Test Methods to Merge Within Species
Briana Mittleman
4/2/2020
Last updated: 2020-04-03
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/TrialFiltersMeta.txt.sb-9845453e-R58Y0Q/
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/HandC.TvN
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/annotationBias.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: analysis/pol2.Rmd
Untracked: code/._AlignmentScores.sh
Untracked: code/._BothFCMM.sh
Untracked: code/._BothFCMMPrim.sh
Untracked: code/._BothFCnewOInclusive.sh
Untracked: code/._ChimpStarMM2.sh
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._ClosestorthoEx.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._Filter255MM.sh
Untracked: code/._FilterPrimSec.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._MismatchNumbers.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._SAF2Bed.py
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._StarMM2.sh
Untracked: code/._TestFC.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2Bedbothstrand.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chimpMultiCov.sh
Untracked: code/._chimpMultiCov255.sh
Untracked: code/._chimpMultiCovInclusive.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopGeneral.ph
Untracked: code/._extractPhylopGeneral.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._filterPrimaryread.py
Untracked: code/._filterSecondaryread.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixFCheadforExp.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._humanMultiCov.sh
Untracked: code/._humanMultiCov255.sh
Untracked: code/._humanMultiCov_inclusive.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapMMandOrthoexon.sh
Untracked: code/._overlapPASandOrthoexon.sh
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantLiftedPASPrimary.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/AlignmentScores.err
Untracked: code/AlignmentScores.out
Untracked: code/AlignmentScores.sh
Untracked: code/BothFCMM.err
Untracked: code/BothFCMM.out
Untracked: code/BothFCMM.sh
Untracked: code/BothFCMMPrim.err
Untracked: code/BothFCMMPrim.out
Untracked: code/BothFCMMPrim.sh
Untracked: code/BothFCnewOInclusive.sh
Untracked: code/BothFCnewOInclusive.sh.err
Untracked: code/BothFCnewOInclusive.sh.out
Untracked: code/ChimpStarMM2.err
Untracked: code/ChimpStarMM2.out
Untracked: code/ChimpStarMM2.sh
Untracked: code/ClassifyLeafviz.sh
Untracked: code/ClosestorthoEx.err
Untracked: code/ClosestorthoEx.out
Untracked: code/ClosestorthoEx.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/Filter255.err
Untracked: code/Filter255.out
Untracked: code/Filter255MM.sh
Untracked: code/FilterPrimSec.err
Untracked: code/FilterPrimSec.out
Untracked: code/FilterPrimSec.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/IntersectMMandOrtho.err
Untracked: code/IntersectMMandOrtho.out
Untracked: code/IntersectPASandOrtho.err
Untracked: code/IntersectPASandOrtho.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/MismatchNumbers.err
Untracked: code/MismatchNumbers.out
Untracked: code/MismatchNumbers.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/SAF2Bed.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/StarMM2.err
