5’ Spice Site strength
Briana Mittleman
2/21/2020
Last updated: 2020-02-22
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ClassifyLeafviz.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OverlapBenchmark/
Untracked: data/PAS/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalHvC/
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Unstaged changes:
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/dAPAandapaQTL_DF.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/speciesSpecific.Rmd
Modified: analysis/speciesSpecific_DF.Rmd
Modified: analysis/upsetter_DF.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7e2fb38 | brimittleman | 2020-02-22 | add ss res |
| html | 5bcde2f | brimittleman | 2020-02-21 | Build site. |
| Rmd | 2e8ed44 | brimittleman | 2020-02-21 | add Splice site strength |
library(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ──────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ─────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()There is a hypothesis that increased 5’ splice site strength is assocaited with decreased usage of intronic PAS. This is relate to competition and binding of the U1 snurp. I will ask if there are differences in 5’ splcie sites for humans and chimp.
Need to be careful about orthology here. To be conservative. I will only look at regions that map downstream of an ortho exon.
First step is to map each intronic PAS to a human intron annotation.
I created a transcript minus exon file for my previos project. I will lift this file over andcheck it. I can remake it
mkdir ../data/SpliceSite
liftOver /project2/gilad/briana/apaQTL/data/intron_analysis/transcriptsMinusExons.sort.bed ../data/liftover_files/hg19ToHg38.over.chain.gz ../data/SpliceSite/transcriptMinusexon_hg38.bed ../data/SpliceSite/UnliftedIntron.bed
These look really good, they line up well.
Pull out intronic PAS
PAS_metaIntron=read.table("../data/PAS_doubleFilter/PAS_10perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F) %>% filter(loc=="intron")
PAS=read.table("../data/PAS_doubleFilter/PAS_doublefilter_either_HumanCoordHummanUsage.bed", col.names = c("chr", "start", "end", "PAS", "score", "strand"),stringsAsFactors = F) %>% semi_join(PAS_metaIntron, by="PAS")
write.table(PAS, "../data/SpliceSite/IntronicPAS_humanCoord.bed", col.names = F, row.names = F, quote = F, sep="\t")sbatch assignPeak2Intronicregion.shGet the 5’ splice sites for all of these.
(lose ~800)
PAS2Intron=read.table("../data/SpliceSite/IntronincPAS2Introns_humanCoord.bed",col.names = c("IntronChr", "IntronStart", "IntronEnd", "Gene", "Score", "Strand", "PASChr", "PASStart","PASEnd", "PAS", "humanUsage", "passtrand"),stringsAsFactors = F)
Lost= PAS %>% anti_join(PAS2Intron, by="PAS")
write.table(Lost, "../data/SpliceSite/LostinIntersect.bed", col.names = F, row.names = F, quote =F, sep = "\t")Lose some with multiple isoforms. Downstream of a gene may be an intron in one. It is probably not possible to get perfect annotation.
PAS2Intron_pos= PAS2Intron %>% filter(Strand=="+") %>% mutate(start=IntronStart-3, end= IntronStart +6) %>% select(IntronChr, start,end, PAS,humanUsage, Strand)
PAS2Intron_neg=PAS2Intron %>% filter(Strand=="-") %>% mutate(start=IntronEnd-6, end= IntronEnd +3) %>% select(IntronChr, start,end, PAS,humanUsage, Strand)
PAS_5SS_both= PAS2Intron_neg %>% bind_rows(PAS2Intron_pos)
write.table(PAS_5SS_both, "../data/SpliceSite/IntronicPAS_SS_humanCoord.bed", col.names = F, row.names = F, quote=F, sep="\t")Sort and assign to ortho exon. I need a small amount of overlap with the human ortho exon file. This comes from Kenneth’s work. /project2/gilad/kenneth/OrthoExonPartialMapping/human.noM.gtf
Ortho exon needs to be converted to bed to intersect.
