Explore genes with dAPA double filter
Briana Mittleman
1/21/2020
Last updated: 2020-01-26
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.5.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ClassifyLeafviz.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OverlapBenchmark/
Untracked: data/PAS/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalHvC/
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Unstaged changes:
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/speciesSpecific.Rmd
Modified: analysis/speciesSpecific_DF.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 10d6e05 | brimittleman | 2020-01-26 | add gene level phyloP |
| html | 26e6362 | brimittleman | 2020-01-24 | Build site. |
| Rmd | d867ef6 | brimittleman | 2020-01-24 | by loc |
| html | 5910b06 | brimittleman | 2020-01-24 | Build site. |
| Rmd | ea17340 | brimittleman | 2020-01-24 | add phylo/dnds/go |
| html | 5800231 | brimittleman | 2020-01-22 | Build site. |
| Rmd | 117fd63 | brimittleman | 2020-01-22 | redo differential analysis with double filt |
library(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(ggpubr)Loading required package: magrittr
Attaching package: 'magrittr'The following object is masked from 'package:purrr':
set_namesThe following object is masked from 'package:tidyr':
extractlibrary(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsI want to look more at the genes we found with dAPA.
Question 1:
Do genes with differential APA have different numbers of PAS in each species?
DiffUsage=read.table("../data/DiffIso_Nuclear_DF/SignifianceEitherPAS_2_Nuclear.txt", header = T, stringsAsFactors = F)
PASMeta=read.table("../data/PAS_doubleFilter/PAS_10perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F) %>% dplyr::select(PAS, chr, start,end, gene, loc)
DiffUsagePAS=DiffUsage %>% inner_join(PASMeta, by=c("gene","chr", "start", "end"))Number of PAS in each species:
PAS=read.table("../data/PAS_doubleFilter/PAS_10perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", stringsAsFactors = F, header = T)
PAS_sm=PAS %>% dplyr::select(gene, Chimp, Human)
PAS_m= melt(PAS_sm, id.var="gene", variable.name="species", value.name="meanUsage") %>% filter(meanUsage >=0.1) %>% group_by(species, gene) %>% summarise(nPAS=n())Filter these by those with dAPA:
PAS_m_dAPA= PAS_m %>% mutate(dAPA=ifelse(gene %in% DiffUsagePAS$gene, "Y", "N"))ggplot(PAS_m_dAPA,aes(by=dAPA, y=nPAS,x=species, fill=dAPA)) + geom_boxplot() + stat_compare_means(method = "t.test") + scale_fill_brewer(palette = "Dark2") + labs(y="Number of PAS detected at 10% usage", title="Number of PAS detected by gene with differential usage") 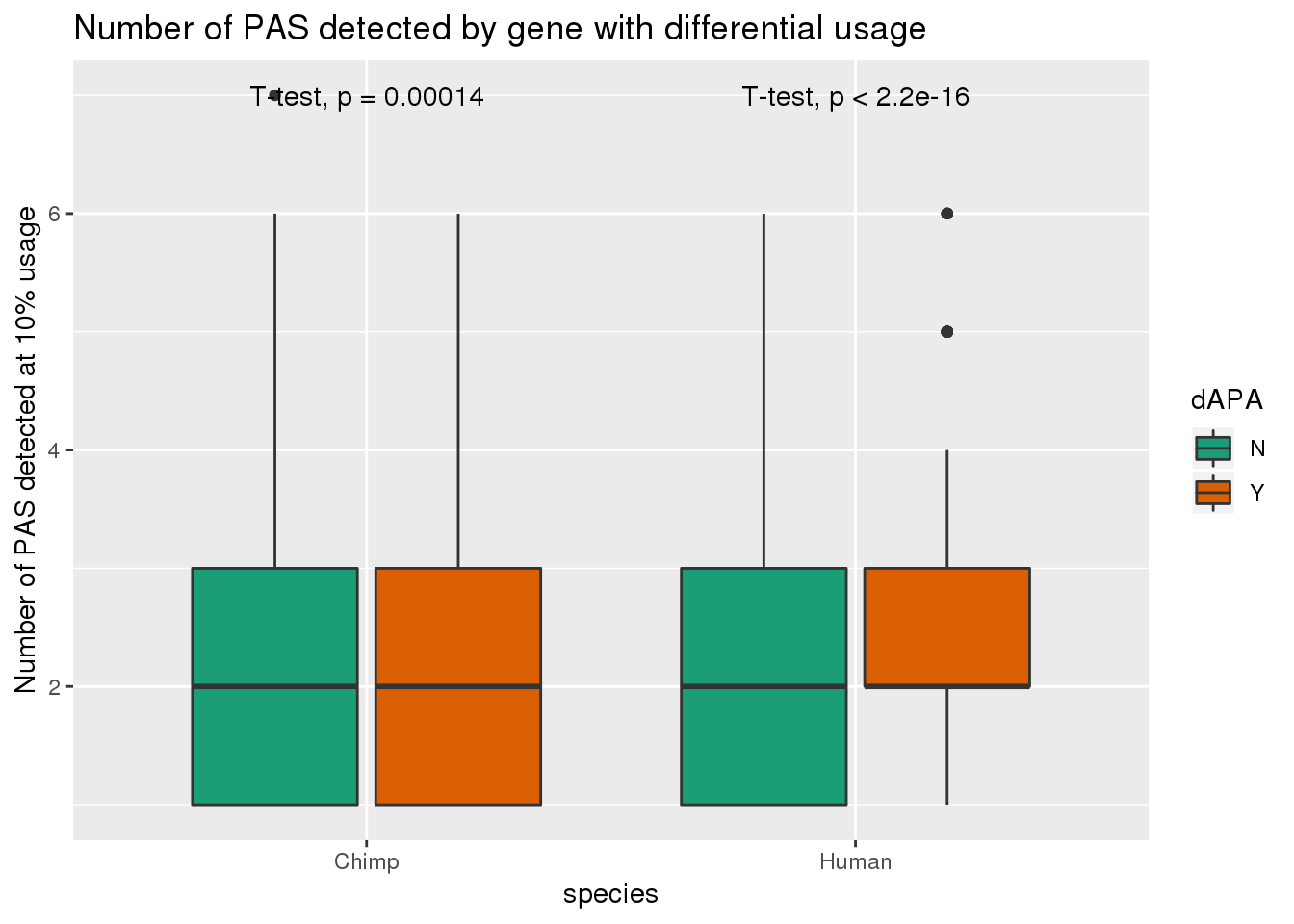
| Version | Author | Date |
|---|---|---|
| 5800231 | brimittleman | 2020-01-22 |
Question 2: Where are the differentially used PAS?
