Information Content
Briana Mittleman
4/24/2020
Last updated: 2020-05-20
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/TrialFiltersMeta.txt.sb-9845453e-R58Y0Q/
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-284518db-RGf0kd/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/AREstabilityScores.Rmd
Untracked: analysis/AllLoc_effectSizeCor.Rmd
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/HandC.TvN
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/annotationBias.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: code/._AlignmentScores.sh
Untracked: code/._BothFCMM.sh
Untracked: code/._BothFCMMPrim.sh
Untracked: code/._BothFCnewOInclusive.sh
Untracked: code/._ChimpStarMM2.sh
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._ClosestorthoEx.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._Filter255MM.sh
Untracked: code/._FilterPrimSec.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._MismatchNumbers.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._SAF2Bed.py
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._StarMM2.sh
Untracked: code/._TestFC.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2Bedbothstrand.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chimpMultiCov.sh
Untracked: code/._chimpMultiCov255.sh
Untracked: code/._chimpMultiCovInclusive.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopGeneral.ph
Untracked: code/._extractPhylopGeneral.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._filterPrimaryread.py
Untracked: code/._filterSecondaryread.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixFCheadforExp.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._humanMultiCov.sh
Untracked: code/._humanMultiCov255.sh
Untracked: code/._humanMultiCov_inclusive.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapMMandOrthoexon.sh
Untracked: code/._overlapPASandOrthoexon.sh
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantLiftedPASPrimary.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ALLPAS_sequenceDF.err
Untracked: code/ALLPAS_sequenceDF.out
Untracked: code/AlignmentScores.err
Untracked: code/AlignmentScores.out
Untracked: code/AlignmentScores.sh
Untracked: code/BothFCMM.err
Untracked: code/BothFCMM.out
Untracked: code/BothFCMM.sh
Untracked: code/BothFCMMPrim.err
Untracked: code/BothFCMMPrim.out
Untracked: code/BothFCMMPrim.sh
Untracked: code/BothFCnewOInclusive.sh
Untracked: code/BothFCnewOInclusive.sh.err
Untracked: code/BothFCnewOInclusive.sh.out
Untracked: code/ChimpStarMM2.err
Untracked: code/ChimpStarMM2.out
Untracked: code/ChimpStarMM2.sh
Untracked: code/ClassifyLeafviz.sh
Untracked: code/ClosestorthoEx.err
Untracked: code/ClosestorthoEx.out
Untracked: code/ClosestorthoEx.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DTUTR.sh
Untracked: code/DiffDom_RNAmotif_4.err
Untracked: code/DiffDom_RNAmotif_4.out
Untracked: code/DiffDom_RNAmotif_4.sh
Untracked: code/DiffDom_RNAmotif_4_splitDE.err
Untracked: code/DiffDom_RNAmotif_4_splitDE.out
Untracked: code/DiffDom_RNAmotif_4_splitDE.sh
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/Filter255.err
Untracked: code/Filter255.out
Untracked: code/Filter255MM.sh
Untracked: code/FilterPrimSec.err
Untracked: code/FilterPrimSec.out
Untracked: code/FilterPrimSec.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindDomXCutoff.py
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/GetTopminus2Usage.py
Untracked: code/H3K36me3DTplot.err
Untracked: code/H3K36me3DTplot.out
Untracked: code/H3K36me3DTplot.sh
Untracked: code/H3K36me3DTplot_DiffIso.err
Untracked: code/H3K36me3DTplot_DiffIso.out
Untracked: code/H3K36me3DTplot_DiffIso.sh
Untracked: code/H3K36me3DTplot_Specific.err
Untracked: code/H3K36me3DTplot_Specific.out
Untracked: code/H3K36me3DTplot_Specific.sh
Untracked: code/H3K36me3DTplot_distalPAS.err
Untracked: code/H3K36me3DTplot_distalPAS.out
Untracked: code/H3K36me3DTplot_distalPAS.sh
Untracked: code/H3K36me3DTplot_transcript.err
Untracked: code/H3K36me3DTplot_transcript.out
Untracked: code/H3K36me3DTplot_transcript.sh
Untracked: code/H3K36me3DTplotwide.err
Untracked: code/H3K36me3DTplotwide.out
Untracked: code/H3K36me3DTplotwide.sh
Untracked: code/H3K9me3DTplot_transcript.err
Untracked: code/H3K9me3DTplot_transcript.out
Untracked: code/H3K9me3DTplot_transcript.sh
Untracked: code/H3K9me3_processandDT.sh
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/InfoContentShannon.py
Untracked: code/InfoContentbyInd.py
Untracked: code/IntersectMMandOrtho.err
Untracked: code/IntersectMMandOrtho.out
Untracked: code/IntersectPASandOrtho.err
Untracked: code/IntersectPASandOrtho.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/MismatchNumbers.err
Untracked: code/MismatchNumbers.out
Untracked: code/MismatchNumbers.sh
Untracked: code/NuclearDTUTR.err
Untracked: code/NuclearDTUTRt.out
Untracked: code/NuclearPlotsDEandDiffDom_4.err
Untracked: code/NuclearPlotsDEandDiffDom_4.out
Untracked: code/NuclearPlotsDEandDiffDom_4.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/PlotNuclearUsagebySpecies_DF_4DIC.R
Untracked: code/PlotNuclearUsagebySpecies_DF_DEout.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/RNAmotif_PAS.err
Untracked: code/RNAmotif_PAS.out
Untracked: code/RNAmotif_PAS.sh
Untracked: code/RNAmotif_PAS_chimp.err
Untracked: code/RNAmotif_PAS_chimp.out
Untracked: code/RNAmotif_PAS_chimp.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunNewDom.err
Untracked: code/RunNewDom.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/SAF2Bed.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/StarMM2.err
Untracked: code/StarMM2.out
Untracked: code/StarMM2.sh
Untracked: code/TestFC.err
Untracked: code/TestFC.out
Untracked: code/TestFC.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/UTR2FASTA.py
Untracked: code/Upstream10Bases_general.py
Untracked: code/allPASSeq_df.sh
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/apaQTLsnakefiltPAS.err
Untracked: code/apaQTLsnakefiltPAS.out
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2Bedbothstrand.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chimpMultiCov.err
Untracked: code/chimpMultiCov.out
Untracked: code/chimpMultiCov.sh
Untracked: code/chimpMultiCov255.sh
Untracked: code/chimpMultiCovInclusive.err
Untracked: code/chimpMultiCovInclusive.out
Untracked: code/chimpMultiCovInclusive.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhaastConGeneral.py
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopGeneral.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterPrimaryread.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSecondaryread.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFCheadforExp.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getAlloverlap.py