sort -k1,1 -k2,2n ../data/SpliceSite/IntronicPAS_SS_humanCoord.bed > ../data/SpliceSite/IntronicPAS_SS_humanCoord.sort.bed
bedtools intersect -a ../data/SpliceSite/IntronicPAS_SS_humanCoord.sort.bed -b /project2/gilad/kenneth/OrthoExonPartialMapping/human.noM.gtf -s -wao > ../data/SpliceSite/IntronicPAS_SS_intersectOrthoExon.txt
#looking for 3 base overlap with splice sites
IntersectRes=read.table("../data/SpliceSite/IntronicPAS_SS_intersectOrthoExon.txt",stringsAsFactors = F,sep="\t", col.names = c("chr",'ssstart','ssend','PAS', 'humanusage','passtrand', 'file', 'loc','exonchr', 'enonstart','exonend', 'score', 'strand', 'score2', 'geneinfo', 'overlap')) %>% filter(overlap==3)
IntersectRes_group= IntersectRes %>% group_by(PAS) %>% summarise(nExon=n())
nrow(IntersectRes_group)[1] 8146From 11060 to 8146.
I will only look at the 8146 intronic PAS that map intersect 3 basepairs of the 5’ splice site to an ortho exon.
Filter :
PAS_5SS_both_filt= PAS_5SS_both %>% semi_join(IntersectRes_group, by="PAS")
PAS_5SS_both_filt %>% group_by(PAS) %>% summarise(n=n()) %>% filter(n>1)# A tibble: 171 x 2
PAS n
<chr> <int>
1 chimp10327 2
2 chimp108153 2
3 chimp130492 2
4 chimp130494 2
5 chimp132166 2
6 chimp13702 2
7 chimp13703 2
8 chimp13858 2
9 chimp147711 2
10 chimp151277 2
# … with 161 more rowsPAS_5SS_both_filt %>% group_by(PAS) %>% summarise(n=n()) %>% filter(n>1) %>% nrow()[1] 171171 map to 2 introns. Count each site for now.
Write these out to sort and liftover.
write.table(PAS_5SS_both_filt, "../data/SpliceSite/IntronicPAS_SS_humanCoord_filterOotho.bed", col.names = F, row.names = F, quote = F, sep="\t")sort -k1,1 -k2,2n ../data/SpliceSite/IntronicPAS_SS_humanCoord_filterOotho.bed > ../data/SpliceSite/IntronicPAS_SS_humanCoord_filterOotho.sort.bed
liftOver ../data/SpliceSite/IntronicPAS_SS_humanCoord_filterOotho.sort.bed ../data/chainFiles/hg38ToPanTro6.over.chain ../data/SpliceSite/IntronicPAS_SS_ChimpCoord_filterOotho.sort.bed ../data/SpliceSite/ChimpCoordUnliftedSS.txt Remove unlifted from human
unliftedSS=read.table("../data/SpliceSite/ChimpCoordUnliftedSS.txt",col.names = c("chr", 'start','end','PAS', 'humanscore', 'strand'), stringsAsFactors = F)
#check still 9 bases
liftedSS=read.table("../data/SpliceSite/IntronicPAS_SS_ChimpCoord_filterOotho.sort.bed",col.names = c("chr", 'start','end','PAS', 'humanscore', 'strand'), stringsAsFactors = F) %>% mutate(legnth=end-start)
liftedSS_wrongsize= liftedSS %>% filter(legnth!=9)
nrow(liftedSS_wrongsize)[1] 13nrow(unliftedSS)[1] 18BADSS= as.data.frame(cbind(PAS=c(liftedSS_wrongsize$PAS,unliftedSS$PAS)))Remove the 31 that to not lift or lift to the wrong size.
ChimpSS=liftedSS %>% filter(legnth==9) %>% select(-legnth)
nrow(ChimpSS)[1] 8298HumanSS=PAS_5SS_both_filt %>% anti_join(BADSS, by="PAS")Warning: Column `PAS` joining character vector and factor, coercing into
character vectornrow(HumanSS)[1] 8298I will look at 8298 PAS.