ggplot(DiffUsagePAS,aes(x=loc, fill=loc)) + geom_bar(stat="count") 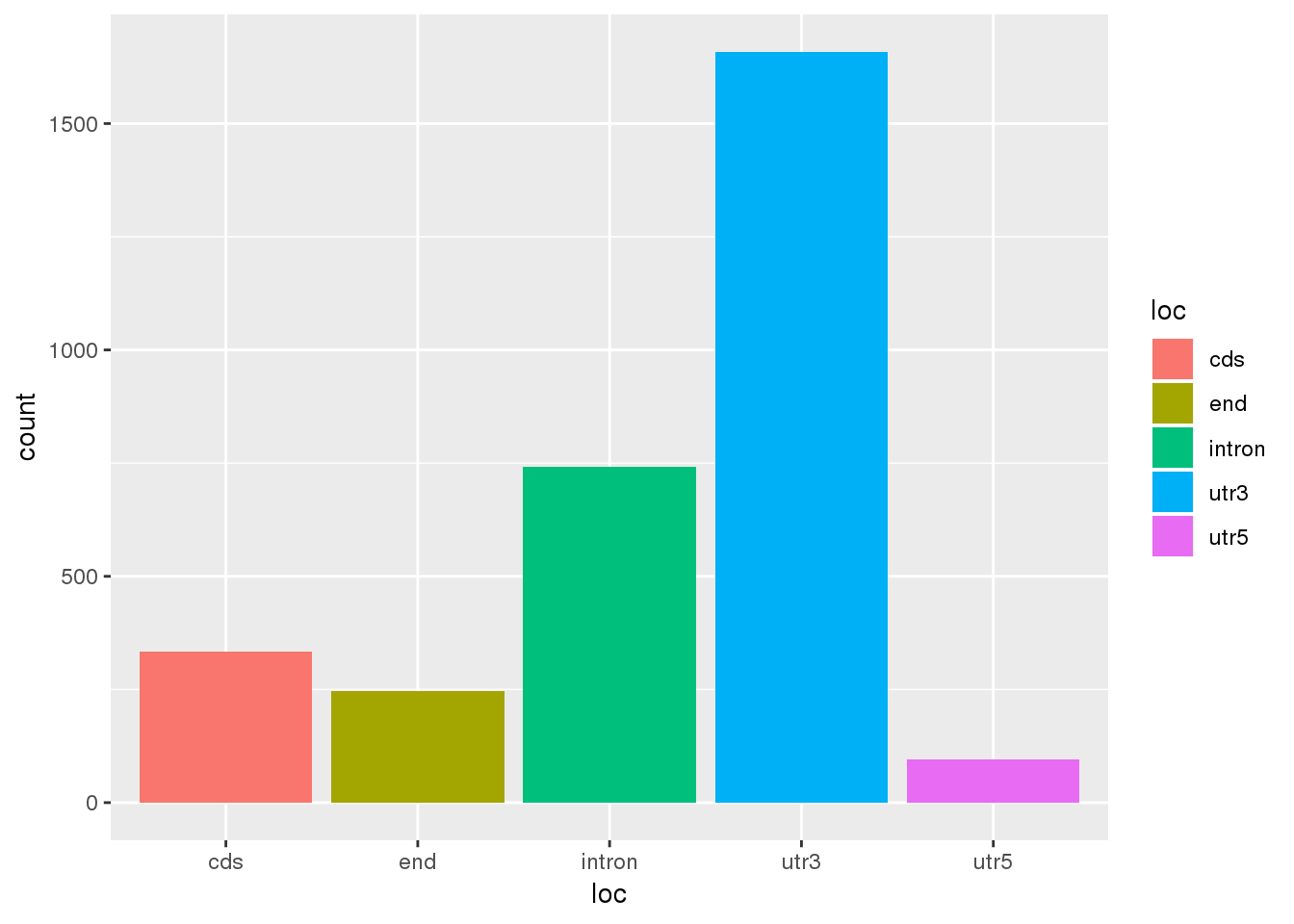
| Version | Author | Date |
|---|---|---|
| 5800231 | brimittleman | 2020-01-22 |
Seperate by location:
#negative deltaPAU is used more in human
DiffUsagePAS_dir= DiffUsagePAS %>% mutate(direction=ifelse(deltaPAU >=0, "Chimp", "Human"))
ggplot(DiffUsagePAS_dir,aes(x=loc, fill=loc)) + geom_bar(stat="count") + facet_grid(~direction)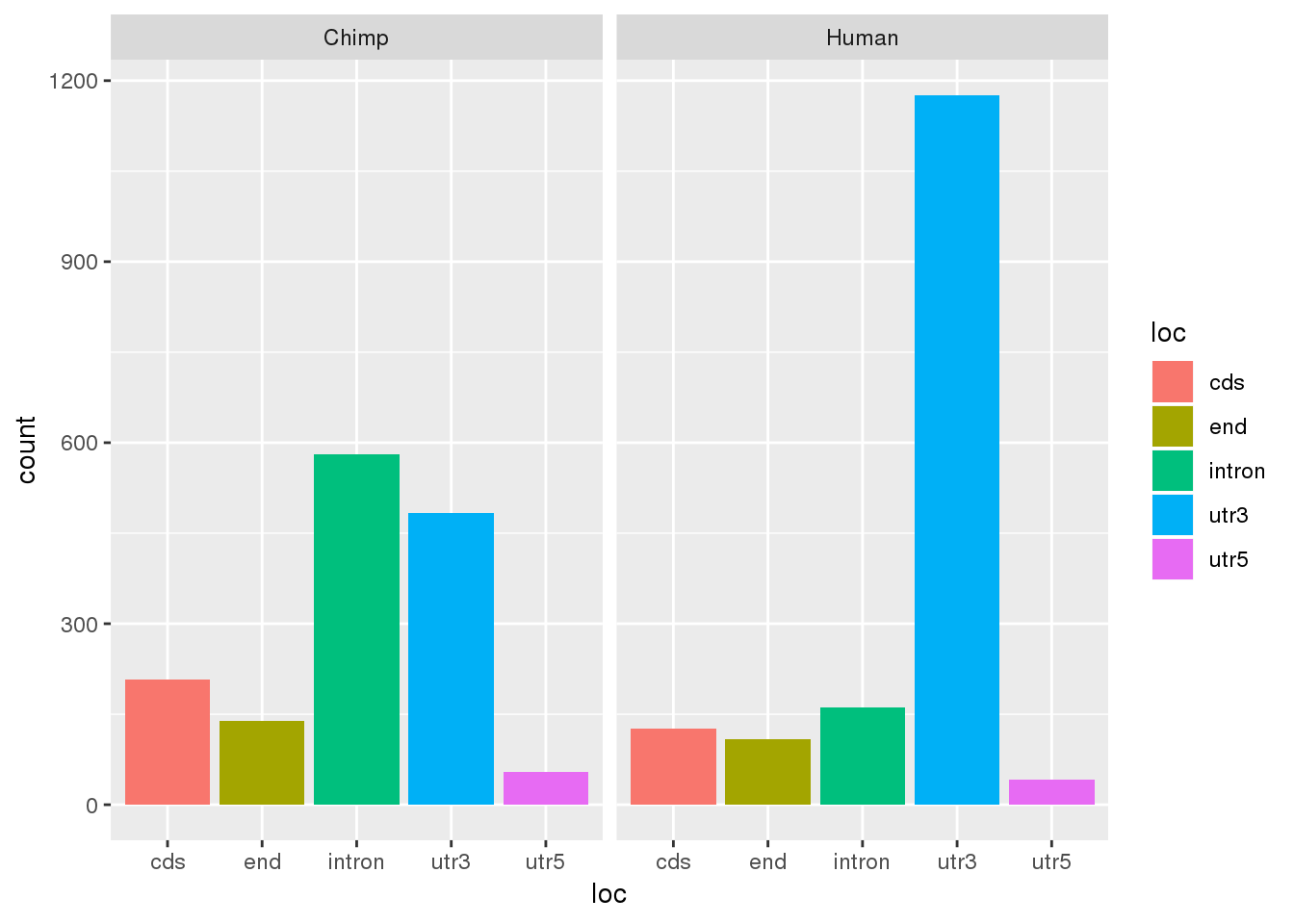
| Version | Author | Date |
|---|---|---|
| 5800231 | brimittleman | 2020-01-22 |
This is opposite of the results using just the dominant PAS. I probably shouldn’t put too much into that.