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/humanMultiCov.err
Untracked: code/humanMultiCov.out
Untracked: code/humanMultiCov.sh
Untracked: code/humanMultiCov255.err
Untracked: code/humanMultiCov255.out
Untracked: code/humanMultiCov255.sh
Untracked: code/humanMultiCovInclusive.err
Untracked: code/humanMultiCovInclusive.out
Untracked: code/humanMultiCov_inclusive.sh
Untracked: code/infoContentSimpson.py
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeDIC.err
Untracked: code/makeDIC.out
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/makedICPlots_DF.sh
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/mergeChimpRNA.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergeandsort_H3K9me3
Untracked: code/mergeandsort_h3k36me3
Untracked: code/mergeandsorth3k36me3.sh
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapMMandOrthoexon.sh
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapPASandOrthoexon.sh
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quantLiftedPASPrimary.err
Untracked: code/quantLiftedPASPrimary.out
Untracked: code/quantLiftedPASPrimary.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runChimpDiffIsoDF.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runHumanDiffIsoDF.sh
Untracked: code/runNewDom.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_dF.err
Untracked: code/run_Humanleafcutter_dF.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePAS_Human.batch
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.batch
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Chimp_tvN_DF.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN_DF.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/test.txt
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._TrialFiltersMeta.txt
Untracked: data/._TrialFiltersMeta.txt.sb-9845453e-R58Y0Q
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/AREelements/
Untracked: data/BaseComp/
Untracked: data/CleanLiftedPeaks_FC_primary/
Untracked: data/CompapaQTLpas/
Untracked: data/DIC_Viz/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffDomandDE_example/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DistTwoDom/
Untracked: data/DomDefGreaterX/
Untracked: data/DomStructure_4/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/DoubleFilterUsageNumeric/
Untracked: data/EvalPantro5/
Untracked: data/H3K36me3/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/HumanMolPheno/
Untracked: data/IndInfoContent/
Untracked: data/InfoContent/
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OrthoExonBed/
Untracked: data/OverlapBenchmark/
Untracked: data/OverlappingPAS/
Untracked: data/PAS/
Untracked: data/PAS_SAF/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/PhastCon/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/Pol2Chip/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/TestAnnoBiasOE/
Untracked: data/TestMM2/
Untracked: data/TestMM2_AS/
Untracked: data/TestMM2_PrimaryRead/
Untracked: data/TestMM2_SeondaryRead/
Untracked: data/TestMM2_mismatch/
Untracked: data/TestMM2_quality/
Untracked: data/TestWithinMergePAS/
Untracked: data/Test_FC_methods/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalFractionPAS/
Untracked: data/TotalHvC/
Untracked: data/TrialFiltersMeta.txt
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/bioGRID/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_extra.txt
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/miRNA/
Untracked: data/multimap/
Untracked: data/orthoUTR/
Untracked: data/paiDecay/
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/testQuant/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dAPAandDomEnrich.png
Untracked: output/dEandDomEnrich.png
Untracked: output/dpnotDE
Untracked: output/dtPlots/
Untracked: output/exandte
Untracked: output/whichSiteplot.pdf
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/DeandNumPAS.Rmd
Modified: analysis/DiffTop2SecondDom.Rmd
Modified: analysis/DirSelectionKhan.Rmd
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/MMExpreiment.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/PTM_analysis.Rmd
Modified: analysis/TotalDomStructure.Rmd
Modified: analysis/TotalVNuclearBothSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/changeMisprimcut.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/dInforContent.Rmd
Modified: analysis/diffExpression.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/mRNADecay.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/phastCon.Rmd
Modified: analysis/pol2.Rmd
Modified: analysis/signalsites_doublefilter.Rmd
Modified: analysis/speciesSpecific.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 3cd8e7c | brimittleman | 2020-05-20 | update figures for new levels |
| html | 6a947a5 | brimittleman | 2020-05-12 | Build site. |
| Rmd | 313ee8a | brimittleman | 2020-05-12 | add number of pas plots |
| html | 33d6feb | brimittleman | 2020-05-07 | Build site. |
| Rmd | d680b86 | brimittleman | 2020-05-07 | add number PAS plot for pp |
| html | 10590ea | brimittleman | 2020-05-07 | Build site. |
| Rmd | ae7d82b | brimittleman | 2020-05-06 | add non colored plot |
| html | cb0a024 | brimittleman | 2020-04-30 | Build site. |
| Rmd | c253b1b | brimittleman | 2020-04-30 | fix same no dapa bug, change simp color |
| html | 81dcd9f | brimittleman | 2020-04-27 | Build site. |
| Rmd | d9ba030 | brimittleman | 2020-04-27 | add equitability |
| html | 7725e4d | brimittleman | 2020-04-27 | Build site. |
| Rmd | b653f27 | brimittleman | 2020-04-27 | add simpson |
| html | 448aa08 | brimittleman | 2020-04-24 | Build site. |
| Rmd | 93d00f3 | brimittleman | 2020-04-24 | add info content |
library(tidyverse)── Attaching packages ─────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(ggpubr)Loading required package: magrittr
Attaching package: 'magrittr'The following object is masked from 'package:purrr':
set_namesThe following object is masked from 'package:tidyr':
extractlibrary(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggpubr':
get_legendThe following object is masked from 'package:ggplot2':
ggsavePackages/functions for this:
vegan: diversity, can calculate shannon or simpson
I will probably do this in python because I can go gene by gene easier:
scipy stats example
This is good because I will be able to change the base and see how it effects the measurements
https://kite.com/python/docs/scipy.stats.entropy
default base is e
from scipy.stats import entropy
import numpy as np
from math import log, e
entropy([1/2, 1/2], base=2)
#shannon
Shannon2 = -np.sum(pA*np.log2(pA))I most likely want to use a uniform prior. for this. I could get more complicated in the future by weighting differences by utr and intron. this would help find “more surpising” results.