Next step is to use bedtools nuc to get the strand specific basepairs.
write.table(ChimpSS, "../data/SpliceSite/Chimp_SS.bed", col.names = F, row.names = F, quote = F, sep="\t")
write.table(HumanSS, "../data/SpliceSite/Human_SS.bed", col.names = F, row.names = F, quote = F, sep="\t")sort -k1,1 -k2,2n ../data/SpliceSite/Chimp_SS.bed > ../data/SpliceSite/Chimp_SS_sort.bed
sort -k1,1 -k2,2n ../data/SpliceSite/Human_SS.bed > ../data/SpliceSite/Human_SS.sort.bed
#bedtools nuc -fi /project2/gilad/briana/genome_anotation_data/genome/Homo_sapiens.GRCh37.75.dna_sm.all.fa -bed ../data/splicesite/AllPASSS.sort.noChr.bed -seq -s > ../data/splicesite/AllPASSS.sort.Nuc.txt
bedtools nuc -fi /project2/gilad/briana/genome_anotation_data/Chimp_genome/panTro6.fa -bed ../data/SpliceSite/Chimp_SS_sort.bed -seq -s > ../data/SpliceSite/Chimp_SS_sort.Nuc.txt
bedtools nuc -fi /project2/gilad/kenneth/References/human/genome/hg38.fa -bed ../data/SpliceSite/Human_SS.sort.bed -seq -s > ../data/SpliceSite/Human_SS.sort.Nuc.txt
#parse
