Species Specific PAS Double Filter
Briana Mittleman
1/21/2020
Last updated: 2020-03-19
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/TrialFiltersMeta.txt.sb-9845453e-R58Y0Q/
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/HandC.TvN
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/annotationBias.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: analysis/orthoexonAnno.Rmd
Untracked: analysis/pol2.Rmd
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._SAF2Bed.py
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopGeneral.ph
Untracked: code/._extractPhylopGeneral.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ClassifyLeafviz.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/SAF2Bed.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopGeneral.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runChimpDiffIsoDF.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runHumanDiffIsoDF.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_dF.err
Untracked: code/run_Humanleafcutter_dF.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Chimp_tvN_DF.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN_DF.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/test.txt
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._TrialFiltersMeta.txt
Untracked: data/._TrialFiltersMeta.txt.sb-9845453e-R58Y0Q
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/BaseComp/
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OrthoExonBed/
Untracked: data/OverlapBenchmark/
Untracked: data/OverlappingPAS/
Untracked: data/PAS/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalHvC/
Untracked: data/TrialFiltersMeta.txt
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/bioGRID/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/DiffUsedIntronic.Rmd
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/ExploredAPA_DF.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/SpliceSiteStrength.Rmd
Modified: analysis/TotalVNuclearBothSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/changeMisprimcut.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/dAPAandapaQTL_DF.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/speciesSpecific.Rmd
Modified: analysis/upsetter_DF.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 4e2a869 | brimittleman | 2020-03-19 | add SS in opp |
| html | 6c39a2a | brimittleman | 2020-01-21 | Build site. |
| Rmd | 010c249 | brimittleman | 2020-01-21 | DF species specific |