simpson- squares the probability
from ecopy import diversity
diversity(x, medod="simpson")
#x- side x species matrix, sites are rows, columns are species - ie column counts, row == paslibrary(vegan)Loading required package: permuteLoading required package: latticeThis is vegan 2.5-3data(BCI)
dim(BCI)[1] 50 225H <- diversity(BCI)
length(H)[1] 50diversity(c(.5,.5,.5))[1] 1.098612diversity(c(.25,.75,.25))[1] 0.9502705#more peak= lower
diversity(c(.5,.5,.5), "simpson")[1] 0.6666667diversity(c(.25,.75,.25),"simpson")[1] 0.56#more peak= lower
diversity(c(.5,.5,.5), "inv")[1] 3diversity(c(.25,.75,.25),"inv")[1] 2.272727Seem like it is most simple to use the mean usages for this.
##Shannon
First test:
use entropy in python with different bases. -base 2 is the classic shannon and it uses the - when probabilities are given (ie uniform prior)
the python code will work with my meta file for now and take species as an input.
\(H=-\sum^{s}_{i=1}p_{i}log_{2}p_{i}\)
\(H=-\sum^{s}_{i=1}p_{i}lnp_{i}\)
mkdir ../data/InfoContent
python InfoContentShannon.py Human
python InfoContentShannon.py Chimp
Results:
HumanResInfo= read.table("../data/InfoContent/Human_InfoContent.txt", header = T,stringsAsFactors = F) %>% rename(Human_Base2=base2, Human_basee= basee)
ChimpResInfo= read.table("../data/InfoContent/Chimp_InfoContent.txt", header = T,stringsAsFactors = F) %>% rename(Chimp_Base2=base2, Chimp_basee= basee)
BothResInfo= HumanResInfo %>% inner_join(ChimpResInfo, by=c("gene", "numPAS")) %>% filter(numPAS > 1)First plot the distributions:
BothResInfo_2= BothResInfo %>% select(gene, contains("Base2")) %>% gather("species", "base2", -gene)
ggplot(BothResInfo_2, aes(x=base2, fill=species)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Set1")+ labs(title="Shannon Information Content")Warning: Removed 1 rows containing non-finite values (stat_density).
wilcox.test(BothResInfo$Human_Base2, BothResInfo$Chimp_Base2, alternative = "greater")
Wilcoxon rank sum test with continuity correction
data: BothResInfo$Human_Base2 and BothResInfo$Chimp_Base2
W = 39254000, p-value < 2.2e-16
alternative hypothesis: true location shift is greater than 0Human shift higher, ie less density:
BothResInfo_e= BothResInfo %>% select(gene, contains("basee")) %>% gather("species", "basee", -gene)
ggplot(BothResInfo_e, aes(x=basee, fill=species)) + geom_density(alpha=.3)Warning: Removed 1 rows containing non-finite values (stat_density).
I want to look at this by dominance:
ggplot(BothResInfo_2,aes(x=base2, fill=species)) + geom_histogram() + facet_grid(~species)`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.Warning: Removed 1 rows containing non-finite values (stat_bin).
Plot human vs chimp:
ggplot(BothResInfo,aes(x=Human_Base2,y= Chimp_Base2 )) + geom_point() + geom_abline(slope=1, intercept = 0) + stat_cor(col="blue") + geom_density_2d(col="blue")Warning: Removed 1 rows containing non-finite values (stat_cor).Warning: Removed 1 rows containing non-finite values (stat_density2d).Warning: Removed 1 rows containing missing values (geom_point).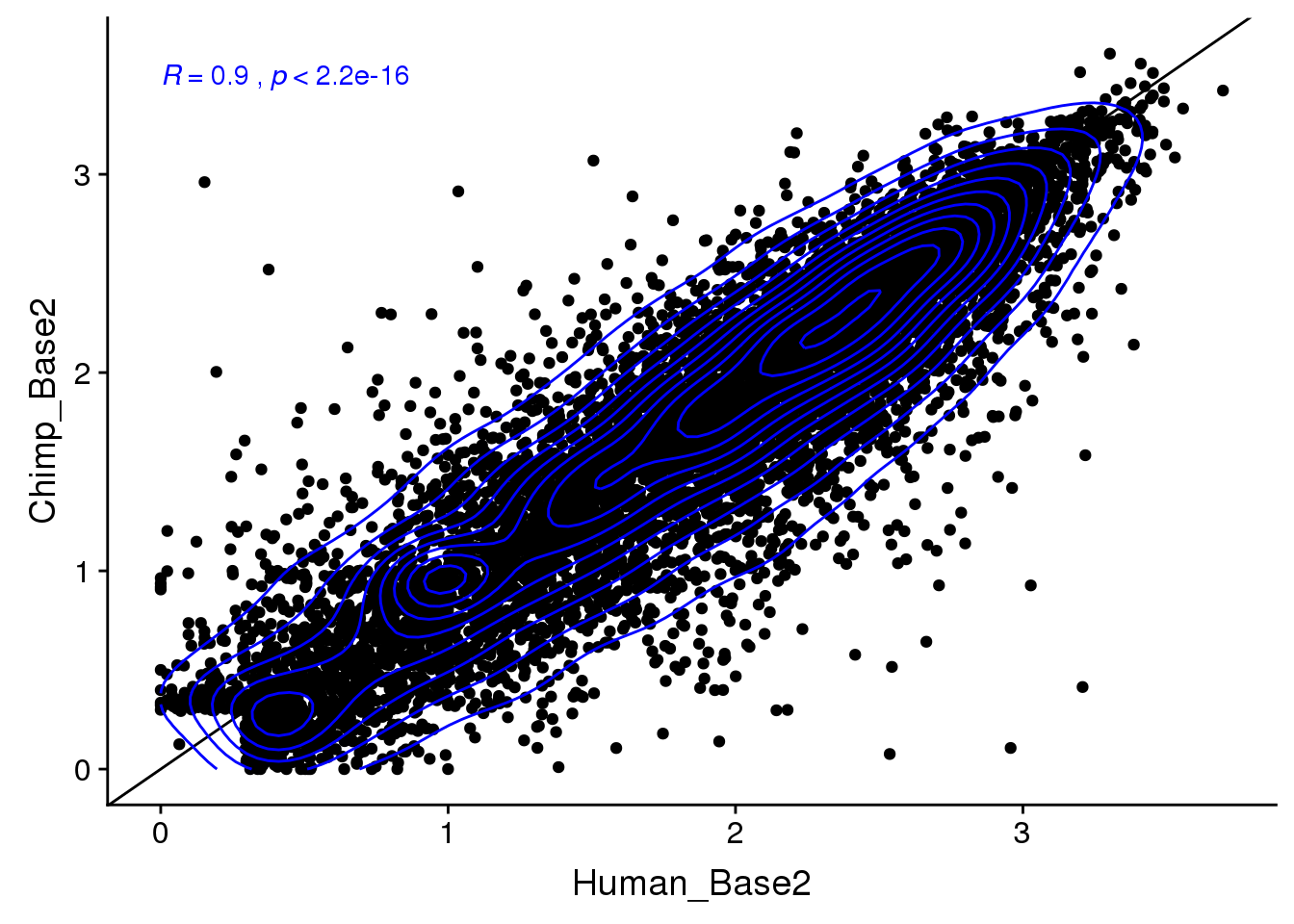
ggplot(BothResInfo,aes(x=Human_Base2,y= Chimp_Base2 ,col=numPAS)) + geom_point(alpha=.4) + geom_abline(slope=1, intercept = 0) +labs(title="Shannon Index Colored by number of PAS")Warning: Removed 1 rows containing missing values (geom_point).