Question 3: Does locaiton of the PAS effect the absolute value of the effect size
ggplot(DiffUsagePAS_dir,aes(x=loc, y=abs(deltaPAU), fill=loc)) + geom_violin() 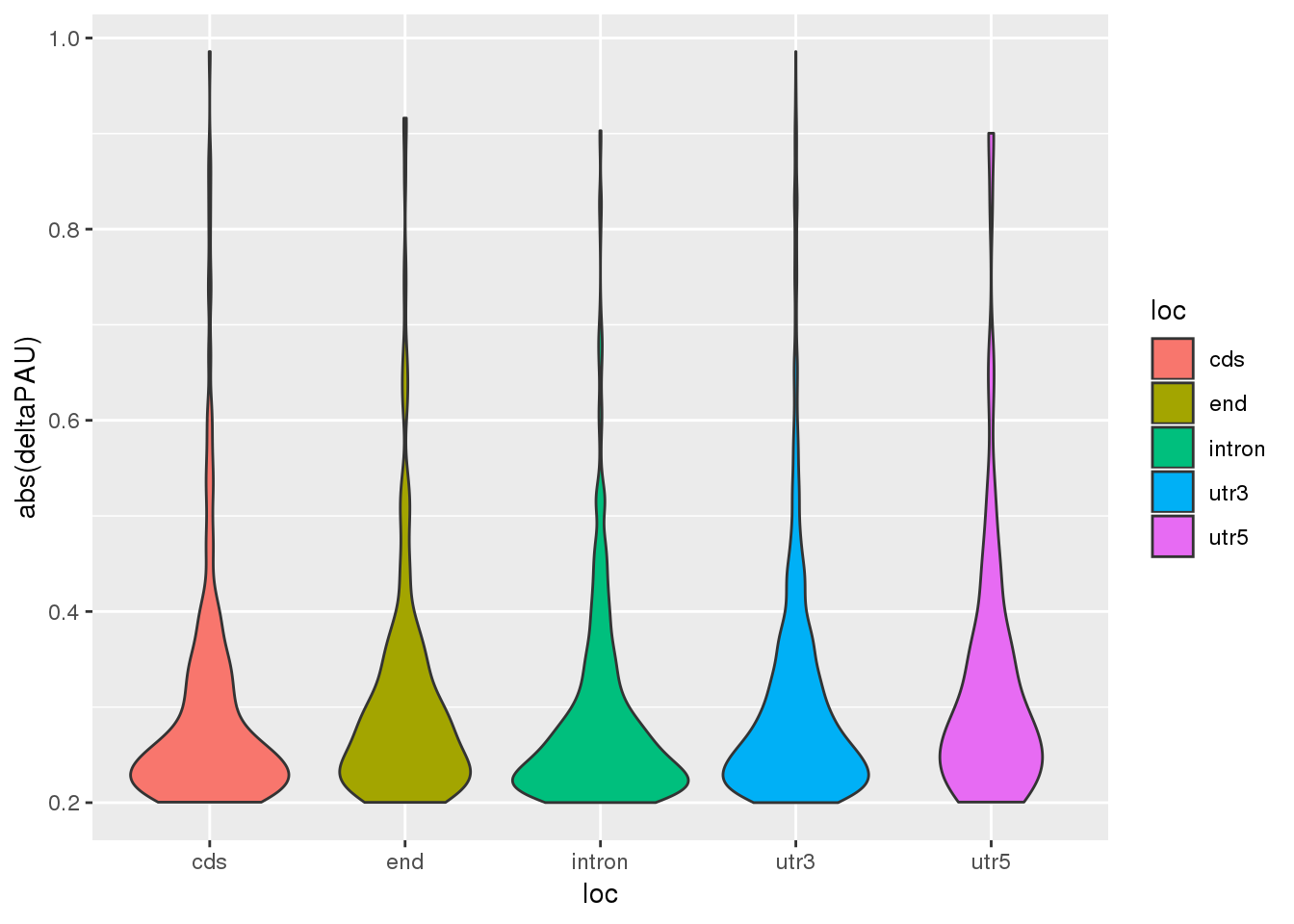
| Version | Author | Date |
|---|---|---|
| 5800231 | brimittleman | 2020-01-22 |
Explore conservation:
https://www.ultraconserved.org
https://useast.ensembl.org/info/genome/compara/conservation_and_constrained.html
phylo p from genomebrowser
mkdir ../data/PhyloP
mkdir ../data/DNDS
PhyloP: Column #1 contains a one-based position coordinate. Column #2 contains a score showing the posterior probability that the phylogenetic hidden Markov model (HMM) of phastCons is in its most conserved state at that base position.