python spliceSite2Fasta.py ../data/SpliceSite/Chimp_SS_sort.Nuc.txt ../data/SpliceSite/Chimp_SS_sort.Nuc.fasta
python spliceSite2Fasta.py ../data/SpliceSite/Human_SS.sort.Nuc.txt ../data/SpliceSite/Human_SS_sort.Nuc.fasta
#run ss maxent
cd /MaxEntCode/fordownload
perl score5.pl ../../../data/SpliceSite/Chimp_SS_sort.Nuc.fasta > ../../../data/SpliceSite/Chimp_SS_sort.Nuc.MaxentScores.txt
perl score5.pl ../../../data/SpliceSite/Human_SS_sort.Nuc.fasta > ../../../data/SpliceSite/Human_SS_sort.Nuc.MaxentScore.txtChimpSS=read.table("../data/SpliceSite/Chimp_SS_sort.bed", col.names = c("chr",'start','end','PAS', 'HumanUsage', 'strand'), stringsAsFactors = F) %>% select(PAS,HumanUsage)
ChimpRES=read.table("../data/SpliceSite/Chimp_SS_sort.Nuc.MaxentScores.txt", col.names =c("Chimpseq", "ChimpScore"))
ChimpSSandRes=ChimpSS %>% bind_cols(ChimpRES)
HumanSS=read.table("../data/SpliceSite/Human_SS.sort.bed", col.names = c("chr",'start','end','PAS', 'HumanUsage', 'strand'), stringsAsFactors = F)%>% select(PAS,HumanUsage)
