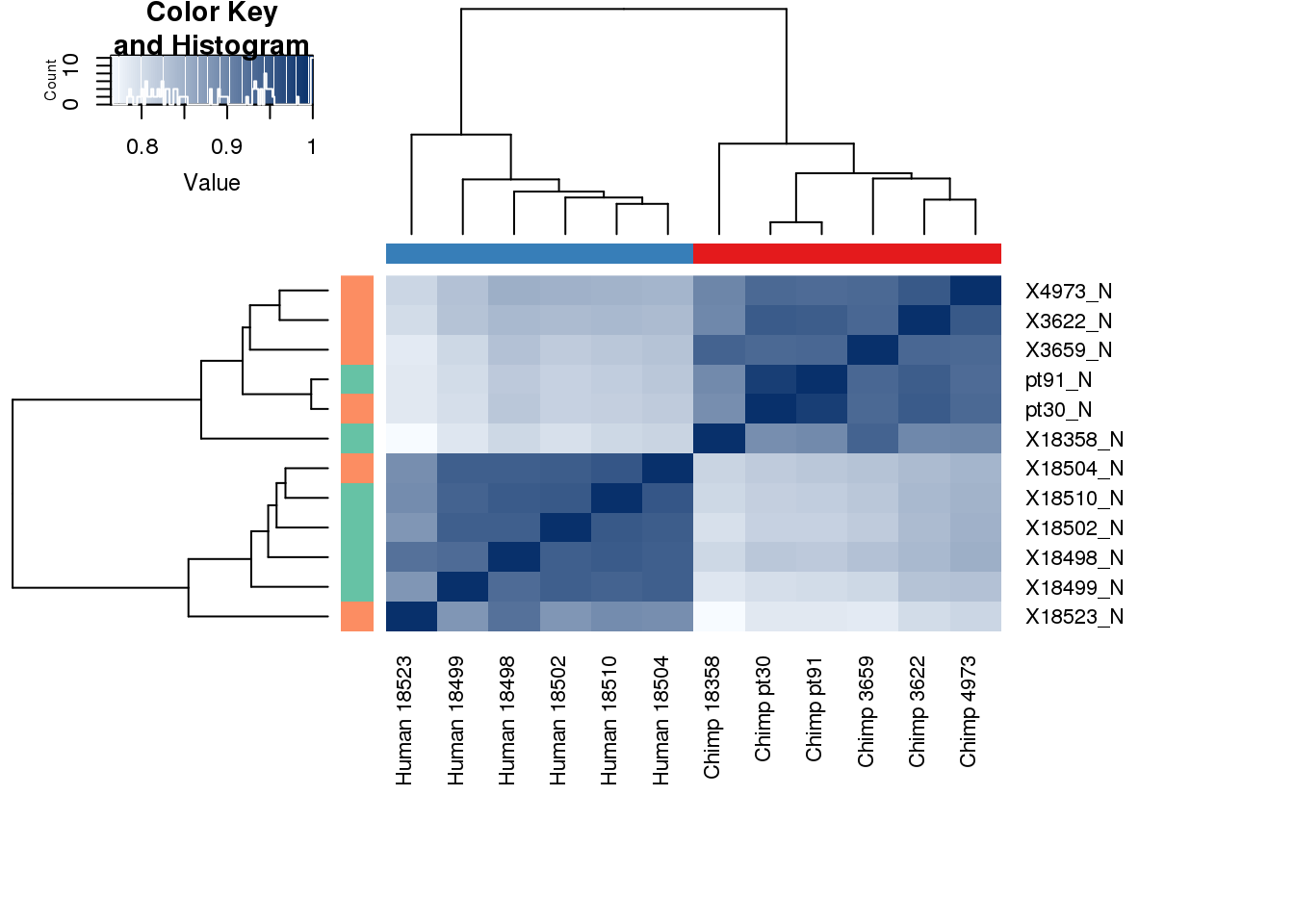
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
Look at the correlation between this and expression:
nameID=read.table("../../genome_anotation_data/ensemble_to_genename.txt",sep="\t", header = T, stringsAsFactors = F,col.names = c("Geneid","gene","source")) %>% dplyr::select(-source)
HumanCounts=read.table("../Human/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fc", header = T, stringsAsFactors = F) %>% dplyr::select(-Chr,-Start,-End,-Strand, -Length)
ChimpCounts=read.table("../Chimp/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fc", header = T, stringsAsFactors = F) %>% dplyr::select(-Chr,-Start,-End,-Strand, -Length)
counts_genes=HumanCounts %>% inner_join(ChimpCounts,by="Geneid") %>% inner_join(nameID, by="Geneid") %>% dplyr::select(-Geneid)
counts_genes_nog=counts_genes %>% dplyr::select(-gene)