I want to get the average score for each of the tested PAS. I can use pybigwig.
python extractPhyloReg.pyphylores=read.table("../data/PhyloP/PAS_phyloP.txt", col.names = c("chr","start","end", "phyloP"), stringsAsFactors = F) %>% drop_na()
NucReswPhy=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T, stringsAsFactors = F) %>% inner_join(phylores, by=c("chr","start","end"))40756 have results of the 40776 Plot:
ggplot(NucReswPhy,aes(y=phyloP, x=SigPAU2,fill=SigPAU2)) + geom_boxplot() + stat_compare_means()+ scale_fill_brewer(palette = "Dark2")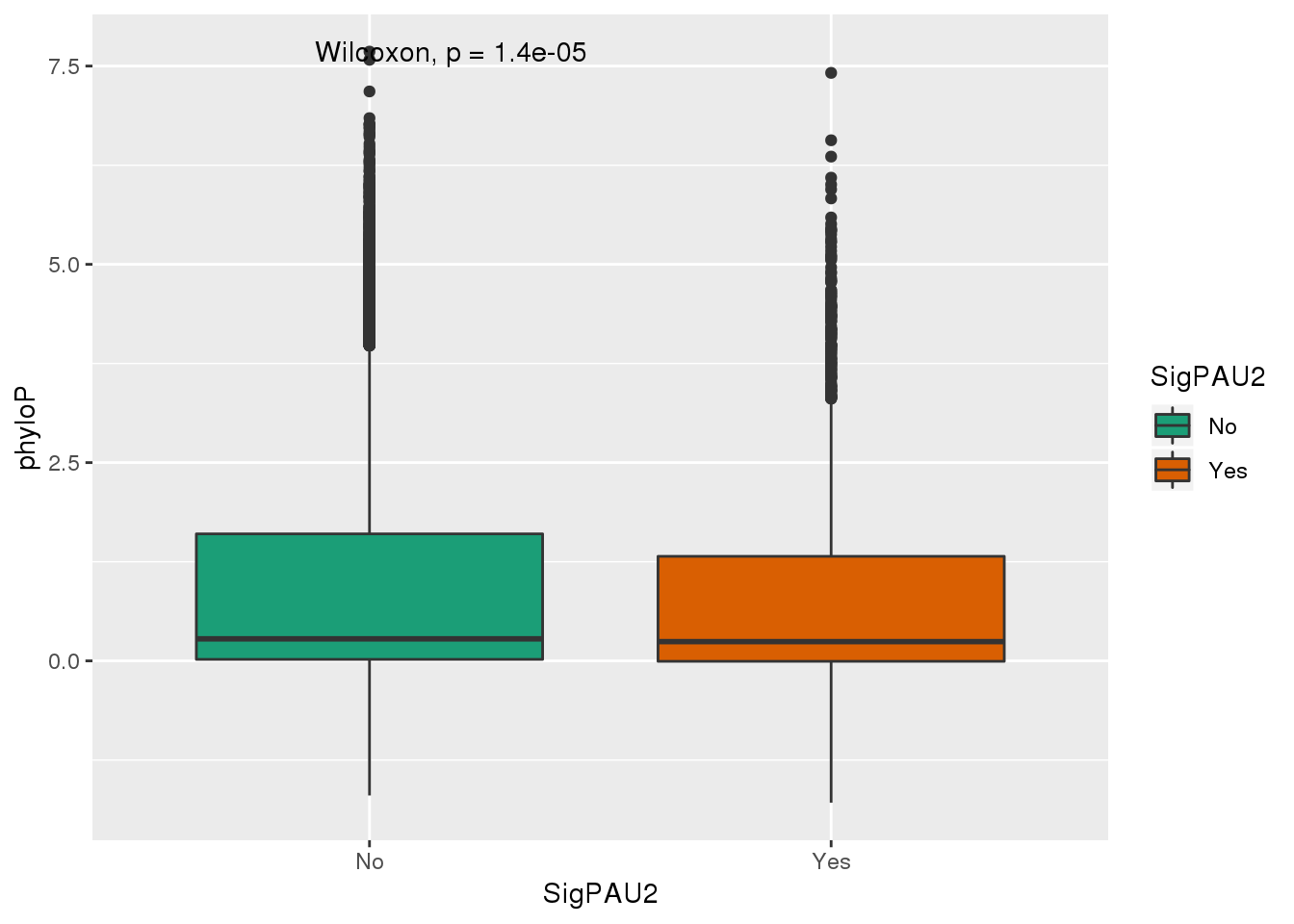
| Version | Author | Date |
|---|---|---|
| 5910b06 | brimittleman | 2020-01-24 |
ggplot(NucReswPhy,aes(x=phyloP, by=SigPAU2, fill=SigPAU2)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Dark2")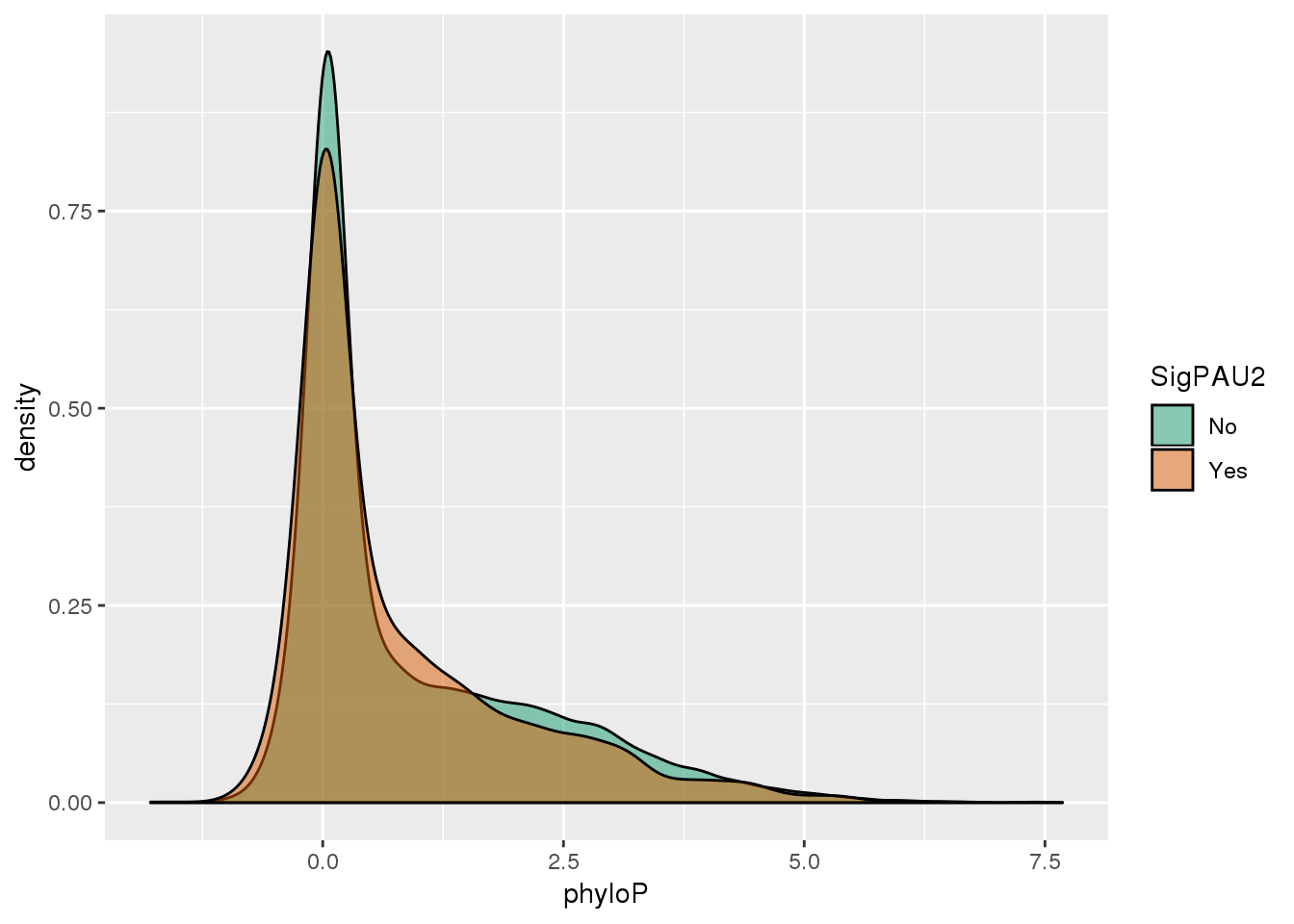
| Version | Author | Date |
|---|---|---|
| 5910b06 | brimittleman | 2020-01-24 |
The significant PAS have on average lower phyloP scores.