HumanRES=read.table("../data/SpliceSite/Human_SS_sort.Nuc.MaxentScore.txt", col.names = c("Humanseq", "HumanScore"))
HumanSSandRes=HumanSS %>% bind_cols(HumanRES)
BothSSandRes= ChimpSSandRes %>% inner_join(HumanSSandRes, by=c('PAS','HumanUsage'))Add mean chimp
ChimpPASUsage=read.table("../data/PAS_doubleFilter/PAS_doublefilter_either_ChimpCoordChimpUsage.sort.bed",col.names = c('chr','start','end',"PAS", 'ChimpUsage','strand' ),stringsAsFactors = F) %>% select(PAS, ChimpUsage)
BothSSandReswUsage=BothSSandRes %>% inner_join(ChimpPASUsage,by='PAS')ggplot(BothSSandReswUsage, aes(x=HumanScore, y=HumanUsage)) +geom_point(col="blue", alpha=.3) + geom_point(data=BothSSandReswUsage, aes(x=ChimpScore, y=ChimpUsage), alpha=.3,col="orange") 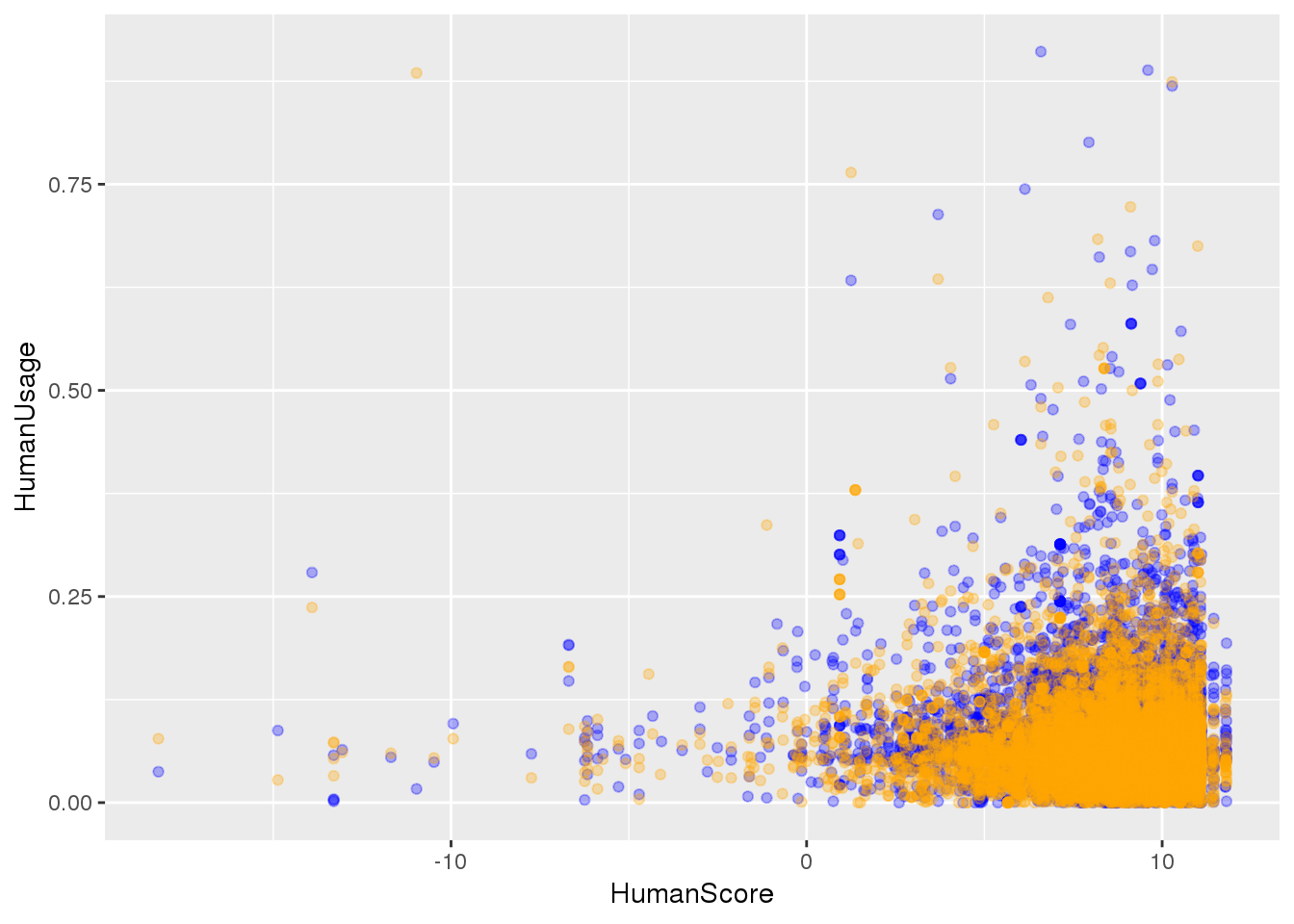
ggplot(BothSSandReswUsage,aes(x=ChimpScore, y=HumanScore)) + geom_point() + geom_density2d(col="green") + geom_smooth(method="lm",col="orange") + annotate("text",label="Pearsons Correlation = .98", x=-10, y=10)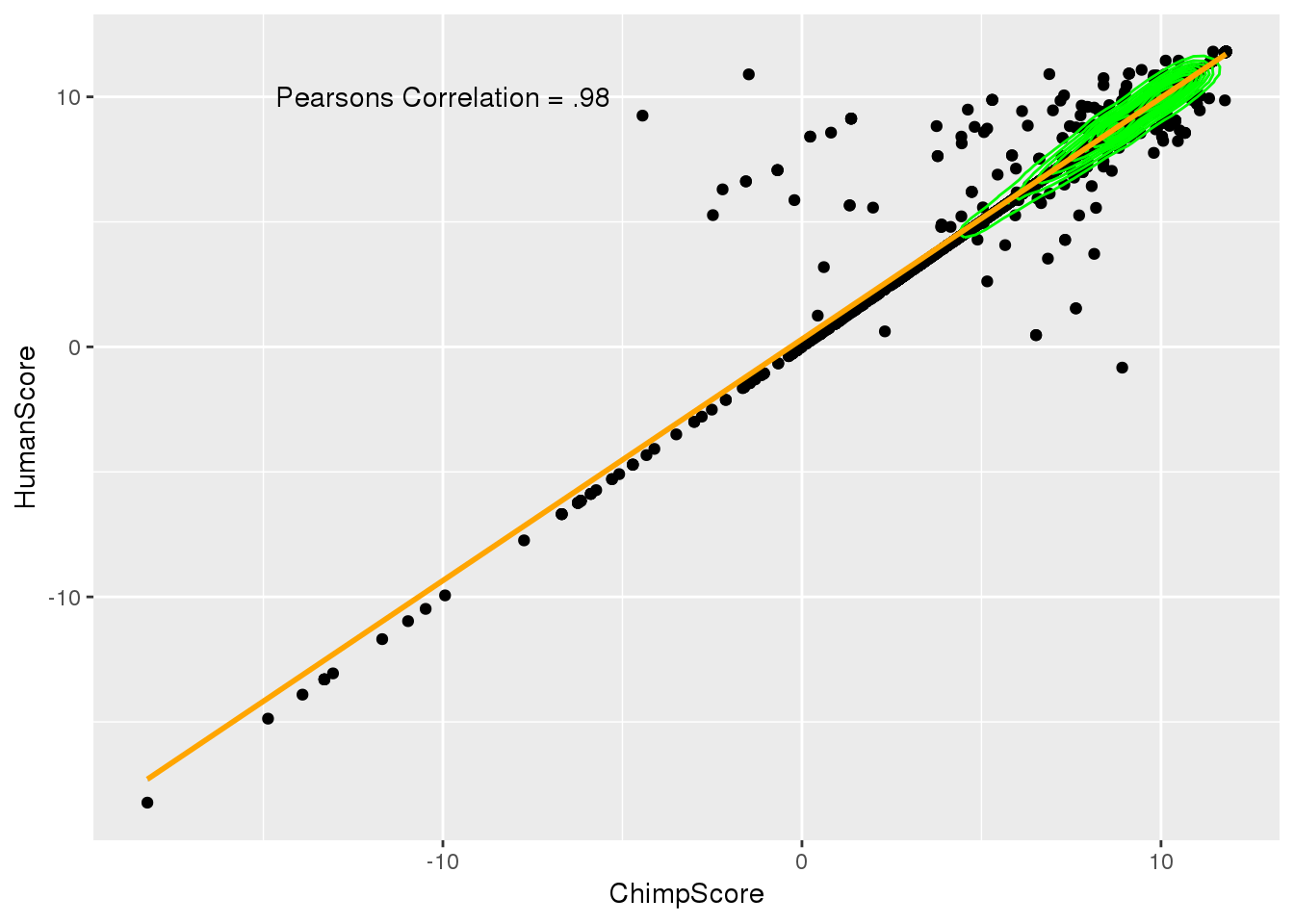
cor.test(BothSSandReswUsage$ChimpScore,BothSSandReswUsage$HumanScore )