In this analysis I want to look at the PAS that are identified at at least 10% in one species but are not identified in the other species. I will work with avergage nuclear. I can then run the differential apa analysis with only the PAS identified in both.
library(tidyverse)── Attaching packages ─────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(ggpubr)Loading required package: magrittr
Attaching package: 'magrittr'The following object is masked from 'package:purrr':
set_namesThe following object is masked from 'package:tidyr':
extractPAS=read.table("../data/PAS_doubleFilter/PAS_10perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F)
HumanSpec= PAS %>% filter(Chimp==0)
nrow(HumanSpec)[1] 233ChimpSpec= PAS %>% filter(Human==0)
nrow(ChimpSpec)[1] 226This is a lot better. This is down from 2500 and 4000 respecively.
Look at the distribution of these accross the gene.
ggplot(HumanSpec,aes(x=loc,fill=loc)) + geom_bar(stat="count") + labs(x="Genic location", y="Number of PAS", title="Location of Human Specific PAS") + scale_fill_brewer(palette = "Dark2")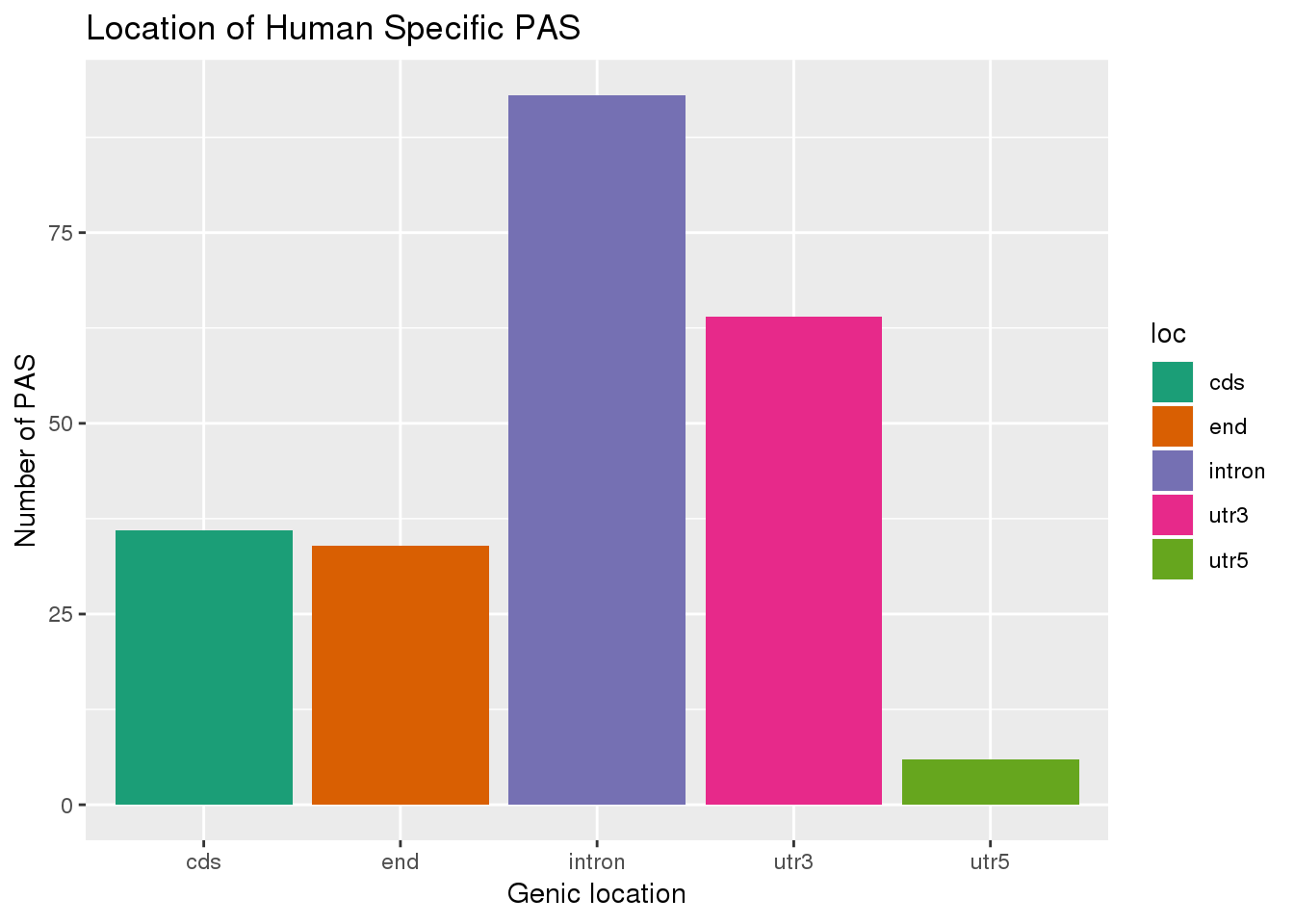
| Version | Author | Date |
|---|---|---|
| 6c39a2a | brimittleman | 2020-01-21 |