Does number explain:
summary(lm(BothResInfo$Human_Base2 ~BothResInfo$numPAS))
Call:
lm(formula = BothResInfo$Human_Base2 ~ BothResInfo$numPAS)
Residuals:
Min 1Q Median 3Q Max
-2.88694 -0.20022 0.06073 0.23527 0.53990
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.209838 0.008559 24.52 <2e-16 ***
BothResInfo$numPAS 0.314404 0.001527 205.93 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.3166 on 8448 degrees of freedom
(1 observation deleted due to missingness)
Multiple R-squared: 0.8339, Adjusted R-squared: 0.8339
F-statistic: 4.241e+04 on 1 and 8448 DF, p-value: < 2.2e-16summary(lm(BothResInfo$Chimp_Base2 ~BothResInfo$numPAS ))
Call:
lm(formula = BothResInfo$Chimp_Base2 ~ BothResInfo$numPAS)
Residuals:
Min 1Q Median 3Q Max
-2.76846 -0.25131 0.06184 0.27657 0.67339
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.108685 0.010226 10.63 <2e-16 ***
BothResInfo$numPAS 0.307373 0.001824 168.49 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.3784 on 8449 degrees of freedom
Multiple R-squared: 0.7706, Adjusted R-squared: 0.7706
F-statistic: 2.839e+04 on 1 and 8449 DF, p-value: < 2.2e-16So this is working but the number of PAS explains most of the variation. Maybe I can normalize this out and look at residuals:
BothResInfoRes= BothResInfo %>% mutate(HumanNorm=residuals(BothResInfo$Human_Base2~BothResInfo$numPAS),ChimpNorm=residuals(BothResInfo$Chimp_Base2~BothResInfo$numPAS))pull in dominance:
HumanRes=read.table("../data/DomDefGreaterX/Human_AllGenes_DiffTop.txt", col.names = c("Human_PAS", "gene","Human_DiffDom"),stringsAsFactors = F)
ChimpRes=read.table("../data/DomDefGreaterX/Chimp_AllGenes_DiffTop.txt", col.names = c("Chimp_PAS", "gene","Chimp_DiffDom"),stringsAsFactors = F)
BothRes=HumanRes %>% inner_join(ChimpRes,by="gene")
BothRes_10=BothRes %>% filter(Chimp_DiffDom >=0.1 | Human_DiffDom>=0.1) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=10)
BothRes_20=BothRes %>% filter(Chimp_DiffDom >=0.2 | Human_DiffDom>=0.2) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=20)
BothRes_30=BothRes %>% filter(Chimp_DiffDom >=0.3 | Human_DiffDom>=0.3) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=30)
BothRes_40=BothRes %>% filter(Chimp_DiffDom >=0.4 | Human_DiffDom>=0.4) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=40)
BothRes_50=BothRes %>% filter(Chimp_DiffDom >=0.5 | Human_DiffDom>=0.5) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=50)
BothRes_60=BothRes %>% filter(Chimp_DiffDom >=0.6 | Human_DiffDom>=0.6) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=60)
BothRes_70=BothRes %>% filter(Chimp_DiffDom >=0.7 | Human_DiffDom>=0.7) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=70)
BothRes_80=BothRes %>% filter(Chimp_DiffDom >=0.8 | Human_DiffDom>=0.8) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=80)
BothRes_90=BothRes %>% filter(Chimp_DiffDom >=0.9 | Human_DiffDom>=0.9) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=90)
BothResAll=BothRes_10 %>% bind_rows(BothRes_20) %>% bind_rows(BothRes_30) %>% bind_rows(BothRes_40) %>% bind_rows(BothRes_50) %>% bind_rows(BothRes_60) %>% bind_rows(BothRes_70) %>% bind_rows(BothRes_80) %>% bind_rows(BothRes_90)I want dominance in 1 or both at .4.