ExpMean=as.data.frame(cbind(gene=counts_genes$gene, meanExp=rowMeans(counts_genes_nog)))
ThreeMean=as.data.frame(cbind(gene=BothbyGene$gene, meanThree=rowMeans(Genematrix)))
ExpandThree=ExpMean %>% inner_join(ThreeMean,by="gene") Warning: Column `gene` joining factors with different levels, coercing to
character vectorPlot this:
ExpandThree$meanExp=as.numeric(as.character(ExpandThree$meanExp))
ExpandThree$meanThree=as.numeric(as.character(ExpandThree$meanThree))
ggplot(ExpandThree,aes(x=log10(meanExp),y=log10(meanThree)))+ geom_point() + geom_smooth(method="lm")Warning: Removed 85 rows containing non-finite values (stat_smooth).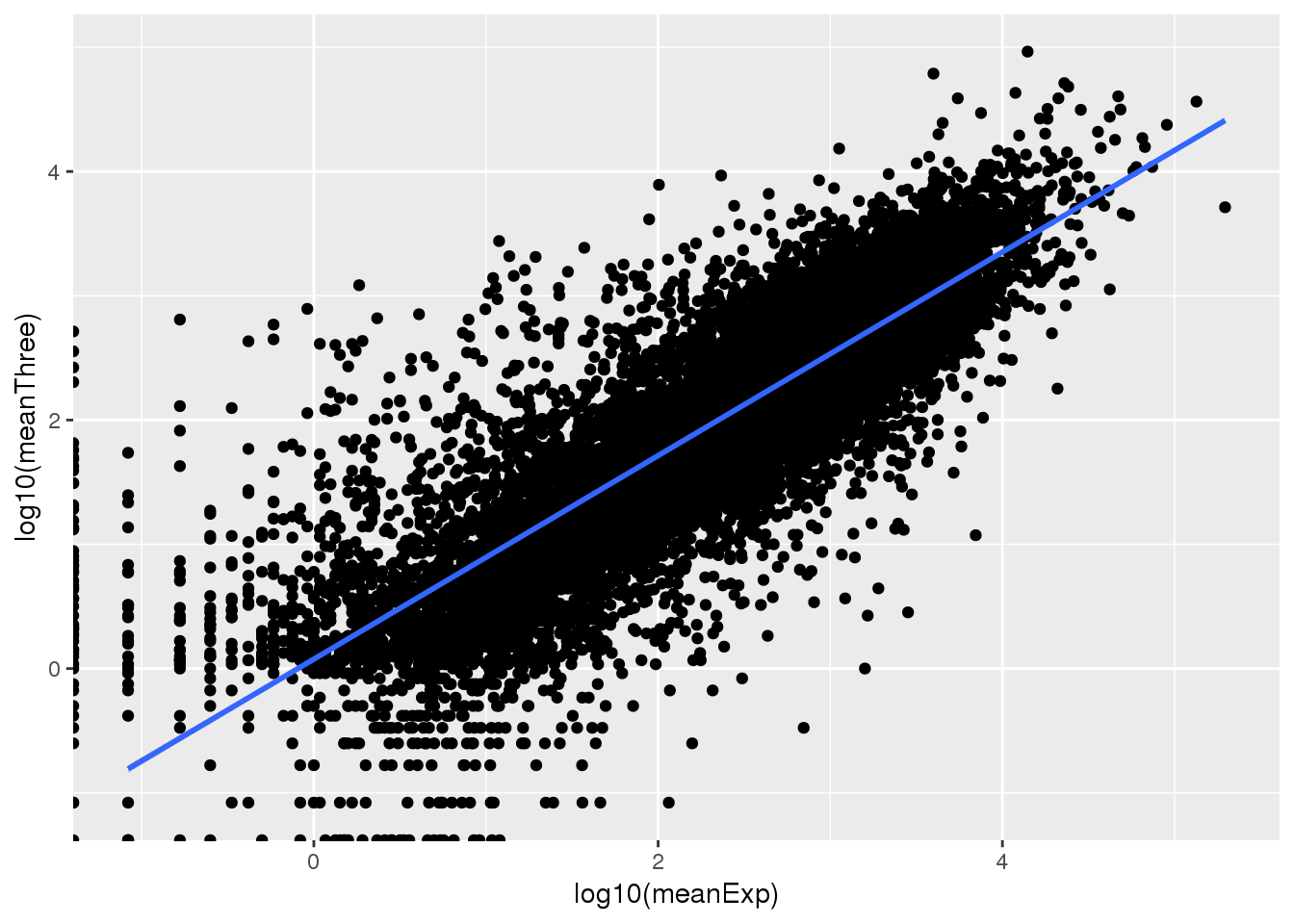
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
This looks pretty good. I can treat the psuedo threeprime as expression to find an expression cuttoff. Next I will normalize and standardize the sum gene counts.