ggplot(HumanSpec,aes(x=loc, y=Human, ,fill=loc)) + geom_boxplot() + labs(x="Genic location", y="Human Average Usage", title="Human Specific PAS") + scale_fill_brewer(palette = "Dark2")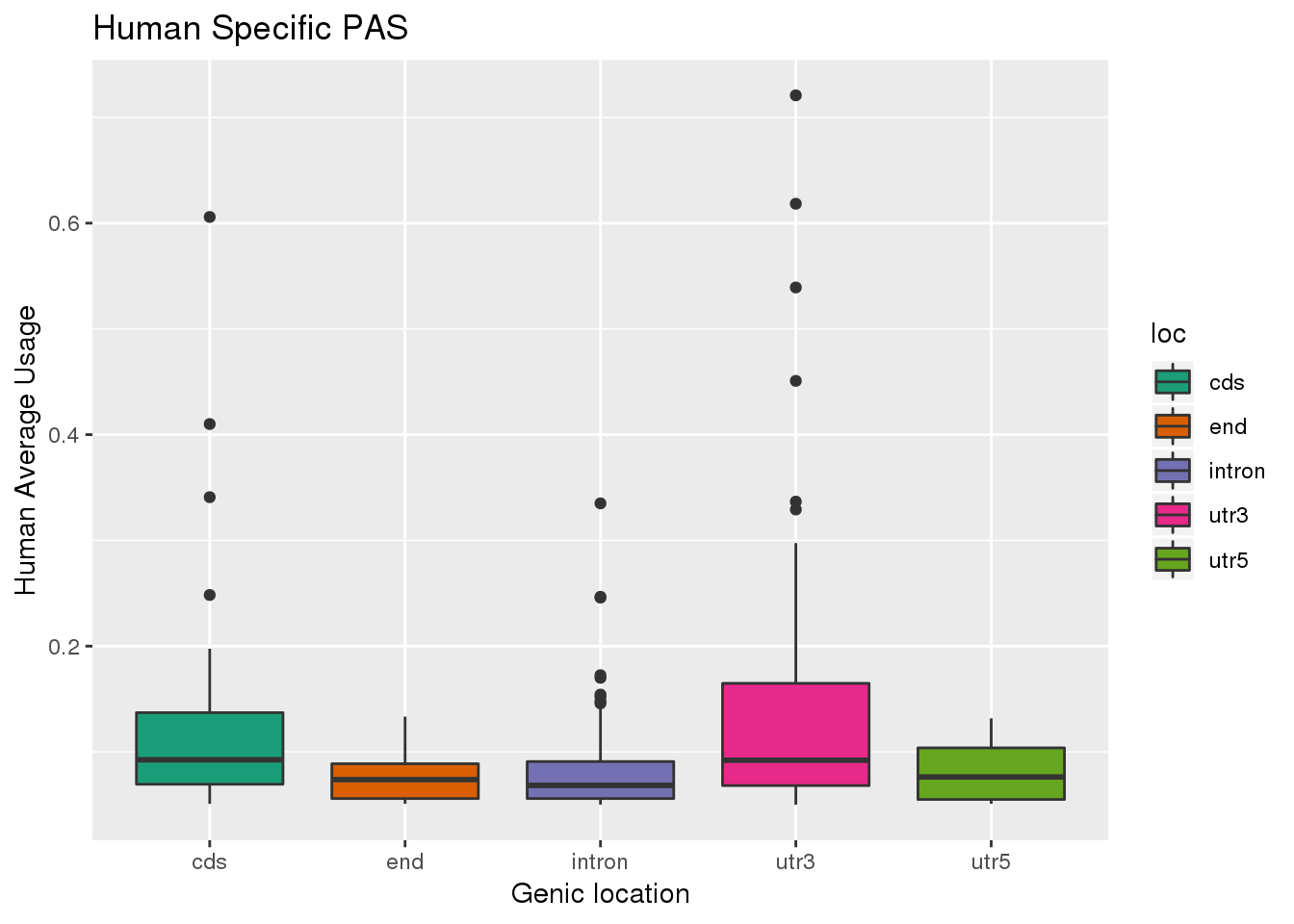
| Version | Author | Date |
|---|---|---|
| 6c39a2a | brimittleman | 2020-01-21 |
ggplot(ChimpSpec,aes(x=loc,fill=loc)) + geom_bar(stat="count") + labs(x="Genic location", y="Number of PAS", title="Location of Chimp Specific PAS")+ scale_fill_brewer(palette = "Dark2")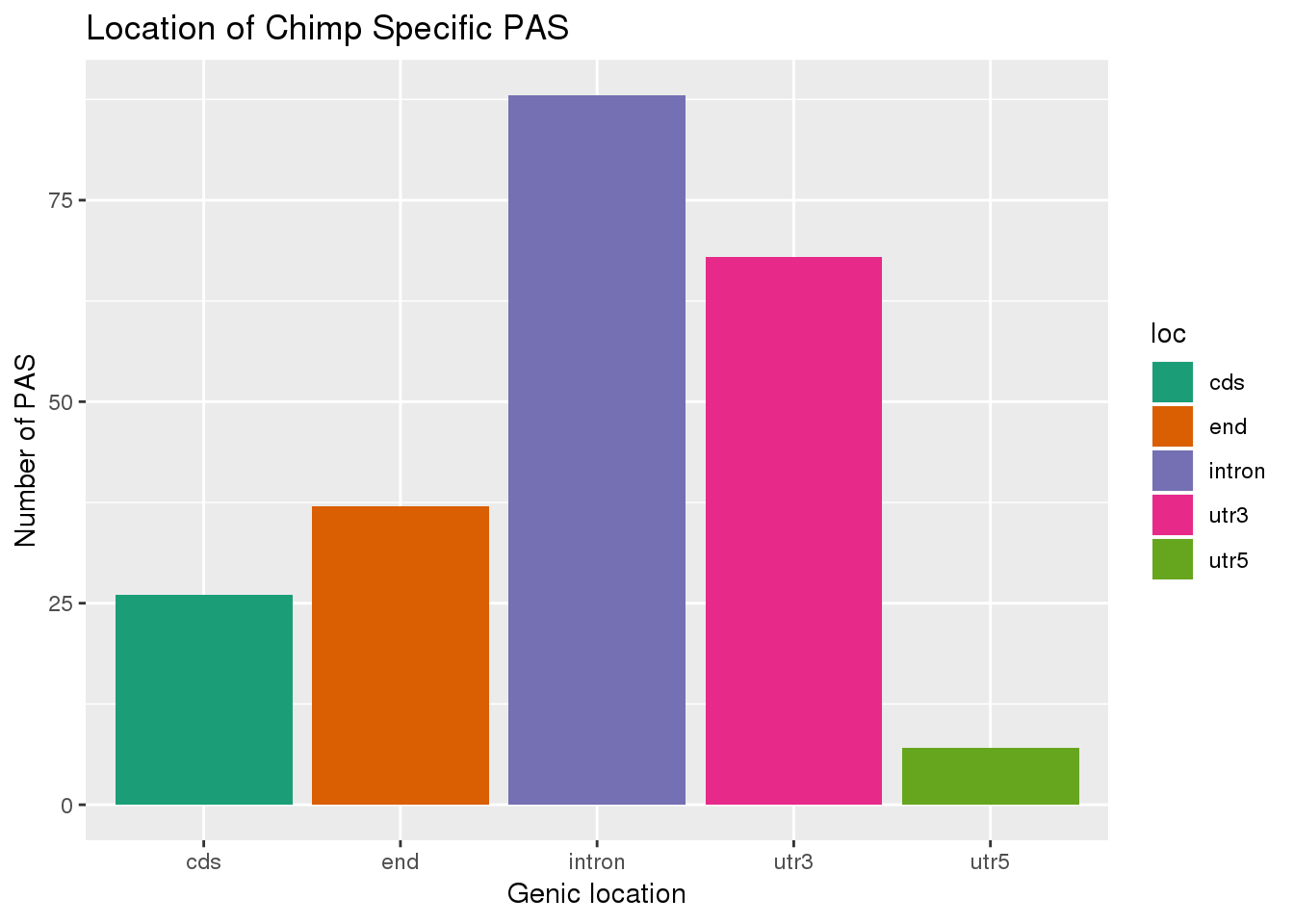
ggplot(ChimpSpec,aes(x=loc, y=Chimp, ,fill=loc)) + geom_boxplot() + labs(x="Genic location", y="Chimp Average Usage", title="Chimp Specific PAS") + scale_fill_brewer(palette = "Dark2")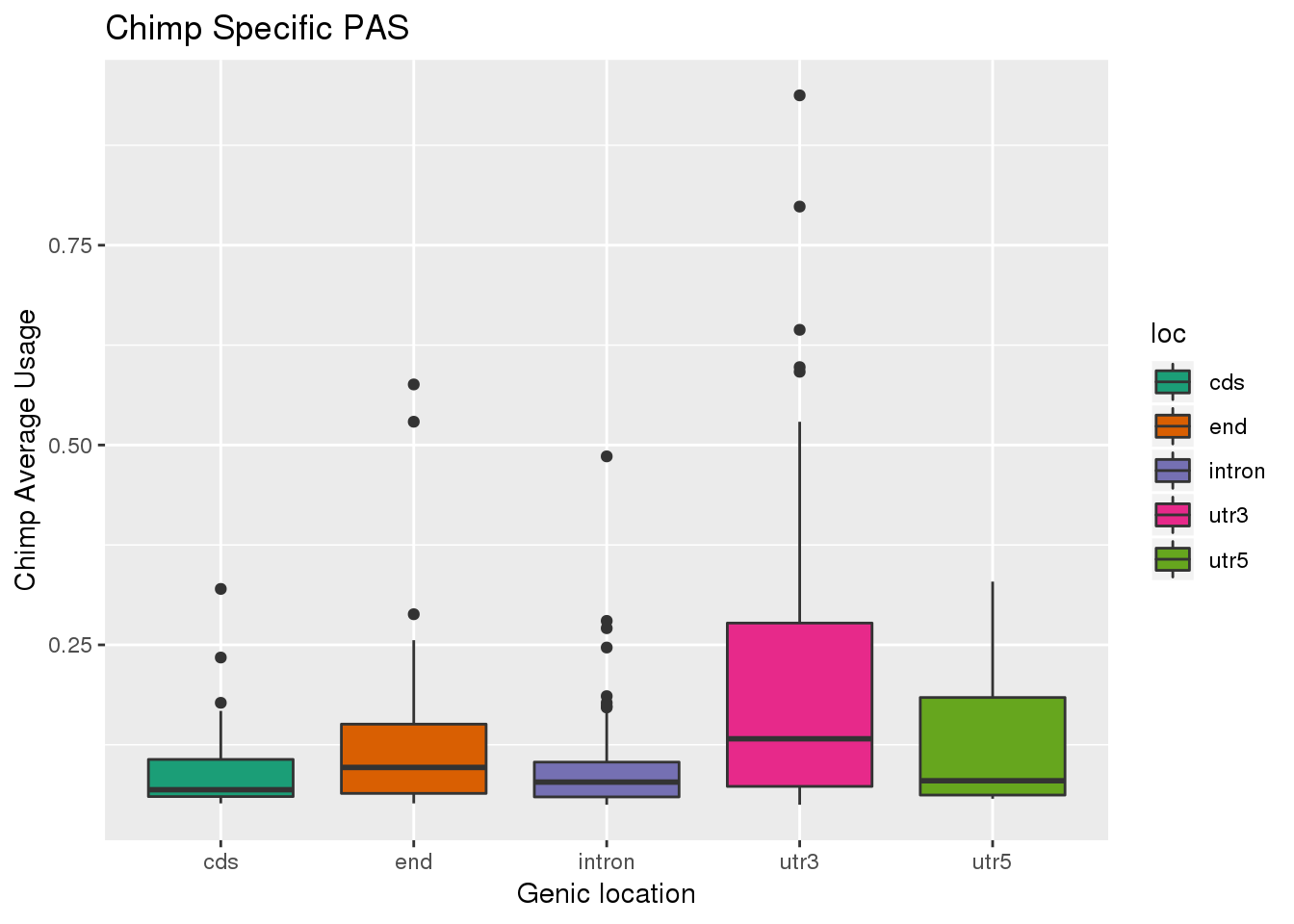
Next I will see if these are due to low expression. I will pull in the average normalized expression and rerun the filter.
nameID=read.table("../../genome_anotation_data/ensemble_to_genename.txt",sep="\t", header = T, stringsAsFactors = F) %>% dplyr::select(Gene_stable_ID,Gene.name)
expr=read.table("../data/DiffExpression/NoramalizedExpression.txt",header = T,stringsAsFactors = F) %>% rename('Gene_stable_ID'=genes) %>% inner_join(nameID, by="Gene_stable_ID") %>% dplyr::select(Gene.name,Chimp, Human) %>% rename("ChimpExp"=Chimp, "HumanExp"=Human, "gene"=Gene.name)