Pearson's product-moment correlation
data: BothSSandReswUsage$ChimpScore and BothSSandReswUsage$HumanScore
t = 446.07, df = 8688, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.9779605 0.9797205
sample estimates:
cor
0.9788586 ggplot(BothSSandReswUsage,aes(x=ChimpUsage, y=HumanUsage)) + geom_point() + geom_density2d(col="green") + geom_smooth(method="lm",col="orange") + annotate("text", label="Pearsons Correlation= 0.58", x=.65,y=.8)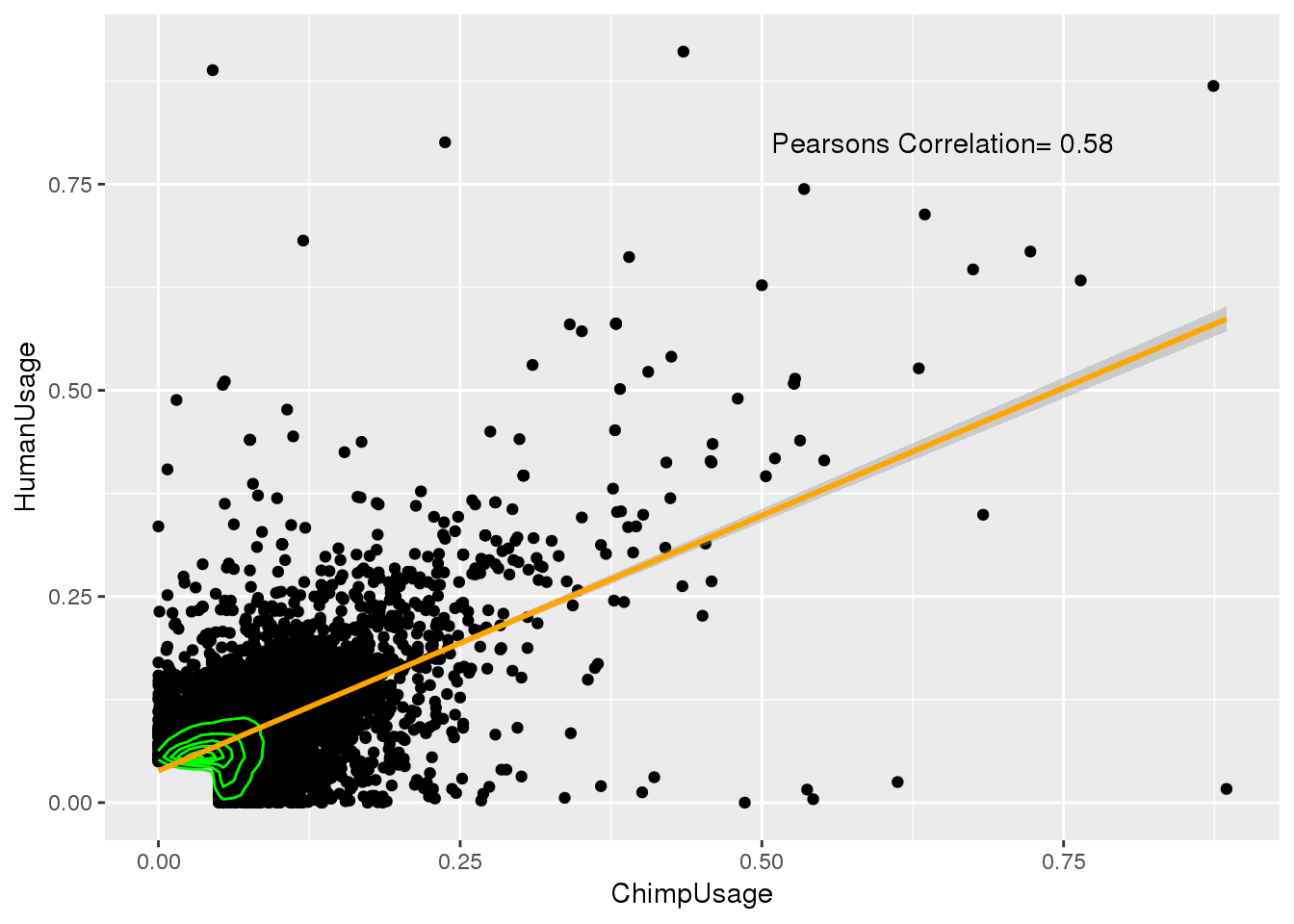
cor.test(BothSSandReswUsage$ChimpUsage,BothSSandReswUsage$HumanUsage )
Pearson's product-moment correlation
data: BothSSandReswUsage$ChimpUsage and BothSSandReswUsage$HumanUsage
t = 65.703, df = 8688, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.5619293 0.5900259
sample estimates:
cor
0.5761478 How many have different score:
BothSSandReswUsage_diff= BothSSandReswUsage %>% filter(ChimpScore!=HumanScore)
nrow(BothSSandReswUsage_diff)[1] 229229/8298 PAS have different scores in human and chimp
I expect higher scores to have lower usage