BothRes_40_each= BothRes_40 %>% mutate(Dom=ifelse(Human_DiffDom>=.4, ifelse(Chimp_DiffDom >=.4, "Both", "Human"), "Chimp"))
BothRes_40_each %>% group_by(Dom) %>% summarise(n())# A tibble: 3 x 2
Dom `n()`
<chr> <int>
1 Both 1565
2 Chimp 906
3 Human 257BothRes_40_each %>% group_by(Set,Dom) %>% summarise(n())# A tibble: 6 x 3
# Groups: Set [2]
Set Dom `n()`
<chr> <chr> <int>
1 Different Both 22
2 Different Chimp 114
3 Different Human 46
4 Same Both 1543
5 Same Chimp 792
6 Same Human 211BothRes_40_eachsm= BothRes_40_each %>% select(gene, Set, Dom)
BothResInfoDom= BothResInfo %>% full_join(BothRes_40_eachsm, by="gene", fill="None") %>% mutate(Set= replace_na(Set, "None"),Dom= replace_na(Dom, "None"))
ggplot(BothResInfoDom,aes(x=Human_Base2,y= Chimp_Base2, col=Dom )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + scale_color_brewer(palette = "Set2") + labs(x="Human Information", y="Chimp Information", title="Shannon Information Index colored by whether gene has a dominant PAS")Warning: Removed 1 rows containing missing values (geom_point).
ggplot(BothResInfoDom,aes(x=Human_Base2,y= Chimp_Base2, col=Set )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + scale_color_brewer(palette = "Set2") +geom_density2d()+ labs(x="Human Information", y="Chimp Information", title="Shannon Information Index colored by Dominance Structure ")Warning: Removed 1 rows containing non-finite values (stat_density2d).
Warning: Removed 1 rows containing missing values (geom_point).
BothResInfoDom$numPAS=as.factor(BothResInfoDom$numPAS)
ggplot(BothResInfoDom,aes(x=Human_Base2,y= Chimp_Base2, col=numPAS )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + labs(x="Human Information", y="Chimp Information", title="Shannon Information Index colored by number of PAS") + facet_grid(~Dom)Warning: Removed 1 rows containing missing values (geom_point).
#+ scale_color_brewer(palette = "Spectral")Dominance and number of PAS:
BothResInfoDom$numPAS=as.numeric(as.character(BothResInfoDom$numPAS))
ggplot(BothResInfoDom,aes(x=Dom, y=numPAS)) +geom_boxplot() +stat_compare_means() + labs(x="Dominance Structure",y="Number of PAS", title="Number of PAS differ by dominance structure")
ggplot(BothResInfoDom,aes(x=Set, y=numPAS)) +geom_boxplot() +stat_compare_means() + labs(x="Dominance Structure",y="Number of PAS", title="Number of PAS differ by dominance structure")
Ratio problem!!!!
but the confounder is biological- number of PAS.
Simpson
Try the simpson index.
skit-bio: http://scikit-bio.org/docs/0.1.3/math.diversity.alpha.html
\(D=\sum_{i=1}^{R}p_{i}^{2}\)
and
\(D=1-\sum_{i=1}^{R}p_{i}^{2}\)
python infoContentSimpson.py Human
python infoContentSimpson.py Chimp
SimpHuman=read.table("../data/InfoContent/Human_SimpsonInfoContent.txt", header = T, stringsAsFactors = F) %>% rename(simpson_Human=simpson) %>% mutate(simpOpp_Human=1-simpson_Human)
SimpChimp=read.table("../data/InfoContent/Chimp_SimpsonInfoContent.txt", header = T, stringsAsFactors = F)%>% rename(simpson_Chimp=simpson)%>% mutate(simpOpp_Chimp=1-simpson_Chimp)
BothSimp= SimpHuman %>% inner_join(SimpChimp, by=c("gene", "numPAS")) %>% filter(numPAS > 1)Gather and plot:
BothSimp_g= BothSimp %>% select(-contains("Opp")) %>% gather("species", "Simpson", -gene, -numPAS)
ggplot(BothSimp_g, aes(x=Simpson, fill=species)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Set1")+labs(title="Simpson Index")
BothOppSimp_g= BothSimp %>% select(-contains("simpson")) %>% gather("species", "SimpsonOpp", -gene, -numPAS)
ggplot(BothOppSimp_g, aes(x=SimpsonOpp, fill=species)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Set1")+labs(title="Simpson Index (1-opp)")
wilcox.test(BothSimp$simpOpp_Human, BothSimp$simpOpp_Chimp, alternative = "greater")
Wilcoxon rank sum test with continuity correction
data: BothSimp$simpOpp_Human and BothSimp$simpOpp_Chimp
W = 40925000, p-value < 2.2e-16
alternative hypothesis: true location shift is greater than 0Histogram:
ggplot(BothSimp_g,aes(x=Simpson, fill=species)) + geom_histogram() + facet_grid(~species)+scale_fill_brewer(palette = "Set1")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
ggplot(BothOppSimp_g, aes(x=SimpsonOpp, fill=species)) + geom_histogram() + facet_grid(~species)+scale_fill_brewer(palette = "Set1")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
here, higher index is lower diversity= more dominance (opposite of shannon)
the opposite one is 1- sum. this is more dominance at lower values like shannon. I will go with this so the sign is the same.