PAS_exp=PAS %>% inner_join(expr,by="gene")
nrow(PAS_exp)[1] 36028nrow(PAS_exp)/nrow(PAS)[1] 0.8513635HumanSpecExp= PAS_exp %>% filter(Chimp==0)
nrow(HumanSpecExp)[1] 153nrow(HumanSpecExp)/nrow(HumanSpec)[1] 0.6566524ChimpSpecExp= PAS_exp %>% filter(Human==0)
nrow(ChimpSpecExp)[1] 143nrow(ChimpSpecExp)/nrow(ChimpSpec)[1] 0.6327434We dont lose as many this way. This is evident the filter worked.
PAS_exp_spe=PAS_exp %>% mutate(HumanSpec=ifelse(gene %in%HumanSpecExp$gene, "yes", "no"), ChimpSpec=ifelse(gene %in% ChimpSpecExp$gene, "yes","no"))
ggplot(PAS_exp_spe,aes(x=HumanSpec,y=HumanExp)) + geom_boxplot() + stat_compare_means(method = "t.test") + labs(x="Presence of Human Specific PAS", y="Average Normalized Expression", title="Expression in Genes with Human Specific PAS")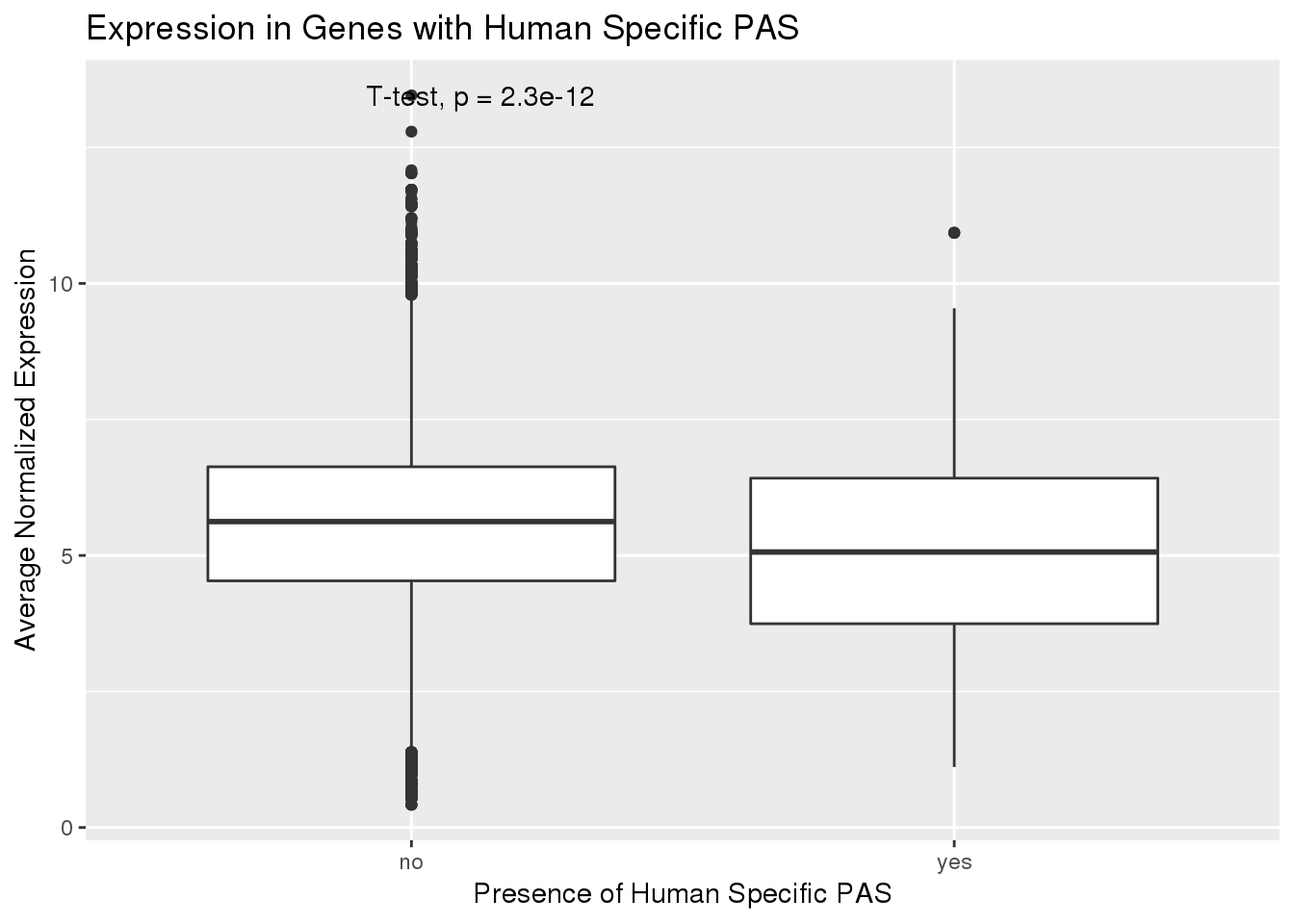
ggplot(PAS_exp_spe,aes(x=ChimpSpec,y=ChimpExp)) + geom_boxplot() + stat_compare_means(method = "t.test") + labs(x="Presence of Chimp Specific PAS", y="Average Normalized Expression", title="Expression in Genes with Chimp Specific PAS")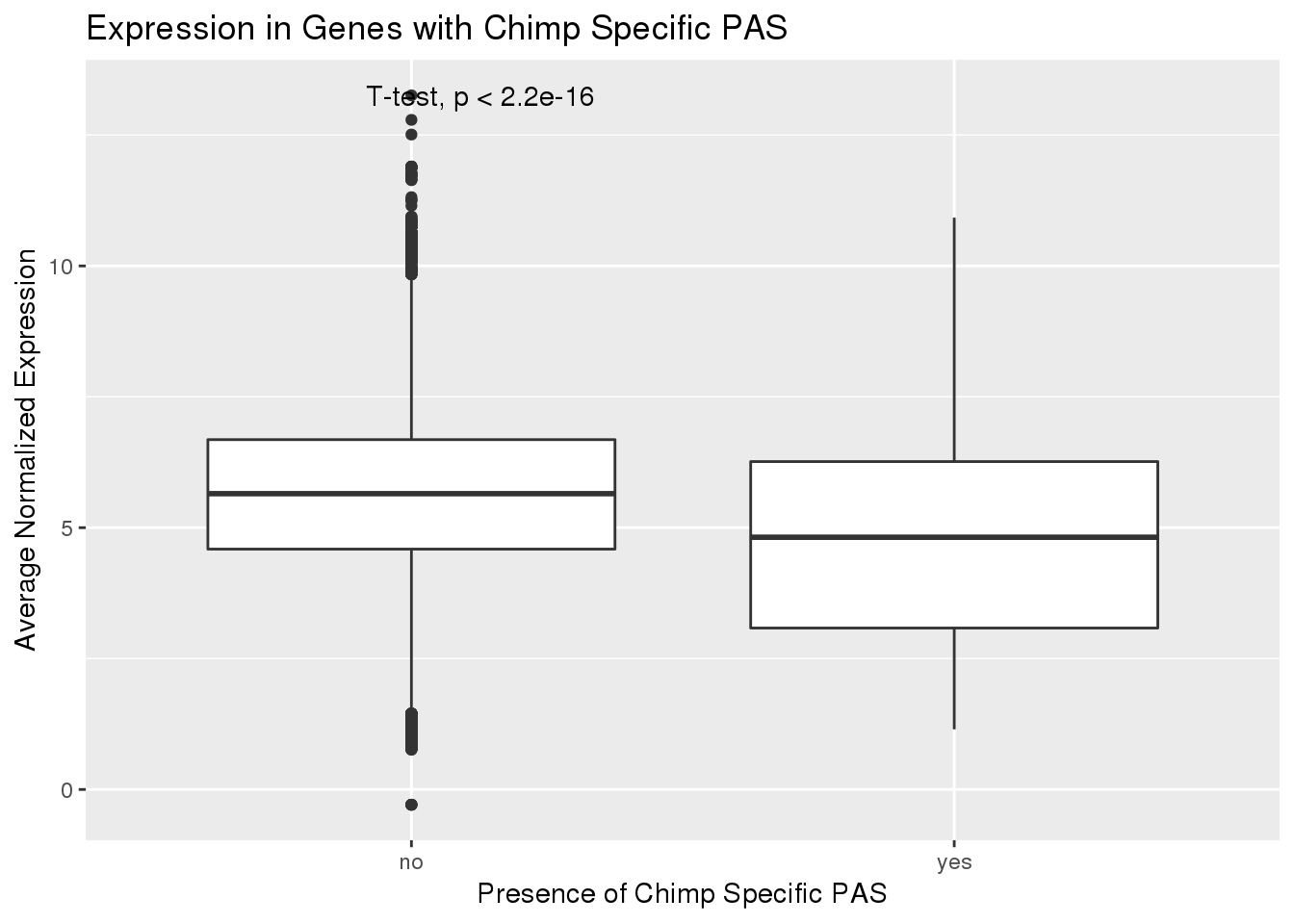 Still a difference but not as strong.