Plot difference in score and diff in usage
BothSSandReswUsage_diff_score= BothSSandReswUsage_diff %>% mutate(DiffScore=HumanScore-ChimpScore, DiffUsage=HumanUsage-ChimpUsage)
ggplot(BothSSandReswUsage_diff_score, aes(x=DiffScore, y=DiffUsage)) + geom_point() + geom_smooth(method="lm")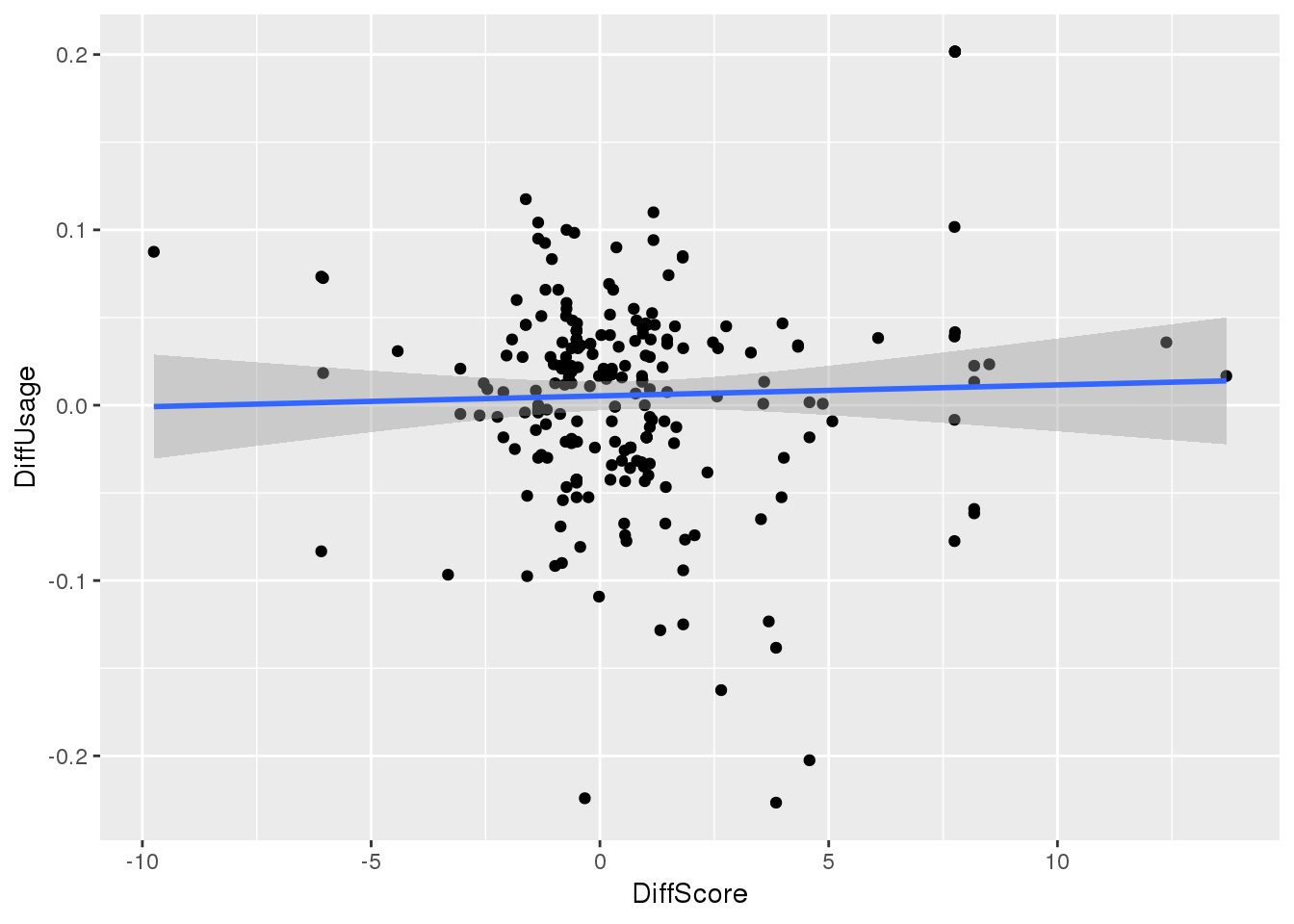
summary(lm(BothSSandReswUsage_diff_score$DiffScore ~ BothSSandReswUsage_diff_score$DiffUsage))
Call:
lm(formula = BothSSandReswUsage_diff_score$DiffScore ~ BothSSandReswUsage_diff_score$DiffUsage)
Residuals:
Min 1Q Median 3Q Max
-10.6097 -1.5064 -0.4609 0.6594 12.9320
Coefficients:
Estimate Std. Error t value
(Intercept) 0.7341 0.1951 3.762
BothSSandReswUsage_diff_score$DiffUsage 1.4361 3.1798 0.452
Pr(>|t|)
(Intercept) 0.000215 ***
BothSSandReswUsage_diff_score$DiffUsage 0.651963
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 2.94 on 227 degrees of freedom
Multiple R-squared: 0.0008978, Adjusted R-squared: -0.003504
F-statistic: 0.204 on 1 and 227 DF, p-value: 0.652No correlation
Are any of these the differentially used PAS.