Log2
log_counts_genes <- as.data.frame(log2(Genematrix))
head(log_counts_genes) X18498_N X18499_N X18502_N X18504_N X18510_N X18523_N X18358_N
A1BG 8.651052 6.906891 7.826548 6.339850 7.098032 8.184875 9.348728
A1BG-AS1 6.906891 5.209453 6.686501 5.087463 6.491853 6.189825 7.257388
A2M 4.807355 1.584963 1.000000 4.169925 2.000000 2.000000 9.257388
A4GALT 8.348728 3.000000 5.426265 4.807355 6.643856 7.434628 6.629357
AAAS 7.189825 5.285402 7.149747 6.930737 7.169925 6.539159 7.044394
AACS 9.211888 9.084808 8.455327 9.038919 9.721099 9.539159 9.449149
X3622_N X3659_N X4973_N pt30_N pt91_N
A1BG 7.055282 8.581201 6.044394 8.379378 7.693487
A1BG-AS1 4.700440 6.658211 3.321928 4.643856 3.807355
A2M 9.881114 10.090112 8.392317 8.588715 8.134426
A4GALT 2.000000 6.714246 1.584963 2.000000 2.000000
AAAS 7.357552 7.599913 6.965784 7.539159 7.139551
AACS 8.607330 8.535275 8.422065 8.562242 8.483816plotDensities(log_counts_genes, col=pal[as.numeric(metadata$Species)], legend="topright")
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
CPM
cpm <- cpm(Genematrix, log=TRUE)
plotDensities(cpm, col=pal[as.numeric(metadata$Species)], legend="topright")
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
Use log2 cmp:
## Create edgeR object (dge) to calculate TMM normalization
dge_original <- DGEList(counts=as.matrix(Genematrix), genes=rownames(Genematrix), group = as.character(t(labels)))