Still a difference but not as strong.
Ask if signal sites account for this:
PASSS=read.table("../data/PAS_doubleFilter/PAS_10perc_either_HumanCoord_BothUsage_meta_doubleFilter_withSSTop2.txt", header = T, stringsAsFactors = F) %>% dplyr::select(PAS,HumanTopSS ,ChimpTopSS)
HumanSpec_SS=HumanSpec %>% inner_join(PASSS, by="PAS") %>% rename("HumanSS"= HumanTopSS, "ChimpSS"=ChimpTopSS) %>% mutate(OnlyHuman=ifelse(HumanSS=="Yes" & ChimpSS=="No", "Yes", "No"), OnlyChimp=ifelse(HumanSS=="No" & ChimpSS=="Yes", "Yes", "No"))
ChimpSpec_SS=ChimpSpec %>% inner_join(PASSS, by="PAS")%>% rename("HumanSS"= HumanTopSS, "ChimpSS"=ChimpTopSS) %>% mutate(OnlyHuman=ifelse(HumanSS=="Yes" & ChimpSS=="No", "Yes", "No"), OnlyChimp=ifelse(HumanSS =="No" & ChimpSS=="Yes", "Yes", "No"))Summary:
HumanSpec_SS$OnlyHuman= as.factor(HumanSpec_SS$OnlyHuman)
summary(HumanSpec_SS$OnlyHuman) No Yes
220 13 HumanSpec_SS$OnlyChimp= as.factor(HumanSpec_SS$OnlyChimp)
summary(HumanSpec_SS$OnlyChimp) No Yes
231 2 HumanSpec_SS %>% filter(OnlyHuman=="Yes") PAS disc gene loc chr start end Chimp
1 human77129 Human CAPS2 intron chr12 75359873 75360073 0
2 human83275 Human RAB35 utr3 chr12 120095725 120095925 0
3 human92962 Human GPR180 utr3 chr13 94632911 94633111 0
4 human117628 Human C15orf40 utr3 chr15 83004841 83005041 0
5 human145598 Human L3MBTL4 intron chr18 6034146 6034346 0
6 human155700 Human ZNF333 intron chr19 14709065 14709265 0
7 human155707 Human ZNF333 utr3 chr19 14723160 14723360 0
8 human195030 Human RNF24 utr3 chr20 3931407 3931607 0
9 human210052 Human ST13 utr3 chr22 40826487 40826687 0
10 human237433 Human LMLN intron chr3 197962257 197962457 0
11 human269418 Human LINC01184 utr3 chr5 127966440 127966640 0
12 human272855 Human LOC102546294 intron chr5 148282965 148283165 0
13 human295414 Human LOC100507557 intron chr6 145855927 145856127 0
Human strandFix HumanSS ChimpSS OnlyHuman OnlyChimp
1 0.09000000 - Yes No Yes No
2 0.07250000 - Yes No Yes No
3 0.13166667 + Yes No Yes No