Positive scores — Measure conservation, which is slower evolution than expected, at sites that are predicted to be conserved. Negative scores — Measure acceleration, which is faster evolution than expected, at sites that are predicted to be fast-evolving.
I can look at those with negative values:
x=nrow(NucReswPhy %>% filter(SigPAU2=="Yes", phyloP<0))
m= nrow(NucReswPhy %>% filter(phyloP<0))
n=nrow(NucReswPhy %>% filter(phyloP>=0))
k=nrow(NucReswPhy %>% filter(SigPAU2=="Yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 707#actual:
x[1] 788#pval
phyper(x,m,n,k,lower.tail=F)[1] 0.0001570509This means these regions are more likely to be fast evolving.
Look at this by location: (is it driven by region)
NucReswPhy_meta= NucReswPhy %>% inner_join(PASMeta, by=c("chr", "start", "end", "gene"))
ggplot(NucReswPhy_meta,aes(x=phyloP, by=SigPAU2, fill=SigPAU2)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Dark2") + facet_grid(~loc)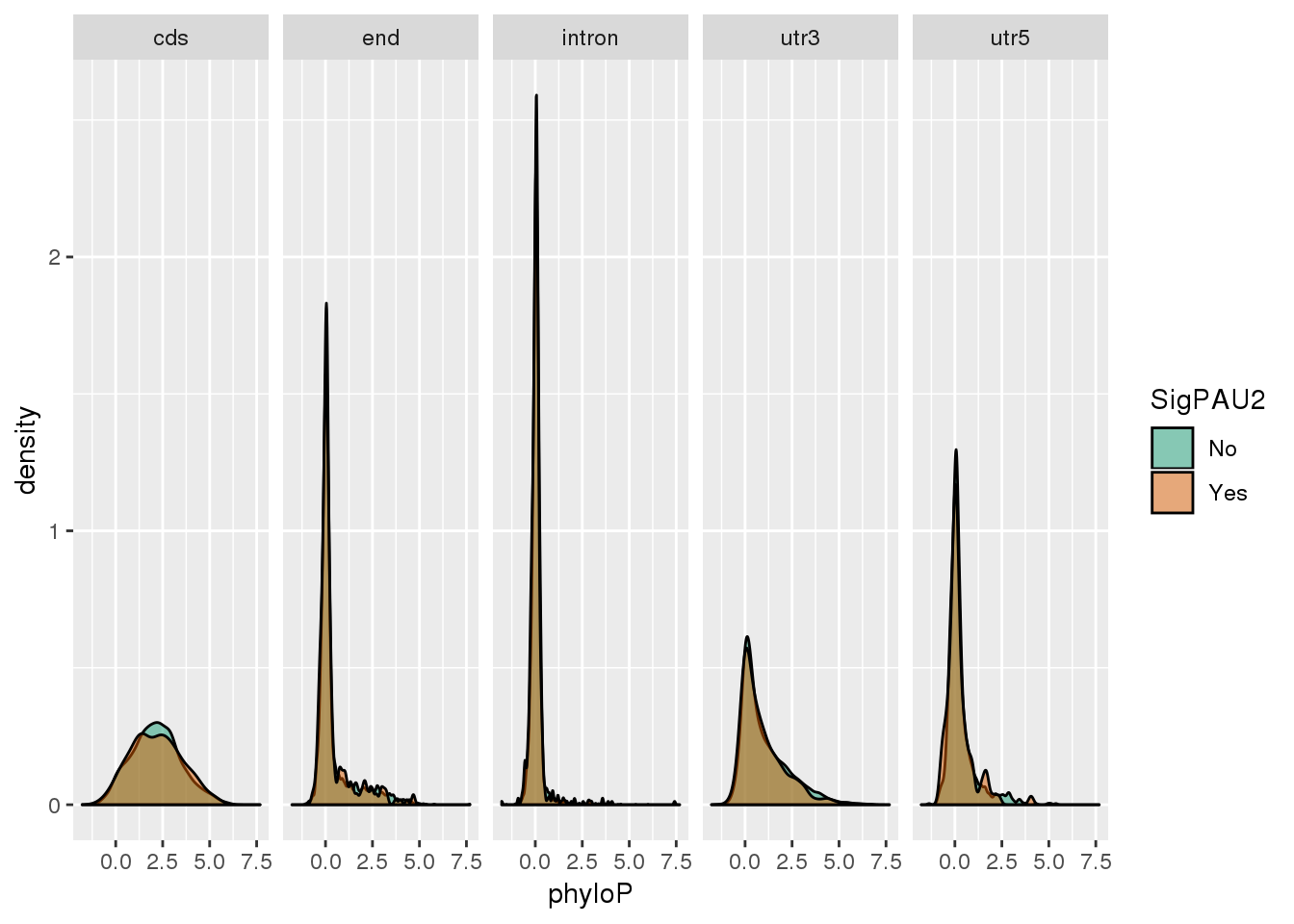
| Version | Author | Date |
|---|---|---|
| 26e6362 | brimittleman | 2020-01-24 |
NucReswPhy_meta_group=NucReswPhy_meta %>% group_by(loc,SigPAU2) %>% summarise(n=n(),meanPhylo=mean(phyloP))
NucReswPhy_meta_group# A tibble: 10 x 4
# Groups: loc [5]
loc SigPAU2 n meanPhylo
<chr> <chr> <int> <dbl>
1 cds No 7141 2.16
2 cds Yes 333 2.16
3 end No 3564 0.450
4 end Yes 247 0.403
5 intron No 10478 0.0630
6 intron Yes 737 0.0702
7 utr3 No 15351 1.04
8 utr3 Yes 1659 0.933
9 utr5 No 1151 0.300
10 utr5 Yes 95 0.230 Compare this to the genes that are expressed for this I will need to make a bedfile with the genes. I will pull them in as well as the gene annotation:
DAPAGeneSig=read.table("../data/DiffIso_Nuclear_DF/SignifianceEitherGENES_Nuclear.txt", stringsAsFactors = F, header = T)
DAPAGene=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", stringsAsFactors = F, header = T) %>% dplyr::select(gene) %>% unique() %>% mutate(Sig=ifelse(gene %in% DAPAGeneSig$gene,"Yes","No"))To be safe ill use the longest transcripts from table browser refseq.