dge_original <- calcNormFactors(dge_original)
tmm_cpm <- cpm(dge_original, normalized.lib.sizes=TRUE, log=TRUE, prior.count = 0.25)
head(cpm) X18498_N X18499_N X18502_N X18504_N X18510_N X18523_N
A1BG 5.341650 4.5993068 4.825291 3.911623 3.751466 4.9774440
A1BG-AS1 3.615999 2.9313152 3.698829 2.688464 3.157697 3.0125468
A2M 1.600132 -0.2806202 -1.087332 1.814585 -0.714223 -0.6153132
A4GALT 5.041173 0.8670626 2.472727 2.419003 3.306094 4.2341398
AAAS 3.894256 3.0051013 4.155261 4.495314 3.822198 3.3532893
AACS 5.899931 6.7668568 5.450058 6.592507 6.354400 6.3255013
X18358_N X3622_N X3659_N X4973_N pt30_N pt91_N
A1BG 5.865947 3.8087270 5.334818 2.9688165 5.0434796 4.3424445
A1BG-AS1 3.792472 1.5453264 3.433898 0.4596994 1.4233049 0.6597173
A2M 5.774967 6.6147191 6.838555 5.2833627 5.2514988 4.7792050
A4GALT 3.177039 -0.6493577 3.488798 -0.8146537 -0.6983404 -0.7119011
AAAS 3.583164 4.1066540 4.361269 3.8706954 4.2109429 3.7959070
AACS 5.965997 5.3455790 5.289151 5.3129416 5.2251829 5.1260744Look at a PCA plot of the log2cpm
#PCA function (original code from Julien Roux)
#Load in the plot_scores function
plot_scores <- function(pca, scores, n, m, cols, points=F, pchs =20, legend=F){
xmin <- min(scores[,n]) - (max(scores[,n]) - min(scores[,n]))*0.05