Untracked: code/StarMM2.out
Untracked: code/StarMM2.sh
Untracked: code/TestFC.err
Untracked: code/TestFC.out
Untracked: code/TestFC.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2Bedbothstrand.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chimpMultiCov.err
Untracked: code/chimpMultiCov.out
Untracked: code/chimpMultiCov.sh
Untracked: code/chimpMultiCov255.sh
Untracked: code/chimpMultiCovInclusive.err
Untracked: code/chimpMultiCovInclusive.out
Untracked: code/chimpMultiCovInclusive.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopGeneral.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterPrimaryread.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSecondaryread.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFCheadforExp.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getAlloverlap.py
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/humanMultiCov.err
Untracked: code/humanMultiCov.out
Untracked: code/humanMultiCov.sh
Untracked: code/humanMultiCov255.err
Untracked: code/humanMultiCov255.out
Untracked: code/humanMultiCov255.sh
Untracked: code/humanMultiCovInclusive.err
Untracked: code/humanMultiCovInclusive.out
Untracked: code/humanMultiCov_inclusive.sh
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapMMandOrthoexon.sh
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapPASandOrthoexon.sh
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quantLiftedPASPrimary.err
Untracked: code/quantLiftedPASPrimary.out
Untracked: code/quantLiftedPASPrimary.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runChimpDiffIsoDF.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runHumanDiffIsoDF.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_dF.err
Untracked: code/run_Humanleafcutter_dF.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Chimp_tvN_DF.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN_DF.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/test.txt
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._TrialFiltersMeta.txt
Untracked: data/._TrialFiltersMeta.txt.sb-9845453e-R58Y0Q
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/BaseComp/
Untracked: data/CleanLiftedPeaks_FC_primary/
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OrthoExonBed/
Untracked: data/OverlapBenchmark/
Untracked: data/OverlappingPAS/
Untracked: data/PAS/
Untracked: data/PAS_SAF/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/TestAnnoBiasOE/
Untracked: data/TestMM2/
Untracked: data/TestMM2_AS/
Untracked: data/TestMM2_PrimaryRead/
Untracked: data/TestMM2_SeondaryRead/
Untracked: data/TestMM2_mismatch/
Untracked: data/TestMM2_quality/
Untracked: data/TestWithinMergePAS/
Untracked: data/Test_FC_methods/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalHvC/
Untracked: data/TrialFiltersMeta.txt
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/bioGRID/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/multimap/
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/testQuant/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/DiffUsedIntronic.Rmd
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/ExploredAPA_DF.Rmd