This will be easiest in a python dictionary.
python parseHg38.py
sort -k1,1 -k2,2n ../../genome_anotation_data/hg38_refseq_anno/hg38_ncbiRefseq_GenesParsed.bed > ../../genome_anotation_data/hg38_refseq_anno/hg38_ncbiRefseq_GenesParsed_sort.bedgenes=read.table("../../genome_anotation_data/hg38_refseq_anno/hg38_ncbiRefseq_GenesParsed_sort.bed", col.names = c("chr", "start", "end","name","score","strand"),stringsAsFactors = F) %>% filter(name %in% DAPAGene$gene ) %>% rename("gene"=name)
genesWithSig= genes %>% inner_join(DAPAGene, by="gene")
write.table(genes, "../data/PhyloP/NuclearSigGenes.bed", col.names = F, row.names = F, quote=F, sep="\t")Get the mean phyloP scores.
python extractPhyloRegGene.py phyloresG=read.table("../data/PhyloP/PAS_phyloP_genes.txt", col.names = c("chr","start","end", "phyloP"), stringsAsFactors = F) %>% drop_na()
GenesPhy=genesWithSig %>% inner_join(phyloresG, by=c("chr","start","end"))I lose 2237 from NAs in the values.
ggplot(GenesPhy,aes(x=phyloP, by=Sig, fill=Sig)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Dark2")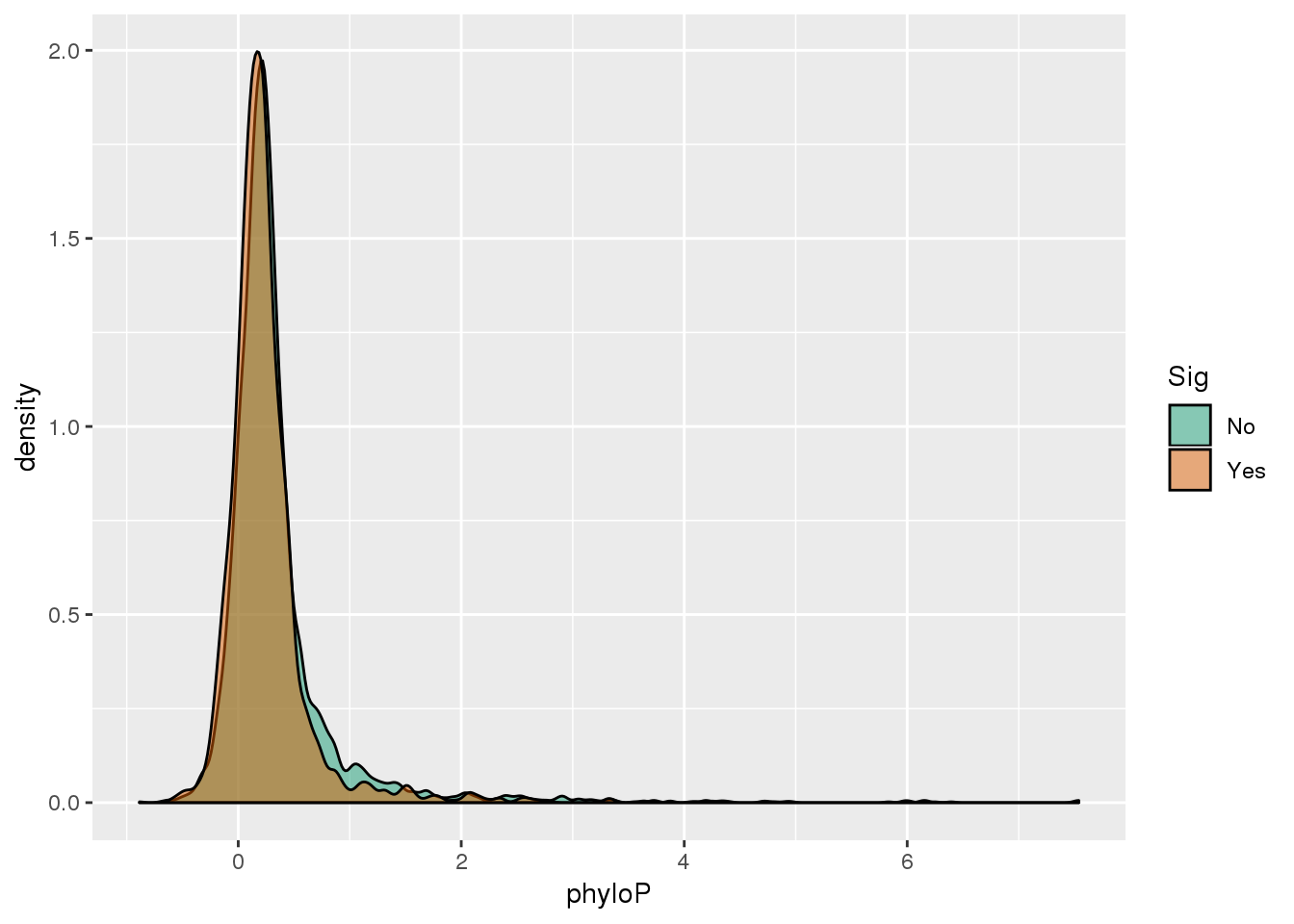
ggplot(GenesPhy,aes(y=phyloP, x=Sig,fill=Sig)) + geom_boxplot() + stat_compare_means()+ scale_fill_brewer(palette = "Dark2")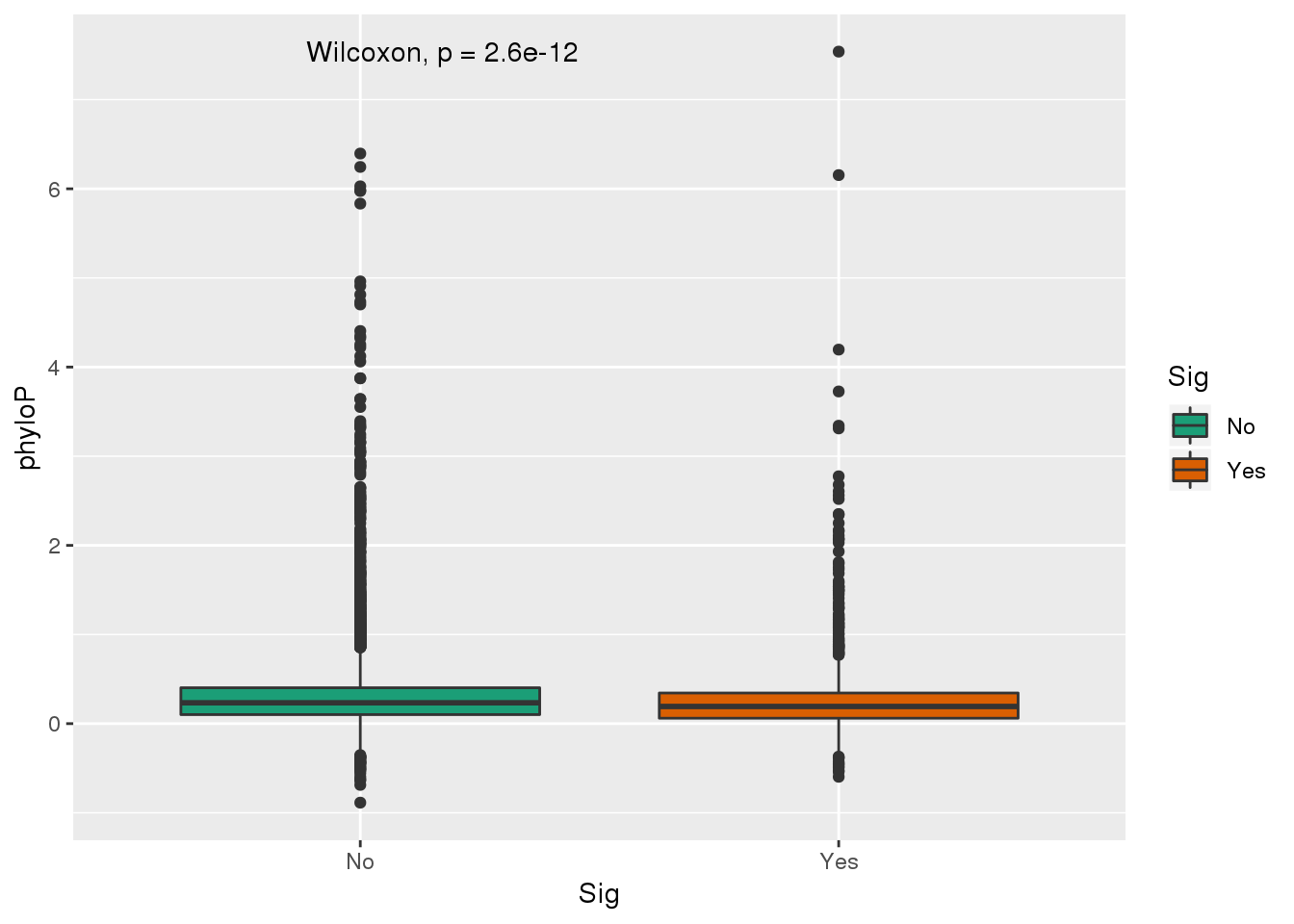 These are also genes with a shift. See if more likely to have - value.
These are also genes with a shift. See if more likely to have - value.
x=nrow(GenesPhy %>% filter(Sig=="Yes", phyloP<0))
m= nrow(GenesPhy %>% filter(phyloP<0))