if (legend == T){ ## let some room (35%) for a legend
xmax <- max(scores[,n]) + (max(scores[,n]) - min(scores[,n]))*0.50
}
else {
xmax <- max(scores[,n]) + (max(scores[,n]) - min(scores[,n]))*0.05
}
ymin <- min(scores[,m]) - (max(scores[,m]) - min(scores[,m]))*0.05
ymax <- max(scores[,m]) + (max(scores[,m]) - min(scores[,m]))*0.05
plot(scores[,n], scores[,m], xlab=paste("PC", n, ": ", round(summary(pca)$importance[2,n],3)*100, "% variance explained", sep=""), ylab=paste("PC", m, ": ", round(summary(pca)$importance[2,m],3)*100, "% variance explained", sep=""), xlim=c(xmin, xmax), ylim=c(ymin, ymax), type="n")
if (points == F){
text(scores[,n],scores[,m], rownames(scores), col=cols, cex=1)
}
else {
points(scores[,n],scores[,m], col=cols, pch=pchs, cex=1.3)
}
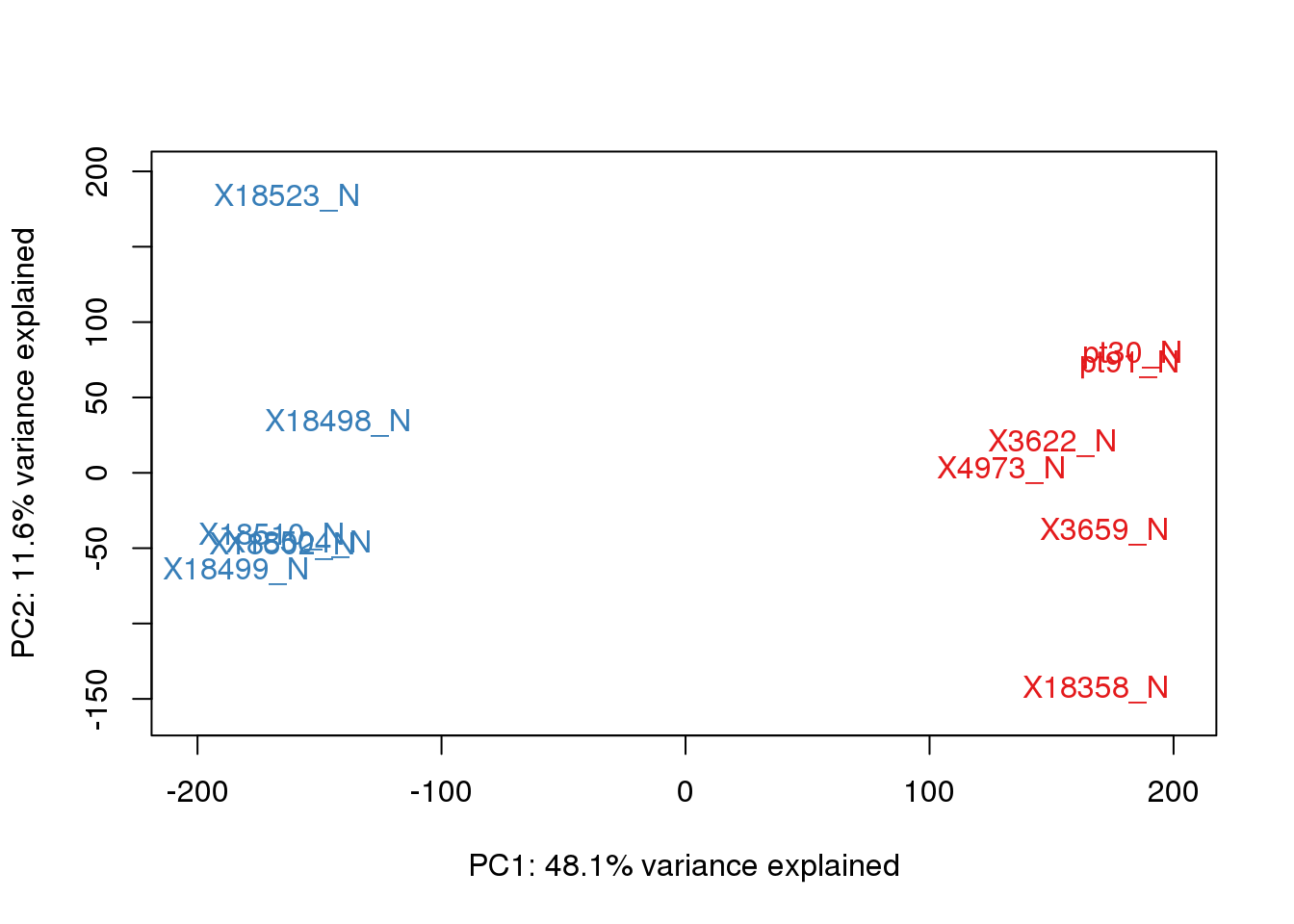
}pca_genes <- prcomp(t(tmm_cpm), scale = F)
scores <- pca_genes$x
for (n in 1:2){
col.v <- pal[as.integer(metadata$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
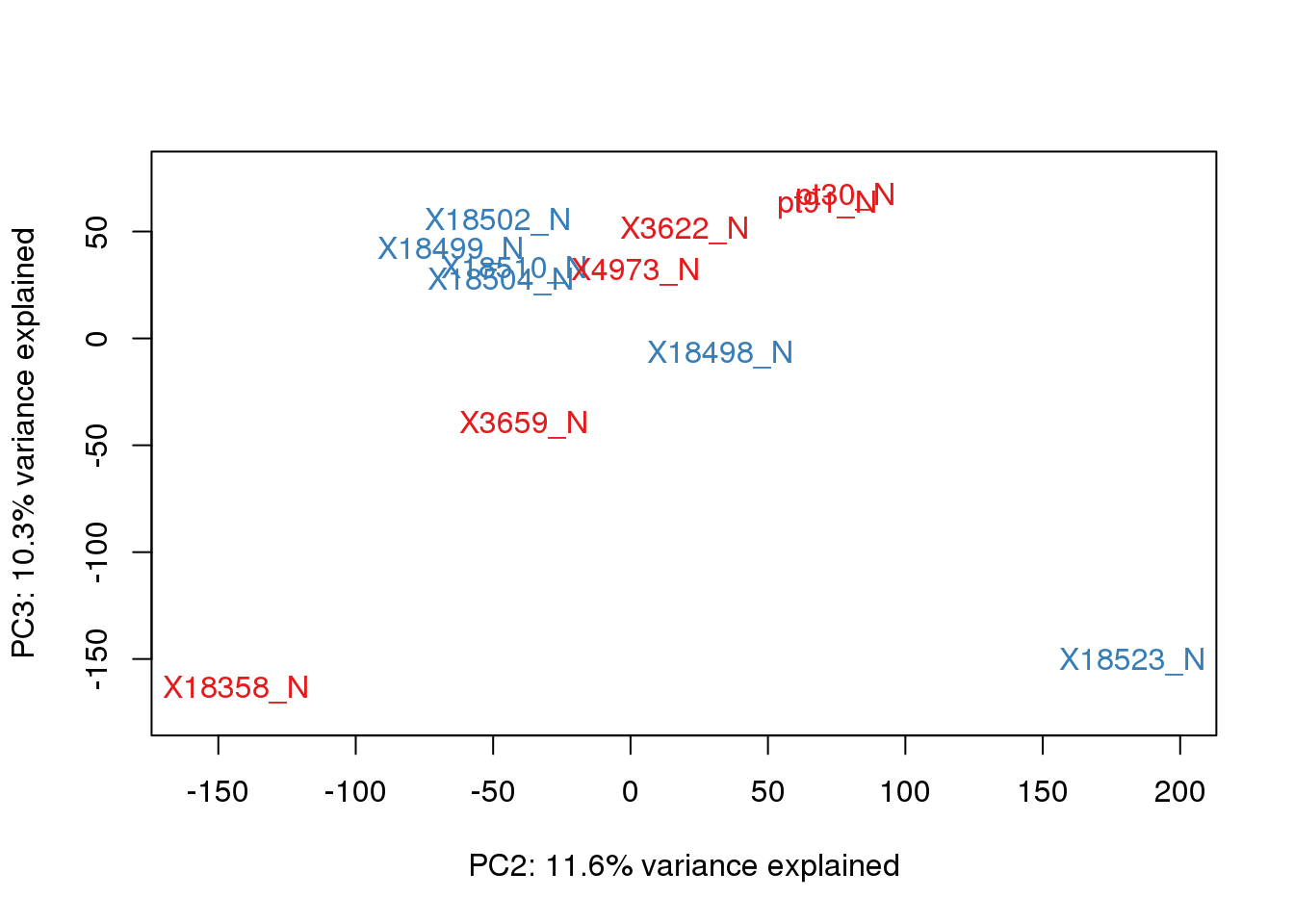
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
Plot the log2cpm
plotDensities(tmm_cpm, col=pal[as.numeric(metadata$Species)], legend="topright")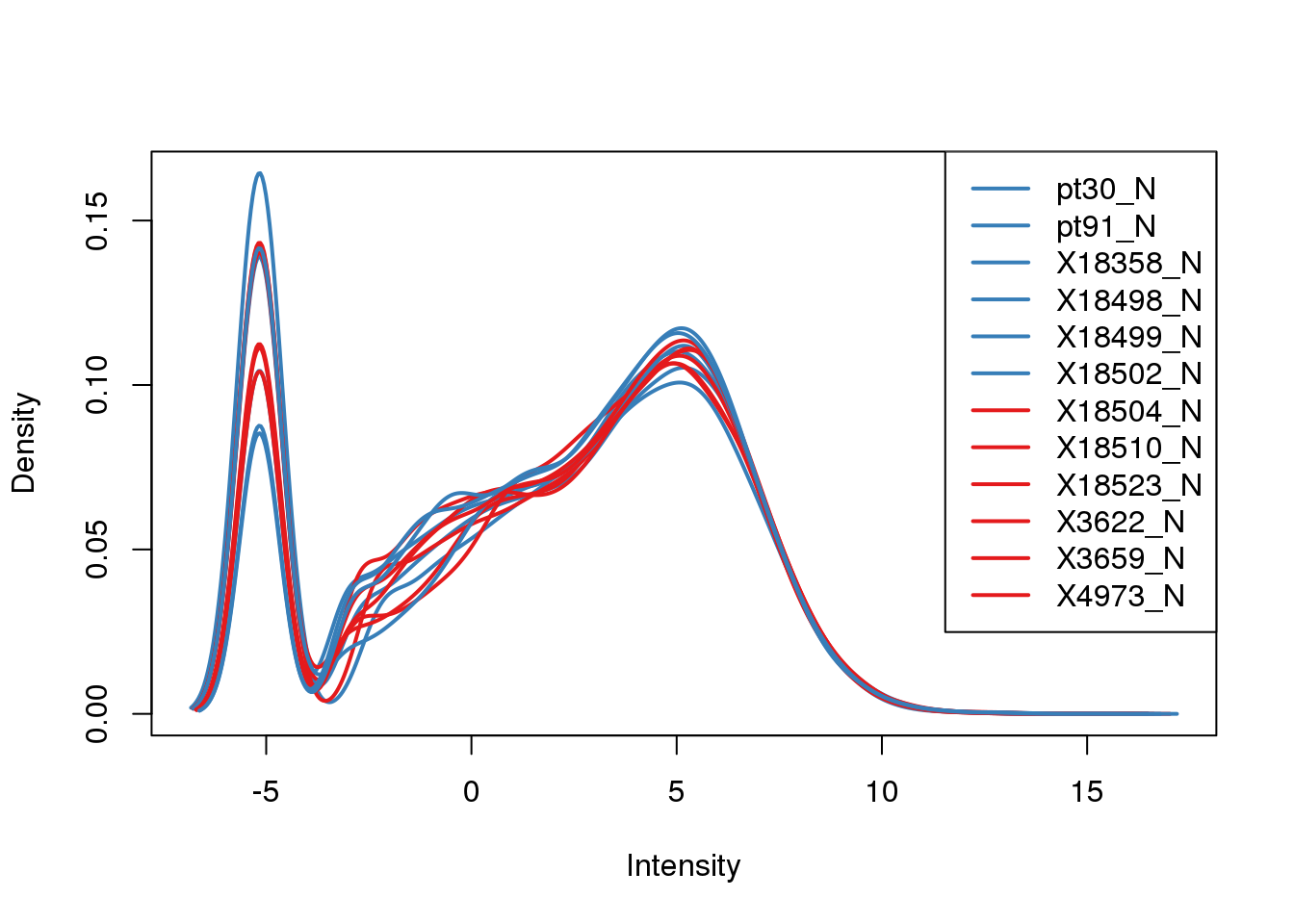
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
I will need to filter out the lowly expressed genes. This will also be the gene filter I use for the PAS.