Meta=read.table("../data/PAS_doubleFilter/PAS_10perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F) %>% select(PAS,chr, loc, start, end)
DiffIsoRes=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T,stringsAsFactors = F) %>% inner_join(Meta, by=c("chr", 'start','end')) %>% select(PAS,SigPAU2 )Join:
BothSSandReswUsage_diff_score_iso=BothSSandReswUsage_diff_score %>% inner_join(DiffIsoRes, by="PAS")
BothSSandReswUsage_diff_score_iso %>% filter(SigPAU2=="Yes") %>% select(PAS,HumanScore, HumanUsage, ChimpScore,ChimpUsage) PAS HumanScore HumanUsage ChimpScore ChimpUsage
1 human34001 9.44 0.11000000 8.27 0.000000000
2 human43606 7.21 0.06833333 7.94 0.115000000
3 human43627 7.21 0.12333333 7.94 0.023333333
4 human70865 10.90 0.07833333 -1.48 0.042500000
5 human86581 6.49 0.08500000 7.31 0.049166667
6 human95658 7.40 0.12666667 7.96 0.028333333
7 human100575 9.46 0.08250000 11.08 0.036666667
8 human114714 8.95 0.06750000 10.03 0.040000000
9 human201213 7.07 0.20583333 -0.68 0.104166667
10 human182838 10.47 0.09250000 8.40 0.166666667
11 human183710 6.62 0.07750000 -1.56 0.055000000
12 human183711 6.62 0.06083333 -1.56 0.047500000
13 human183867 9.46 0.00500000 9.79 0.229166667
14 human213120 7.64 0.07583333 7.23 0.042500000
15 human238691 7.63 0.01666667 3.78 0.243333333
16 human303540 9.72 0.06416667 11.00 0.013333333
17 human324822 9.13 0.58083333 1.37 0.379166667
18 human324822 9.13 0.58083333 1.37 0.379166667
19 human324822 9.13 0.58083333 1.37 0.379166667
20 human324822 9.13 0.58083333 1.37 0.379166667
21 human338065 11.81 0.09916667 11.45 0.009166667nrow(BothSSandReswUsage_diff_score_iso %>% filter(SigPAU2=="Yes"))[1] 21Examples conforming to expectation :
human100575 human86581 human114714 human182838 human238691 human303540
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.3.0 stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2
[5] readr_1.3.1 tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1
[9] tidyverse_1.2.1 workflowr_1.6.0
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38 colorspace_1.3-2
[5] generics_0.0.2 htmltools_0.3.6 yaml_2.2.0 utf8_1.1.4
[9] rlang_0.4.0 later_0.7.5 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 modelr_0.1.2 readxl_1.1.0 plyr_1.8.4
[17] munsell_0.5.0 gtable_0.2.0 cellranger_1.1.0 rvest_0.3.2
[21] evaluate_0.12 labeling_0.3 knitr_1.20 httpuv_1.4.5
[25] fansi_0.4.0 broom_0.5.1 Rcpp_1.0.2 promises_1.0.1
[29] scales_1.0.0 backports_1.1.2 jsonlite_1.6 fs_1.3.1
[33] hms_0.4.2 digest_0.6.18 stringi_1.2.4 grid_3.5.1
[37] rprojroot_1.3-2 cli_1.1.0 tools_3.5.1 magrittr_1.5
[41] lazyeval_0.2.1 crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2
[45] MASS_7.3-51.1 xml2_1.2.0 lubridate_1.7.4 assertthat_0.2.0
[49] rmarkdown_1.10 httr_1.3.1 rstudioapi_0.10 R6_2.3.0
[53] nlme_3.1-137 git2r_0.26.1 compiler_3.5.1