n=nrow(GenesPhy %>% filter(phyloP>=0))
k=nrow(GenesPhy %>% filter(Sig=="Yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 202#actual:
x[1] 246#pval
phyper(x,m,n,k,lower.tail=F)[1] 7.212381e-05Enriched for genes with - scores.
DN (non synonymous) /DS (synonymous): from ensamble site - ratio of substitution rate (quick and dirty way to look at evo), ration >1 usually evidence for positive selection. values are in ../data/DNDS/HumanChimp_DNDS.csv
Remove NA values
DNDS= read.csv("../data/DNDS/HumanChimp_DNDS.csv", header = T,stringsAsFactors = F) %>% drop_na() %>% group_by(Gene.name) %>% slice(1) %>% ungroup() %>% mutate(DNDSratio= dN.with.Chimpanzee/dS.with.Chimpanzee) %>% dplyr::select(Gene.name, dN.with.Chimpanzee,dS.with.Chimpanzee,DNDSratio) %>% rename("gene"=Gene.name)Join with all results then subset based on significance:
I will get all genes,
NucResGenes=read.table("../data/DiffIso_Nuclear_DF/SignifianceEitherGENES_Nuclear.txt",header = T)
NucResAll=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T, stringsAsFactors = F) %>% dplyr::select(gene) %>% unique() %>% mutate(SigPASinGene=ifelse(gene %in% NucResGenes$gene, "yes", "no"))
NucResDNDS= NucResAll %>% inner_join(DNDS,by="gene") We do not have information for 1236 of the genes. I will assess results on the 7308 with data. There are also genes with ratio problems due to zero in the ds column. If it is infinity, i can make it 1 for now because there are only fixed non syn mutations fixing. If both are 0 I will make it 0.