Start with log2cpm>2 for 8 of the 12 indiv. Filter the counts
keep.exprs=rowSums(tmm_cpm>2) >8
counts_filtered= Genematrix[keep.exprs,]
plotDensities(counts_filtered, col=pal[as.numeric(metadata$Species)], legend="topright")
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
Make a new dge list and filter:
labels <- paste(metadata$Species, metadata$Line, sep=" ")
dge_in_cutoff <- DGEList(counts=as.matrix(counts_filtered), genes=rownames(counts_filtered), group = as.character(t(labels)))
dge_in_cutoff <- calcNormFactors(dge_in_cutoff)
cpm_in_cutoff <- cpm(dge_in_cutoff, normalized.lib.sizes=TRUE, log=TRUE, prior.count = 0.25)
head(cpm_in_cutoff) X18498_N X18499_N X18502_N X18504_N X18510_N X18523_N X18358_N
A1BG 5.329663 4.631635 4.907843 3.908975 3.666584 5.080271 5.914427
AAAS 3.870285 3.013698 4.231888 4.498912 3.738314 3.437223 3.612862
AACS 5.890160 6.808219 5.536123 6.605652 6.286849 6.433791 6.014800
AAGAB 6.897081 6.540471 6.287438 6.708810 6.727998 5.851978 6.571066
AAK1 7.173705 7.038504 7.187389 6.461596 6.756658 7.299552 7.137050
AAMDC 3.918769 4.452891 4.589439 4.375697 3.310955 3.994837 4.211144
X3622_N X3659_N X4973_N pt30_N pt91_N
A1BG 3.940920 5.403320 3.058774 5.219852 4.475256
AAAS 4.242667 4.423008 3.977759 4.380532 3.922213
AACS 5.491146 5.357427 5.432327 5.402580 5.264779
AAGAB 6.138503 6.203235 5.949545 5.997346 5.922666
AAK1 6.458894 6.673281 7.758506 6.217116 6.284230
AAMDC 4.838552 4.741566 5.398303 4.879696 5.346957GenesCutoff=rownames(cpm_in_cutoff)
NormalizedGenesCuttoff=as.data.frame(cbind(Gene_stable_ID=GenesCutoff, cpm_in_cutoff))Plot the historgram:
hist(cpm_in_cutoff, xlab = "Log2(CPM)", main = "Log2(CPM) values for genes meeting the filtering criteria", breaks = 100 )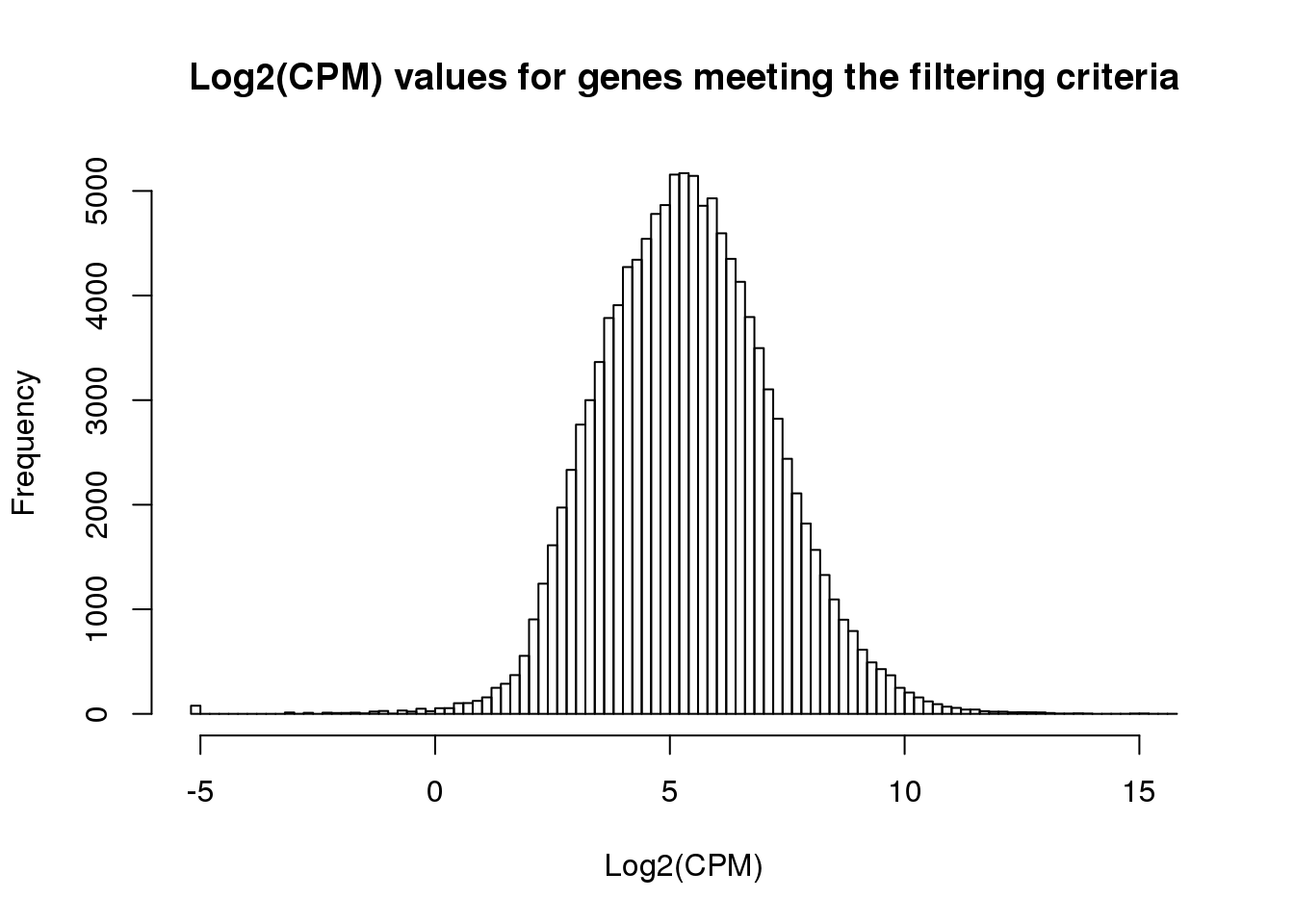
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
This looks relatively normal. I will next look at the voom transformed values with quantile normalization.