Modified: analysis/MMExpreiment.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/SpliceSiteStrength.Rmd
Modified: analysis/TotalVNuclearBothSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/changeMisprimcut.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/dAPAandapaQTL_DF.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/speciesSpecific.Rmd
Modified: analysis/upsetter_DF.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | fa2203f | brimittleman | 2020-04-03 | add res from merge |
| html | 4722051 | brimittleman | 2020-04-02 | Build site. |
| Rmd | 9ba1428 | brimittleman | 2020-04-02 | add start of merge analysis |
library(scales)
library(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ───────────────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ──────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ readr::col_factor() masks scales::col_factor()
✖ purrr::discard() masks scales::discard()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsaveExploration
I noticed there are overlapping PAS within species. I deal with overlapping PAS between species by merging the PAS into one when 125 of the bases overlap.
For the within species I will look at a merge on the PAS that pass liftover.
../data/cleanPeaks_lifted/Chimp_PASregions_humanCoord.sort.bed ../data/cleanPeaks_lifted/Chimp_PASregions.bed ../data/cleanPeaks_lifted/Human_PASregions.sort.bed
I will test the human coordinates and the chimp coordinates for the chimp pas.
I will use bedtools merge
mkdir ../data/TestWithinMergePAS
bedtools merge -s -c 4,4 -o distinct,count -i ../data/cleanPeaks_lifted/Chimp_PASregions_humanCoord.sort.bed > ../data/TestWithinMergePAS/Chimp_PASregions_humanCoord.merge.bed
sort -k1,1 -k2,2n ../data/cleanPeaks_lifted/Chimp_PASregions.bed > ../data/cleanPeaks_lifted/Chimp_PASregions.sort.bed
bedtools merge -s -c 4,4 -o distinct,count -i ../data/cleanPeaks_lifted/Chimp_PASregions.sort.bed > ../data/TestWithinMergePAS/Chimp_PASregions_chimpCoord.merge.bed
bedtools merge -s -c 4,4 -o distinct,count -i ../data/cleanPeaks_lifted/Human_PASregions.sort.bed > ../data/TestWithinMergePAS/Human_PASregions.merge.bed
Chimp
Compare 2 chimp conditions:
ChimpHumanCoord=read.table("../data/TestWithinMergePAS/Chimp_PASregions_humanCoord.merge.bed",col.names = c("chr",'start','end','PAS', 'nPAS'),stringsAsFactors = F)
ChimpPAS=read.table("../data/TestWithinMergePAS/Chimp_PASregions_chimpCoord.merge.bed",col.names = c("chr",'start','end','PAS', 'nPAS'),stringsAsFactors = F)ChimpHumanCoord %>% group_by(nPAS) %>% summarise(n())# A tibble: 7 x 2
nPAS `n()`
<int> <int>
1 1 250102
2 2 18158
3 3 1937
4 4 236
5 5 30
6 6 9
7 7 3ggplot(ChimpHumanCoord,aes(x=nPAS)) + geom_bar(stat="count")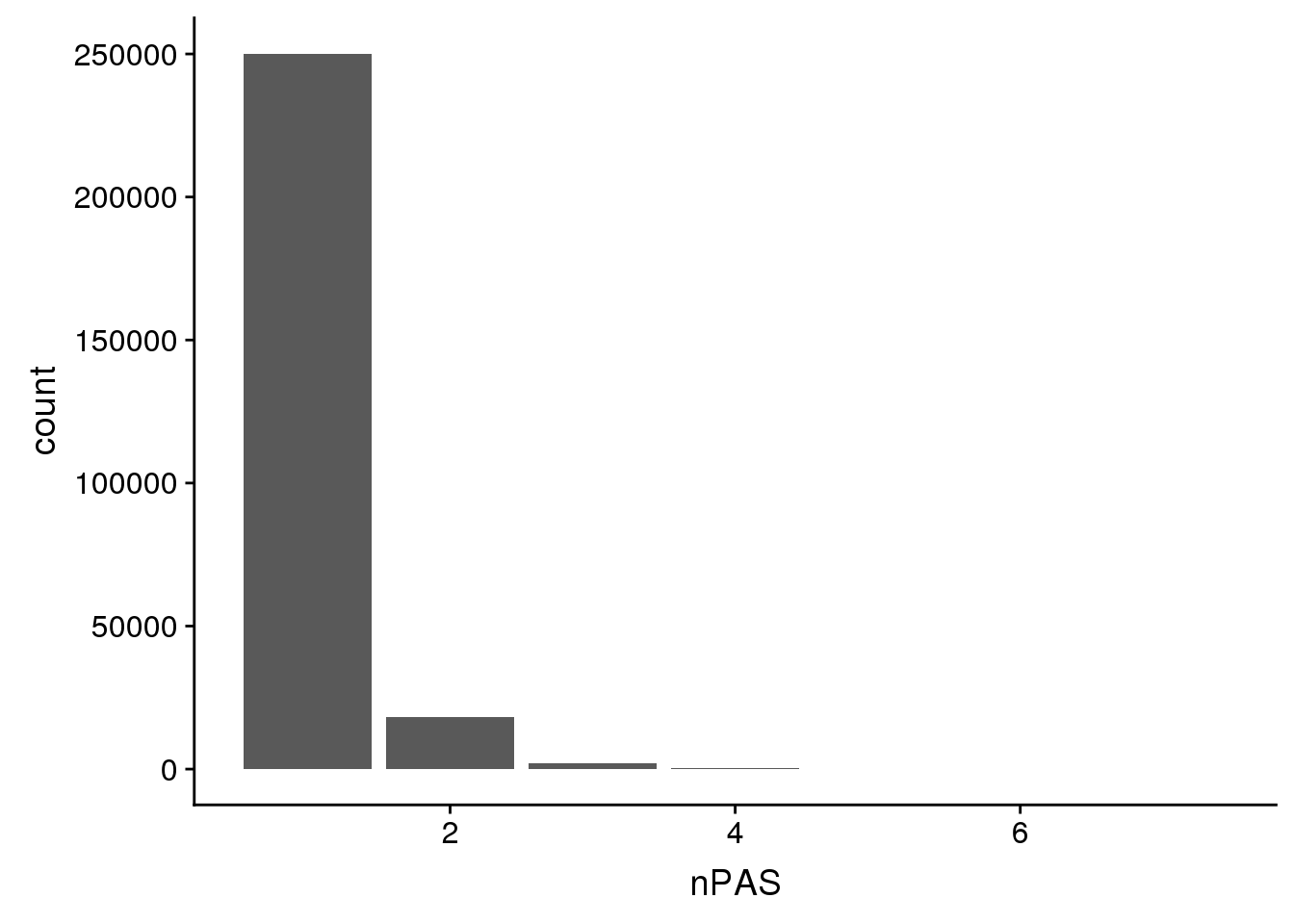
| Version | Author | Date |
|---|---|---|
| 4722051 | brimittleman | 2020-04-02 |
ChimpPAS %>% group_by(nPAS) %>% summarise(n())# A tibble: 7 x 2
nPAS `n()`
<int> <int>
1 1 250102
2 2 18158
3 3 1937
4 4 236
5 5 30
6 6 9
7 7 3ggplot(ChimpPAS,aes(x=nPAS)) + geom_bar(stat="count")
| Version | Author | Date |
|---|---|---|
| 4722051 | brimittleman | 2020-04-02 |
Numbers are the same. Hopfully they are the same overlaps.