NucResDNDS_fix=NucResDNDS %>% mutate(DNDSratio = replace_na(DNDSratio,0))
NucResDNDS_fix[NucResDNDS_fix == Inf] <- 1
summary(NucResDNDS_fix$DNDSratio) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.03571 0.19797 0.34906 0.45455 147.00000 NucResDNDS_fix %>% group_by(SigPASinGene) %>% summarise(n=n())# A tibble: 2 x 2
SigPASinGene n
<chr> <int>
1 no 5481
2 yes 1827Plot this.
ggplot(NucResDNDS_fix,aes(y=log10(DNDSratio+1), x=SigPASinGene, fill=SigPASinGene))+ geom_boxplot() + stat_compare_means( label.y.npc = "middle") + scale_fill_brewer(palette = "Dark2") + labs(x="dAPA in Nuclear") + annotate("text", label="Yes=1827 \n No=5481", y=1.8,x=2)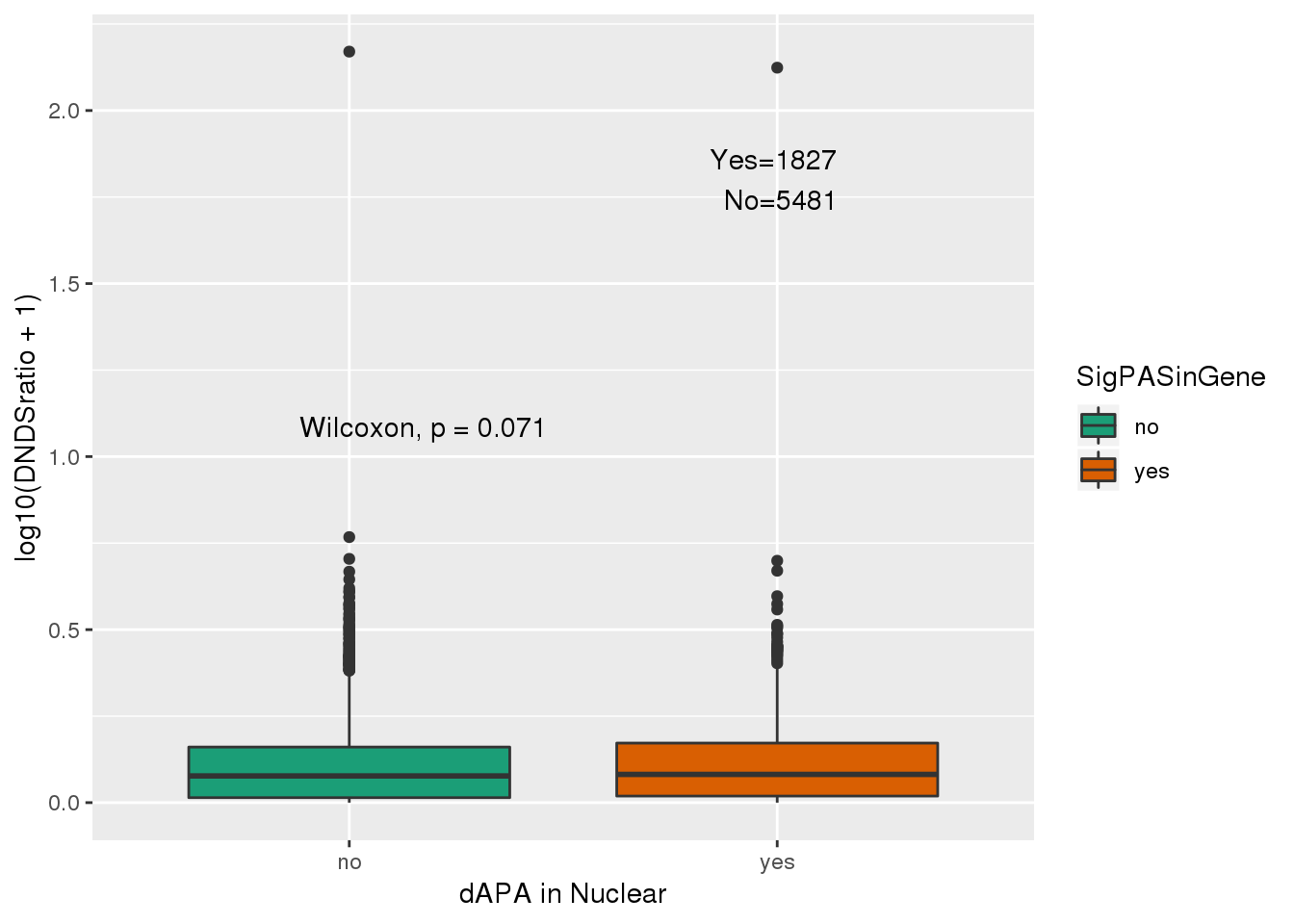 I can ask if they are more likely to be above 1. I can do this with a hypergeo.
I can ask if they are more likely to be above 1. I can do this with a hypergeo.
x=nrow(NucResDNDS_fix %>% filter(SigPASinGene=="yes", DNDSratio>=1))
m= nrow(NucResDNDS_fix %>% filter(DNDSratio>=1))
n=nrow(NucResDNDS_fix %>% filter(DNDSratio<1))
k=nrow(NucResDNDS_fix %>% filter(SigPASinGene=="yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 115#actual:
x[1] 115#pval
phyper(x,m,n,k,lower.tail=F)[1] 0.6666078No enrichment for positive selected genes.
Gene ontology: Need a ranked list of genes. I can do this for the differential apa genes by pvalue.
http://cbl-gorilla.cs.technion.ac.il
NucRes=read.table("../data/DiffIso_Nuclear/SignifianceEitherPAS_2_Nuclear.txt",header = T,stringsAsFactors = F) %>% arrange(p.adjust) %>% dplyr::select(gene) %>% unique()
write.table(NucRes,"../data/DiffIso_Nuclear/SignifianceGenes_orderPval.txt",col.names = F, row.names = F, quote = F)Use gorilla:
Top results:
RNA binding
translation factor activity, RNA binding
protein-containing complex
eukaryotic translation initiation factor
cellular protein-containing complex assembly
intracellular transport
establishment of localization in cell
protein targeting to membrane
nuclear-transcribed mRNA catabolic process, nonsense-mediated decay
translational initiation
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reshape2_1.4.3 ggpubr_0.2 magrittr_1.5 forcats_0.3.0
[5] stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1
[9] tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38
[4] colorspace_1.3-2 generics_0.0.2 htmltools_0.3.6
[7] yaml_2.2.0 utf8_1.1.4 rlang_0.4.0
[10] later_0.7.5 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 RColorBrewer_1.1-2 modelr_0.1.2
[16] readxl_1.1.0 plyr_1.8.4 munsell_0.5.0
[19] gtable_0.2.0 workflowr_1.5.0 cellranger_1.1.0
[22] rvest_0.3.2 evaluate_0.12 labeling_0.3
[25] knitr_1.20 httpuv_1.4.5 fansi_0.4.0
[28] broom_0.5.1 Rcpp_1.0.2 promises_1.0.1
[31] scales_1.0.0 backports_1.1.2 jsonlite_1.6
[34] fs_1.3.1 hms_0.4.2 digest_0.6.18
[37] stringi_1.2.4 grid_3.5.1 rprojroot_1.3-2
[40] cli_1.1.0 tools_3.5.1 lazyeval_0.2.1
[43] crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2
[46] xml2_1.2.0 lubridate_1.7.4 assertthat_0.2.0
[49] rmarkdown_1.10 httr_1.3.1 rstudioapi_0.10
[52] R6_2.3.0 nlme_3.1-137 git2r_0.26.1
[55] compiler_3.5.1