Species <- factor(metadata$Species)
design <- model.matrix(~ 0 + Species)
head(design) SpeciesChimp SpeciesHuman
1 0 1
2 0 1
3 0 1
4 0 1
5 0 1
6 0 1colnames(design) <- gsub("Species", "", dput(colnames(design)))c("SpeciesChimp", "SpeciesHuman")cpm.voom<- voom(counts_filtered, design, normalize.method="quantile", plot=T)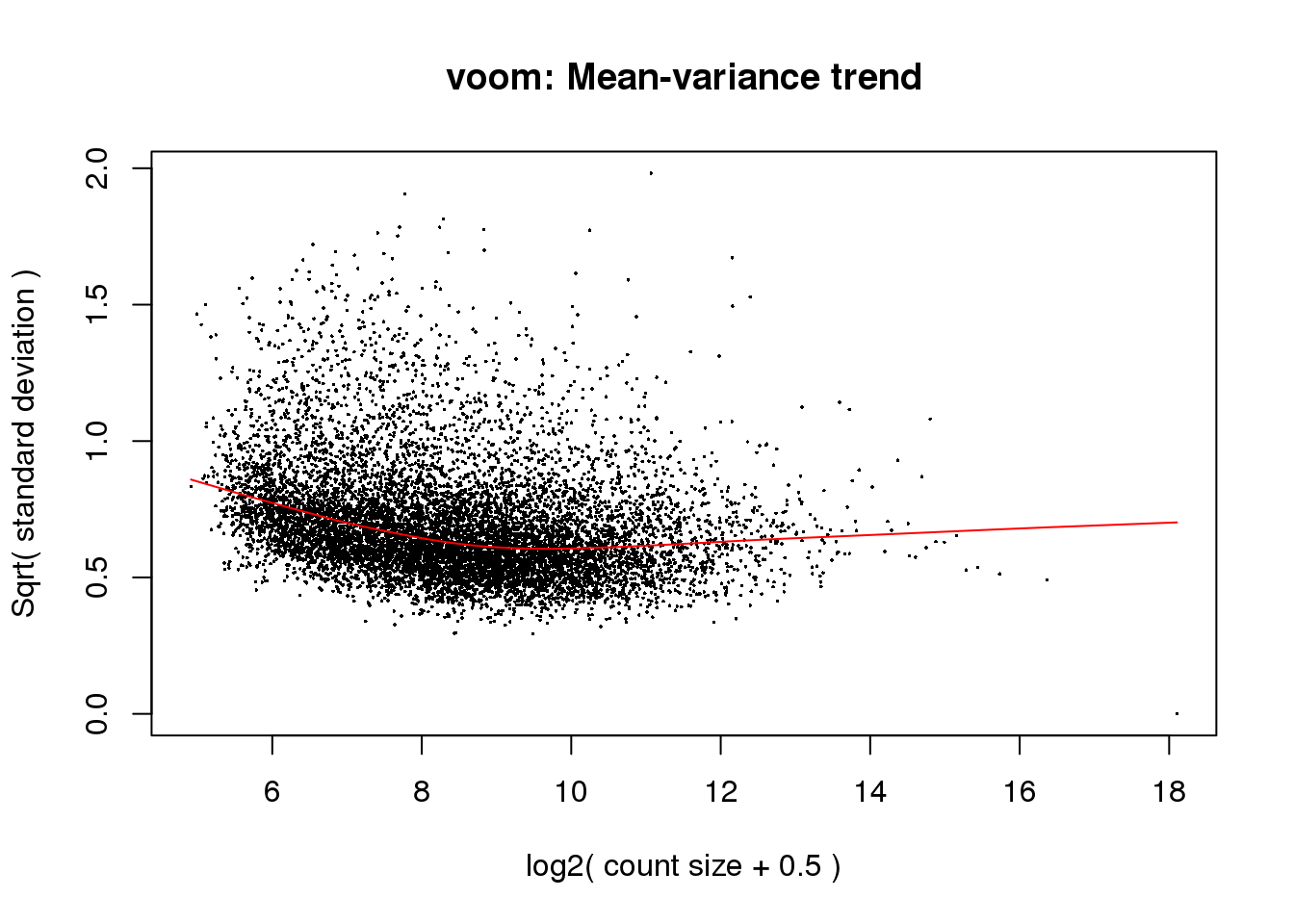
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
boxplot(cpm.voom$E, col = pal[as.numeric(metadata$Species)],las=2)
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
plotDensities(cpm.voom, col = pal[as.numeric(metadata$Species)], legend = "topleft") 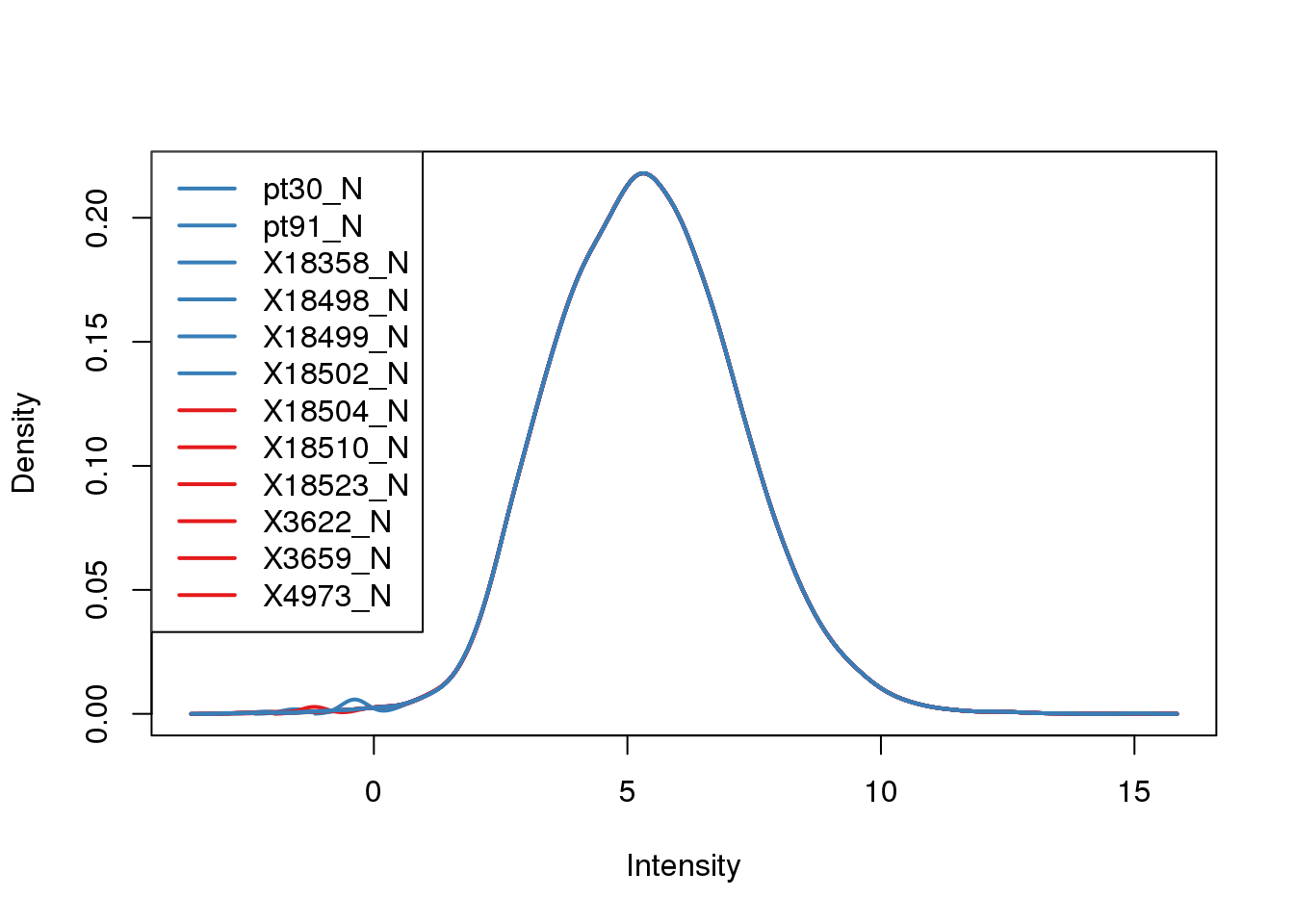
| Version | Author | Date |
|---|---|---|
| f161386 | brimittleman | 2020-01-18 |
This looks like a good cuttoff. I will make a list of the genes that pass the cutoff.
length(GenesCutoff)[1] 9731GenesCutoffDF=as.data.frame(GenesCutoff) %>% rename("genes"=GenesCutoff)
#mkdir ../data/OverlapBenchmark
write.table(GenesCutoffDF,"../data/OverlapBenchmark/genesPassingCuttoff.txt", col.names = T, row.names = F,quote = F)
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] RColorBrewer_1.1-2 scales_1.0.0 edgeR_3.24.0