length(intersect(ChimpHumanCoord$PAS,ChimpPAS$PAS))[1] 270475Ok these are the same. I can do it all in human coordinate.
Explore the size of these bins.
ChimpHumanCoord_details= ChimpHumanCoord %>% mutate(size=end-start)
ChimpHumanCoord_details$nPAS=as.factor(ChimpHumanCoord_details$nPAS)hue_pal()(7)[1] "#F8766D" "#C49A00" "#53B400" "#00C094" "#00B6EB" "#A58AFF" "#FB61D7"chimpsize=ggplot(ChimpHumanCoord_details, aes(x=nPAS, y=size,fill=nPAS))+ geom_boxplot() + geom_hline(yintercept = 200,col="#F8766D")+ geom_hline(yintercept = 400,col="#C49A00") + geom_hline(yintercept = 600,col="#53B400") + geom_hline(yintercept = 800,col="#00C094") + geom_hline(yintercept = 1000,col="#00B6EB") + geom_hline(yintercept = 1200,col="#A58AFF")+ geom_hline(yintercept = 1400,col="#FB61D7")+ labs(title="Size of chimp overlap PAS, colored by expected if overlap = 1 base")Look at some of the top examples in the data. Is there evidence that high level groups should be filtered out? I can also decide whether I take the middle 200 or use the full region.
ChimpHumanCoord_details %>% arrange(desc(nPAS)) %>% head() chr start end
1 chr1 113855646 113856861
2 chr10 48902937 48904077
3 chr2 121765374 121766466
4 chr1 86743781 86744738
5 chr1 116524622 116525562
6 chr12 402215 403226
PAS
1 chimp15807,chimp15808,chimp15809,chimp15810,chimp15811,chimp15812,chimp15813
2 chimp37068,chimp37069,chimp37070,chimp37071,chimp37072,chimp37073,chimp37074
3 chimp186777,chimp186778,chimp186779,chimp186780,chimp186781,chimp186782,chimp186783
4 chimp12419,chimp12420,chimp12421,chimp12422,chimp12423,chimp12424
5 chimp16131,chimp16132,chimp16133,chimp16134,chimp16135,chimp16136
6 chimp63365,chimp63366,chimp63367,chimp63368,chimp63369,chimp63370
nPAS size
1 7 1215
2 7 1140
3 7 1092
4 6 957
5 6 940
6 6 1011This is before I do any combination with the human PAS.
Human
HumanPAS=read.table("../data/TestWithinMergePAS/Human_PASregions.merge.bed",col.names = c("chr",'start','end','PAS', 'nPAS'),stringsAsFactors = F)HumanPAS %>% group_by(nPAS) %>% summarise(n())# A tibble: 7 x 2
nPAS `n()`
<int> <int>
1 1 260836
2 2 20167
3 3 2189
4 4 288
5 5 42
6 6 6
7 7 2ggplot(HumanPAS,aes(x=nPAS)) + geom_bar(stat="count")
There are more overlaps in the human set.