BothInfoTypes=BothSimp %>% inner_join(BothResInfoRes, by=c("gene", "numPAS"))
BothInfoTypes_h=BothInfoTypes %>% select(gene,numPAS, simpOpp_Human, Human_Base2) %>% mutate(species="Human") %>% rename(Simpson= simpOpp_Human, Shannon=Human_Base2)
BothInfoTypes_c=BothInfoTypes %>% select(gene,numPAS, simpOpp_Chimp, Chimp_Base2) %>% mutate(species="Chimp")%>% rename(Simpson= simpOpp_Chimp, Shannon=Chimp_Base2)
BothInfoTypes_both=BothInfoTypes_h %>% bind_rows(BothInfoTypes_c)
ggplot(BothInfoTypes_both,aes(x=Simpson, y=Shannon, by=species, col=species)) +geom_point(alpha=.4) +geom_density2d(col="black") + stat_cor(label.x=0) + geom_smooth(col="black",method = "lm") + facet_grid(~species) + labs(title="Correlation between Indicies") +theme(legend.position = "none")+scale_color_brewer(palette = "Set1")Warning: Removed 1 rows containing non-finite values (stat_density2d).Warning: Removed 1 rows containing non-finite values (stat_cor).Warning: Removed 1 rows containing non-finite values (stat_smooth).Warning: Removed 1 rows containing missing values (geom_point).
There is more variation at the low end here.
Compare human and chimp simpson by PAS number:
ggplot(BothInfoTypes,aes(x=simpOpp_Human,y= simpOpp_Chimp)) + geom_point(alpha=.4) + geom_abline(slope=1, intercept = 0)+labs(title="Simpson Index") + stat_cor(col="blue")+ geom_density_2d(col="blue")
ggplot(BothInfoTypes,aes(x=simpOpp_Human,y= simpOpp_Chimp ,col=numPAS)) + geom_point(alpha=.4) + geom_abline(slope=1, intercept = 0)+labs(title="Simpson Index Colored by number of PAS")
summary(lm(BothInfoTypes$simpOpp_Human ~BothResInfo$numPAS))
Call:
lm(formula = BothInfoTypes$simpOpp_Human ~ BothResInfo$numPAS)
Residuals:
Min 1Q Median 3Q Max
-0.94165 -0.08859 0.00961 0.10528 0.43248
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.4483250 0.0046537 96.34 <2e-16 ***
BothResInfo$numPAS 0.0595955 0.0008302 71.78 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.1722 on 8449 degrees of freedom
Multiple R-squared: 0.3788, Adjusted R-squared: 0.3788
F-statistic: 5153 on 1 and 8449 DF, p-value: < 2.2e-16cor.test(BothInfoTypes$simpOpp_Human,BothResInfo$numPAS)
Pearson's product-moment correlation
data: BothInfoTypes$simpOpp_Human and BothResInfo$numPAS
t = 71.784, df = 8449, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.6020812 0.6285731
sample estimates:
cor
0.615501 summary(lm(BothInfoTypes$simpOpp_Chimp ~BothResInfo$numPAS ))
Call:
lm(formula = BothInfoTypes$simpOpp_Chimp ~ BothResInfo$numPAS)
Residuals:
Min 1Q Median 3Q Max
-0.79002 -0.10111 0.01491 0.11286 0.50115
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.3685223 0.0049596 74.31 <2e-16 ***
BothResInfo$numPAS 0.0651630 0.0008848 73.65 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.1835 on 8449 degrees of freedom
Multiple R-squared: 0.391, Adjusted R-squared: 0.3909
F-statistic: 5424 on 1 and 8449 DF, p-value: < 2.2e-16cor.test(BothInfoTypes$simpOpp_Chimp,BothResInfo$numPAS)
Pearson's product-moment correlation
data: BothInfoTypes$simpOpp_Chimp and BothResInfo$numPAS
t = 73.649, df = 8449, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.6121253 0.6380995
sample estimates:
cor
0.6252856 Number of PAS is less correlated with this index.
Add in the dominanace structure to compare to simpson:
BothResBothInfoDom= BothInfoTypes %>% full_join(BothRes_40_eachsm, by="gene", fill="None") %>% mutate(Set= replace_na(Set, "None"),Dom= replace_na(Dom, "None"))
ggplot(BothResBothInfoDom,aes(x=simpOpp_Human,y= simpOpp_Chimp, col=Dom )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + scale_color_brewer(palette = "Set2") + labs(x="Human Simpson", y="Chimp Simpson", title="Simpson Information Index colored by whether gene has a dominant PAS")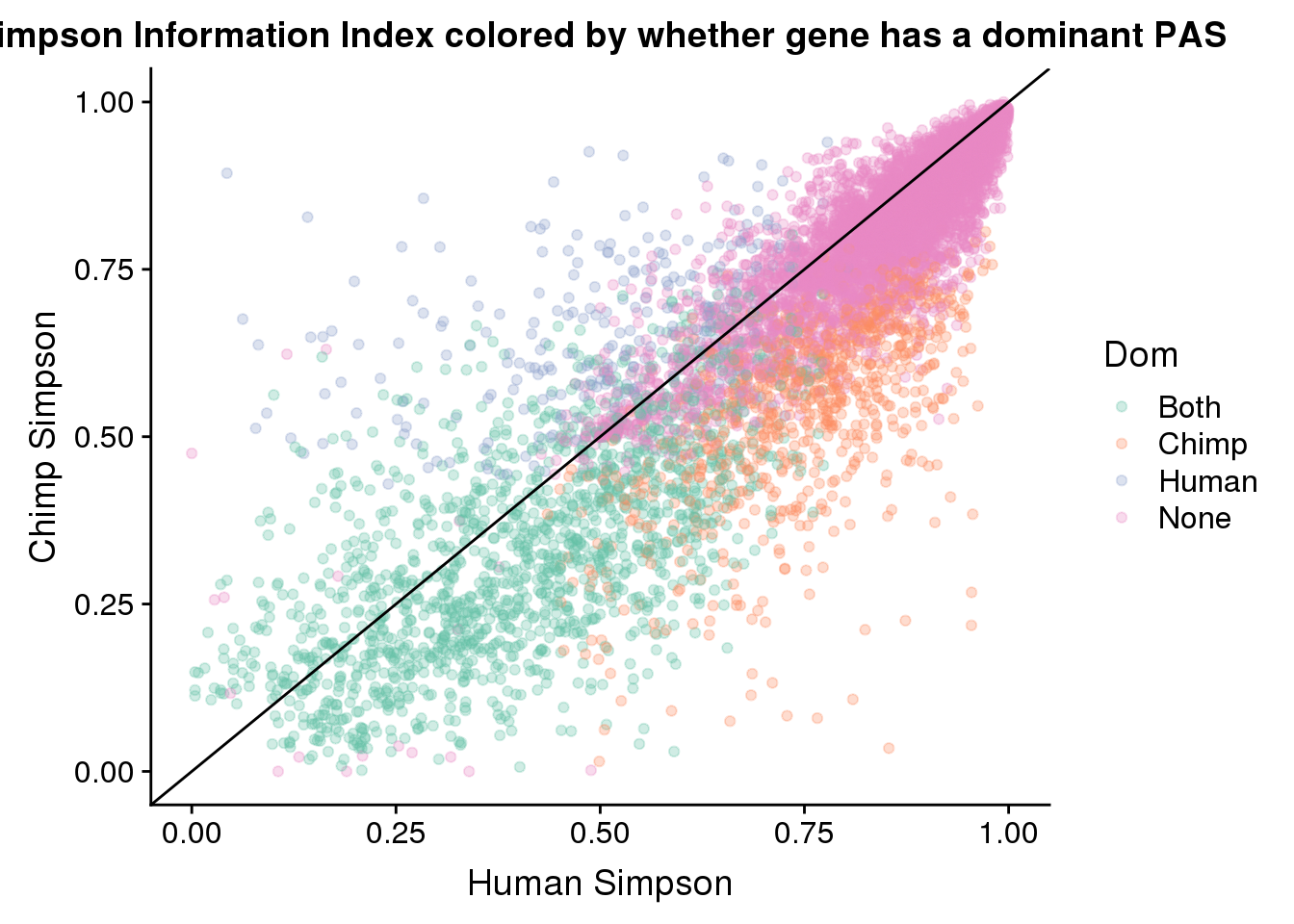
ggplot(BothResBothInfoDom,aes(x=simpOpp_Human,y= simpOpp_Chimp, col=Set )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + scale_color_brewer(palette = "Set2") +geom_density2d()+ labs(x="Human Simpson", y="Chimp Simpson", title="Simpson Information Index colored by Dominance Structure ")
BothResBothInfoDom$numPAS=as.factor(BothResBothInfoDom$numPAS)
ggplot(BothResBothInfoDom,aes(x=simpOpp_Human,y= simpOpp_Chimp, col=numPAS )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + labs(x="Human Simpson", y="Chimp Simpson", title="Simpson Information Index colored by number of PAS ") + facet_grid(~Dom)
#+ scale_color_brewer(palette = "Spectral")Shannon Equitability
Equitability. Shannon diversity divided by the logarithm of number of taxa. This measures the evenness with which individuals are divided among the taxa present.
Shannon’s equitability (EH) measures the evenness of a community and can be easily calculated by diving the value of H with H_max, which equals to lnS(S=number of species encountered). Its value ranges between 0 and 1, with being complete evenness. (0-1)
\(E_{h}=H/log2(NumPAS)\)
BothResBothInfoDomEH=BothResBothInfoDom %>% mutate(human_EH=Human_Base2/log2(as.numeric(as.character(numPAS))), chimp_EH=Chimp_Base2/log2(as.numeric(as.character(numPAS))))
BothEH= BothResBothInfoDomEH %>% select(gene, numPAS, human_EH,chimp_EH) %>% gather("species", "ShannonEH", -gene, -numPAS)
ggplot(BothEH, aes(x=ShannonEH, fill=species)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Set1")+labs(title="Shannon equitability", x="Shannon equitability")Warning: Removed 1 rows containing non-finite values (stat_density).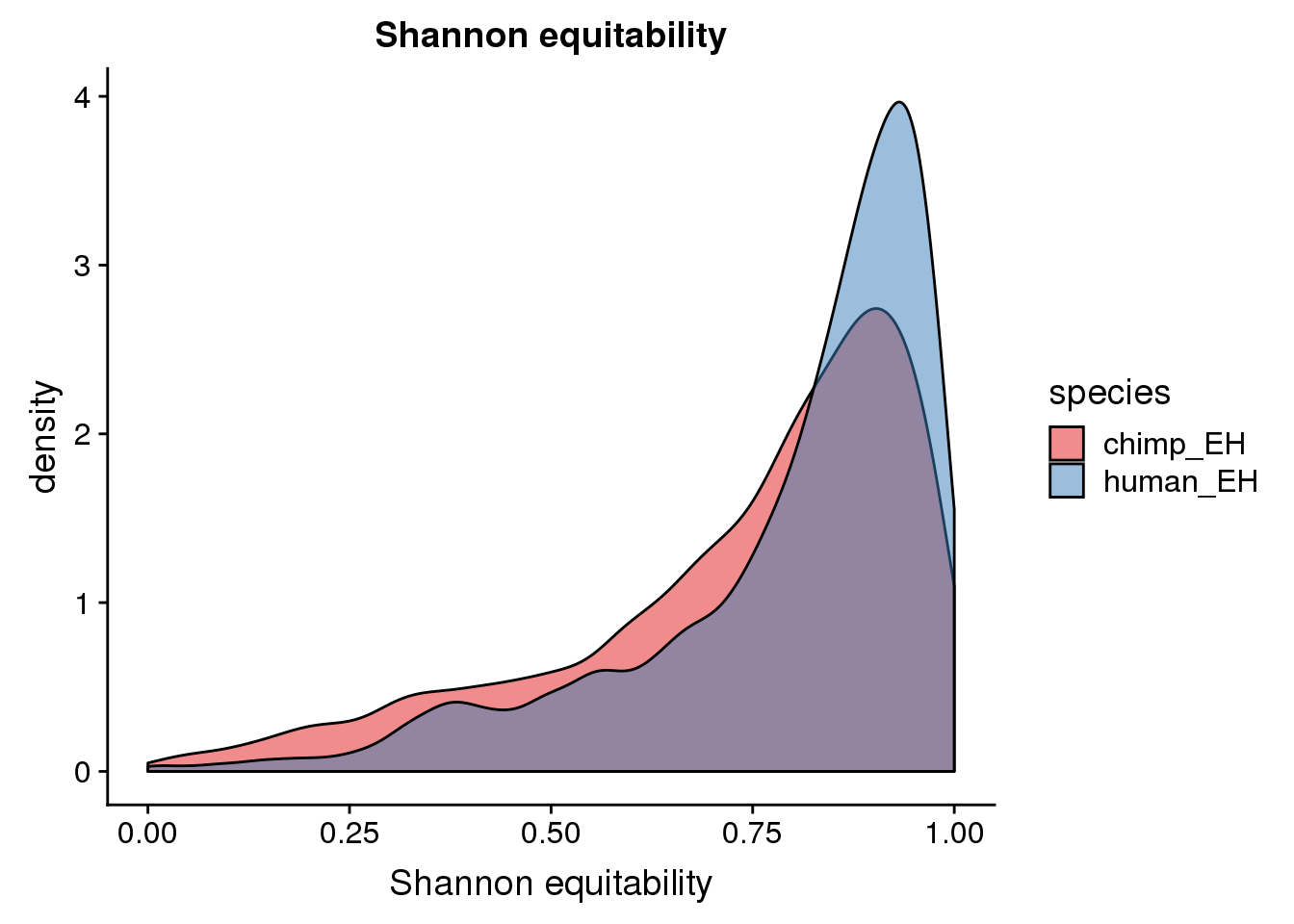
wilcox.test(BothResBothInfoDomEH$human_EH, BothResBothInfoDomEH$chimp_EH)