[4] limma_3.38.2 forcats_0.3.0 stringr_1.3.1
[7] dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1
[10] tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1
[13] tidyverse_1.2.1 R.utils_2.7.0 R.oo_1.22.0
[16] R.methodsS3_1.7.1 gplots_3.0.1 workflowr_1.6.0
loaded via a namespace (and not attached):
[1] locfit_1.5-9.1 Rcpp_1.0.2 lubridate_1.7.4
[4] lattice_0.20-38 gtools_3.8.1 assertthat_0.2.0
[7] rprojroot_1.3-2 digest_0.6.18 R6_2.3.0
[10] cellranger_1.1.0 plyr_1.8.4 backports_1.1.2
[13] evaluate_0.12 httr_1.3.1 pillar_1.3.1
[16] rlang_0.4.0 lazyeval_0.2.1 readxl_1.1.0
[19] rstudioapi_0.10 gdata_2.18.0 whisker_0.3-2
[22] rmarkdown_1.10 labeling_0.3 munsell_0.5.0
[25] broom_0.5.1 compiler_3.5.1 httpuv_1.4.5
[28] modelr_0.1.2 pkgconfig_2.0.2 htmltools_0.3.6
[31] tidyselect_0.2.5 crayon_1.3.4 withr_2.1.2
[34] later_0.7.5 bitops_1.0-6 grid_3.5.1
[37] nlme_3.1-137 jsonlite_1.6 gtable_0.2.0
[40] git2r_0.26.1 magrittr_1.5 KernSmooth_2.23-15
[43] cli_1.1.0 stringi_1.2.4 fs_1.3.1
[46] promises_1.0.1 xml2_1.2.0 generics_0.0.2
[49] tools_3.5.1 glue_1.3.0 hms_0.4.2
[52] yaml_2.2.0 colorspace_1.3-2 caTools_1.17.1.1
[55] rvest_0.3.2 knitr_1.20 haven_1.1.2