HumanPAS_details= HumanPAS %>% mutate(size=end-start)
HumanPAS_details$nPAS=as.factor(HumanPAS_details$nPAS)
humansize=ggplot(HumanPAS_details, aes(x=nPAS, y=size,fill=nPAS))+ geom_boxplot() + geom_hline(yintercept = 200,col="#F8766D")+ geom_hline(yintercept = 400,col="#C49A00") + geom_hline(yintercept = 600,col="#53B400") + geom_hline(yintercept = 800,col="#00C094") + geom_hline(yintercept = 1000,col="#00B6EB") + geom_hline(yintercept = 1200,col="#A58AFF")+ geom_hline(yintercept = 1400,col="#FB61D7")+ labs(title="Size of human overlap PAS, colored by expected if overlap = 1 base")plot_grid(chimpsize,humansize) This shows that the human overlaps at the higher numbers are closer together.
This shows that the human overlaps at the higher numbers are closer together.
HumanPAS_details %>% arrange(desc(nPAS)) %>% head() chr start end
1 chr3 25745548 25746665
2 chr4 25250500 25251656
3 chr10 48902772 48903819
4 chr2 88614337 88615211
5 chr2 121765478 121766460
6 chr2 159805437 159806359
PAS
1 human215546,human215547,human215548,human215549,human215550,human215551,human215552
2 human240331,human240332,human240333,human240334,human240335,human240336,human240337
3 human39096,human39097,human39098,human39099,human39100,human39101
4 human174806,human174807,human174808,human174809,human174810,human174811
5 human178226,human178227,human178228,human178229,human178230,human178231
6 human182346,human182347,human182348,human182349,human182350,human182351
nPAS size
1 7 1117
2 7 1156
3 6 1047
4 6 874
5 6 982
6 6 922Passing filters
Focus on examples that make it in the final set.
overlapres=read.table("../data/OverlappingPAS/MergedPAS_HumanCords.txt", col.names = c("chr", "start", "end", "name", "numb")) %>% mutate(length=end-start)
overlapres %>% arrange(desc(numb)) %>% head() chr start end
1 chr1 26691964 26692567
2 chr19 45408963 45409714
3 chr2 143172078 143172946
4 chr3 112565734 112566493
5 chr8 81701986 81702782
6 chr1 89184993 89185569
name numb length
1 chimp3956,chimp3958,human4143,human4144,human4145 5 603
2 chimp152730,human159918,human159919,human159920,human159922 5 751
3 human180107,human180108,human180109,human180110,human180111 5 868
4 chimp213715,chimp213717,human224753,human224754,human224755 5 759
5 chimp308243,human326513,human326514,human326515,human326516 5 796
6 chimp12728,chimp12729,human13580,human13581 4 576After looking at examples, it does not seem like merging is the right way to handle this. It would lead to inflatted numbers in regions and I do not want that. I will leave this for now.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cowplot_0.9.4 forcats_0.3.0 stringr_1.3.1 dplyr_0.8.0.1
[5] purrr_0.3.2 readr_1.3.1 tidyr_0.8.3 tibble_2.1.1
[9] ggplot2_3.1.1 tidyverse_1.2.1 workflowr_1.6.0 scales_1.0.0
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38 colorspace_1.3-2
[5] generics_0.0.2 htmltools_0.3.6 yaml_2.2.0 utf8_1.1.4
[9] rlang_0.4.0 later_0.7.5 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 modelr_0.1.2 readxl_1.1.0 plyr_1.8.4
[17] munsell_0.5.0 gtable_0.2.0 cellranger_1.1.0 rvest_0.3.2
[21] evaluate_0.12 labeling_0.3 knitr_1.20 httpuv_1.4.5
[25] fansi_0.4.0 broom_0.5.1 Rcpp_1.0.2 promises_1.0.1
[29] backports_1.1.2 jsonlite_1.6 fs_1.3.1 hms_0.4.2
[33] digest_0.6.18 stringi_1.2.4 grid_3.5.1 rprojroot_1.3-2
[37] cli_1.1.0 tools_3.5.1 magrittr_1.5 lazyeval_0.2.1
[41] crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2 xml2_1.2.0
[45] lubridate_1.7.4 assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[49] rstudioapi_0.10 R6_2.3.0 nlme_3.1-137 git2r_0.26.1
[53] compiler_3.5.1