4 0.13166667 - Yes No Yes No
5 0.05083333 - Yes No Yes No
6 0.33500000 + Yes No Yes No
7 0.12250000 + Yes No Yes No
8 0.06833333 - Yes No Yes No
9 0.10916667 - Yes No Yes No
10 0.05750000 + Yes No Yes No
11 0.08166667 - Yes No Yes No
12 0.07333333 - Yes No Yes No
13 0.07666667 + Yes No Yes NoLook at usage of these:
HumanSpec_SSexp=HumanSpec_SS %>% filter(OnlyHuman=="Yes")
ggplot(HumanSpec_SSexp,aes(x=loc, y=Human,fill=loc))+ geom_boxplot() + scale_fill_brewer(palette = "Dark2")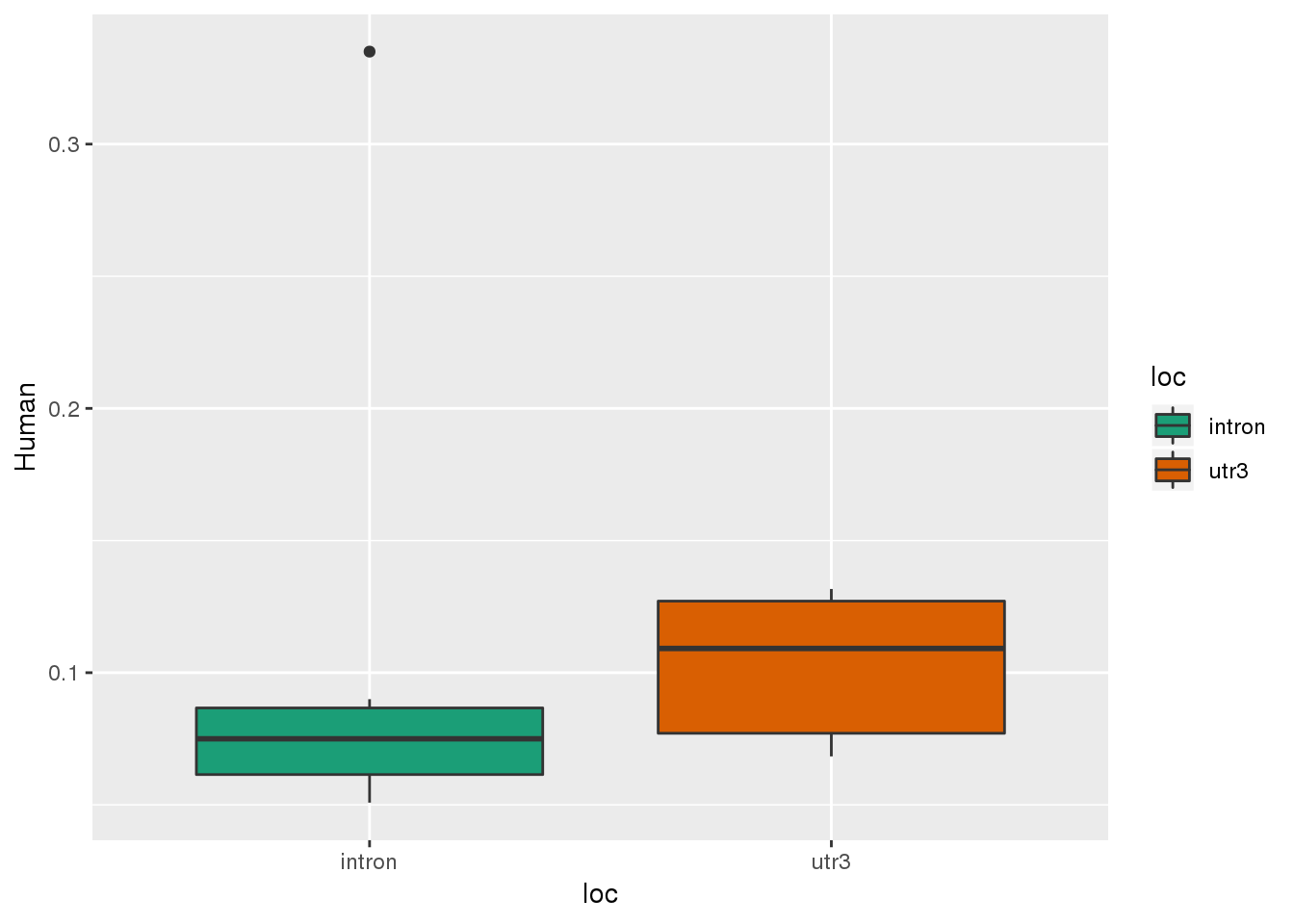
13 sites where there is a gain of a signal site in the human specific. I can look at them in IGV
13 of 233
ChimpSpec_SS$OnlyHuman= as.factor(ChimpSpec_SS$OnlyHuman)
summary(ChimpSpec_SS$OnlyHuman) No Yes
222 4 ChimpSpec_SS$OnlyChimp= as.factor(ChimpSpec_SS$OnlyChimp)
summary(ChimpSpec_SS$OnlyChimp) No Yes
215 11 ChimpSpec_SS %>% filter(OnlyChimp=="Yes") PAS disc gene loc chr start end
1 chimp106755 Chimp MAPKBP1 end chr15 41831602 41831802
2 chimp128457 Chimp LRRC37A2 intron chr17 46516506 46516706
3 chimp154225 Chimp ZNF765-ZNF761 intron chr19 53406381 53406581
4 chimp185396 Chimp RPL31 end chr2 101006807 101007007
5 chimp206436 Chimp EXOG end chr3 38526562 38526763
6 chimp208929 Chimp TMEM110-MUSTN1 intron chr3 52889869 52890069
7 chimp226080 Chimp MAN2B2 utr3 chr4 6621293 6621496
8 chimp242646 Chimp EXOC3 end chr5 469833 470033
9 chimp281737 Chimp GNA12 end chr7 2726153 2726352
10 chimp298751 Chimp ACTR3C intron chr7 150268220 150268420
11 chimp325661 Chimp GARNL3 intron chr9 127345020 127345220
Chimp Human strandFix HumanSS ChimpSS OnlyHuman OnlyChimp
1 0.07083333 0 + No Yes No Yes
2 0.05666667 0 + No Yes No Yes
3 0.15833333 0 + No Yes No Yes
4 0.09666667 0 + No Yes No Yes
5 0.14000000 0 + No Yes No Yes
6 0.06083333 0 - No Yes No Yes
7 0.27500000 0 + No Yes No Yes
8 0.05500000 0 + No Yes No Yes
9 0.14583333 0 - No Yes No Yes
10 0.09166667 0 - No Yes No Yes
11 0.07333333 0 + No Yes No Yes11 sites where there is a gain of a signal site in the chimp specific.
11 of 226
Look at usage of these:
ChimpSpec_SSexp=ChimpSpec_SS %>% filter(OnlyChimp=="Yes")
ggplot(ChimpSpec_SSexp,aes(x=loc, y=Chimp,fill=loc))+ geom_boxplot() + scale_fill_brewer(palette = "Dark2") + labs(title="Chimp location for Chimp specific PAS with added Signal",x="Genic Location", y="Chimp Mean Usage")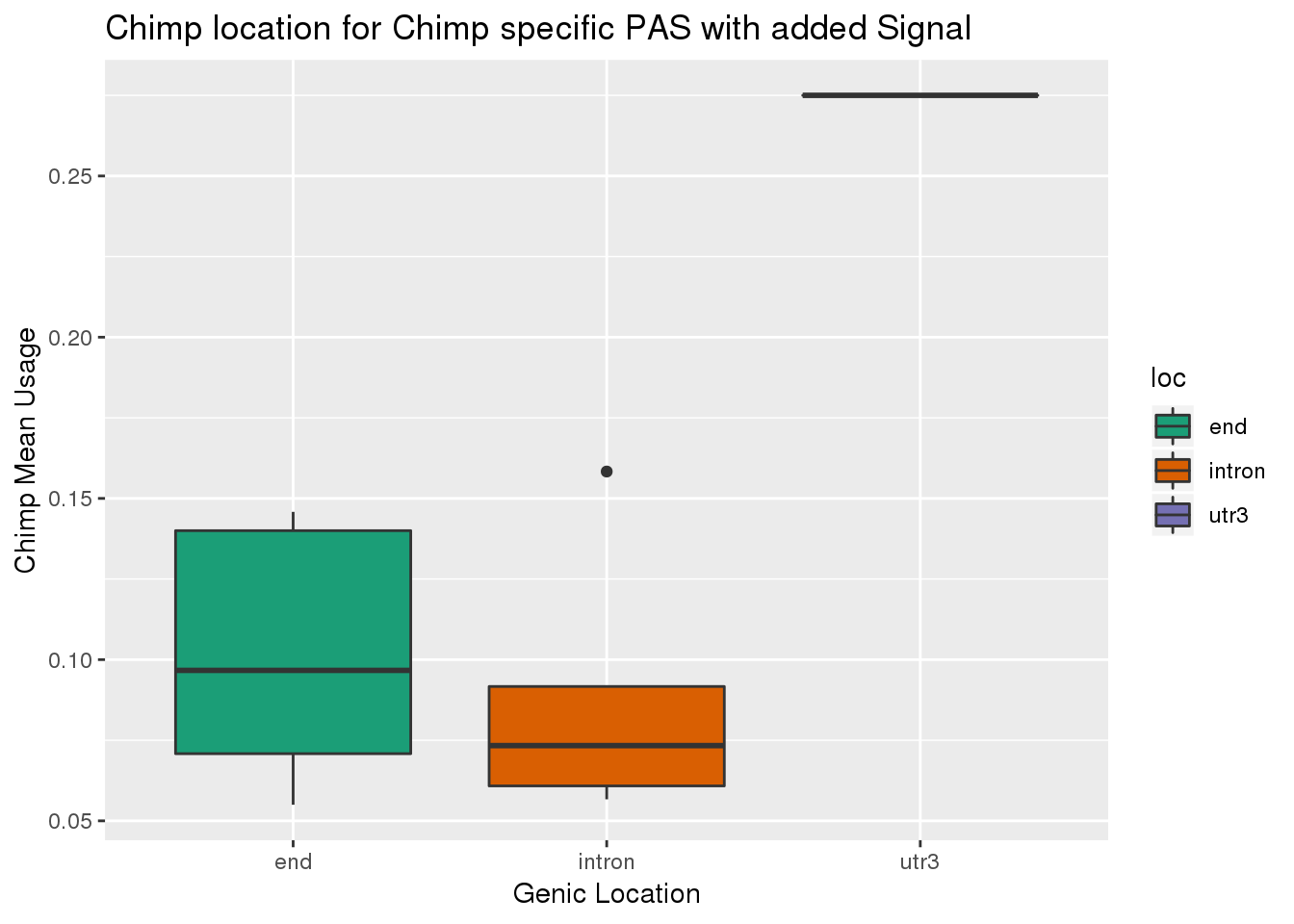
About 5 percent in both.
interesting chimp specific in MAN2b2, there is an AATAAA
Number with opposite relationship.
HumanSpec_SS %>% filter(OnlyChimp=="Yes") PAS disc gene loc chr start end Chimp
1 human207697 Human ZNRF3 intron chr22 28803993 28804193 0
2 human313507 Human METTL2B utr3 chr7 128501913 128502113 0
Human strandFix HumanSS ChimpSS OnlyHuman OnlyChimp
1 0.1516667 + No Yes No Yes
2 0.1791667 + No Yes No YesChimpSpec_SS %>% filter(OnlyHuman=="Yes") PAS disc gene loc chr start end
1 chimp56104 Chimp SPCS2 utr3 chr11 74976869 74977069
2 chimp107271 Chimp ADAL cds chr15 43345803 43346003
3 chimp118114 Chimp SHISA9 end chr16 13565898 13566099
4 chimp192139 Chimp LOC101929947 intron chr2 172543729 172543929
Chimp Human strandFix HumanSS ChimpSS OnlyHuman OnlyChimp
1 0.07666667 0 + Yes No Yes No
2 0.06416667 0 + Yes No Yes No
3 0.15083333 0 + Yes No Yes No
4 0.05833333 0 - Yes No Yes No2 in the opposite for human, 4 opposite for chimp.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggpubr_0.2 magrittr_1.5 forcats_0.3.0 stringr_1.3.1
[5] dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1 tidyr_0.8.3
[9] tibble_2.1.1 ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38
[4] colorspace_1.3-2 generics_0.0.2 htmltools_0.3.6
[7] yaml_2.2.0 rlang_0.4.0 later_0.7.5
[10] pillar_1.3.1 glue_1.3.0 withr_2.1.2
[13] RColorBrewer_1.1-2 modelr_0.1.2 readxl_1.1.0
[16] plyr_1.8.4 munsell_0.5.0 gtable_0.2.0
[19] workflowr_1.6.0 cellranger_1.1.0 rvest_0.3.2
[22] evaluate_0.12 labeling_0.3 knitr_1.20
[25] httpuv_1.4.5 broom_0.5.1 Rcpp_1.0.2
[28] promises_1.0.1 scales_1.0.0 backports_1.1.2
[31] jsonlite_1.6 fs_1.3.1 hms_0.4.2
[34] digest_0.6.18 stringi_1.2.4 grid_3.5.1
[37] rprojroot_1.3-2 cli_1.1.0 tools_3.5.1
[40] lazyeval_0.2.1 crayon_1.3.4 whisker_0.3-2
[43] pkgconfig_2.0.2 xml2_1.2.0 lubridate_1.7.4
[46] assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[49] rstudioapi_0.10 R6_2.3.0 nlme_3.1-137
[52] git2r_0.26.1 compiler_3.5.1