Wilcoxon rank sum test with continuity correction
data: BothResBothInfoDomEH$human_EH and BothResBothInfoDomEH$chimp_EH
W = 42395000, p-value < 2.2e-16
alternative hypothesis: true location shift is not equal to 0BothResBothInfoDomEH$numPAS=as.numeric(as.character(BothResBothInfoDomEH$numPAS))
ggplot(BothResBothInfoDomEH,aes(x=human_EH,y= chimp_EH )) + geom_point(alpha=.4) + geom_abline(slope=1, intercept = 0) +labs(title="Shannon equitability") + stat_cor(col="blue")+ geom_density_2d(col="blue")Warning: Removed 1 rows containing non-finite values (stat_cor).Warning: Removed 1 rows containing non-finite values (stat_density2d).Warning: Removed 1 rows containing missing values (geom_point).
BothResBothInfoDomEH$numPAS=as.numeric(as.character(BothResBothInfoDomEH$numPAS))
ggplot(BothResBothInfoDomEH,aes(x=human_EH,y= chimp_EH ,col=numPAS)) + geom_point(alpha=.4) + geom_abline(slope=1, intercept = 0) +labs(title="Shannon equitability Colored by number of PAS")Warning: Removed 1 rows containing missing values (geom_point).
summary(lm(BothResBothInfoDomEH$human_EH ~BothResBothInfoDomEH$numPAS))
Call:
lm(formula = BothResBothInfoDomEH$human_EH ~ BothResBothInfoDomEH$numPAS)
Residuals:
Min 1Q Median 3Q Max
-0.90216 -0.07951 0.02225 0.10510 0.32953
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.5905281 0.0044589 132.44 <2e-16 ***
BothResBothInfoDomEH$numPAS 0.0399720 0.0007954 50.25 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.165 on 8448 degrees of freedom
(1 observation deleted due to missingness)
Multiple R-squared: 0.2301, Adjusted R-squared: 0.23
F-statistic: 2525 on 1 and 8448 DF, p-value: < 2.2e-16summary(lm(BothResBothInfoDomEH$chimp_EH ~BothResBothInfoDomEH$numPAS ))
Call:
lm(formula = BothResBothInfoDomEH$chimp_EH ~ BothResBothInfoDomEH$numPAS)
Residuals:
Min 1Q Median 3Q Max
-0.82198 -0.10698 0.02453 0.12846 0.40625
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.5058251 0.0053208 95.06 <2e-16 ***
BothResBothInfoDomEH$numPAS 0.0439612 0.0009492 46.31 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.1969 on 8449 degrees of freedom
Multiple R-squared: 0.2025, Adjusted R-squared: 0.2024
F-statistic: 2145 on 1 and 8449 DF, p-value: < 2.2e-16This normalizes the number of PAS.
Correlation between values:
BothInfoTypes_eh_h=BothResBothInfoDomEH %>% select(gene,numPAS, simpOpp_Human, human_EH) %>% mutate(species="Human") %>% rename(Simpson= simpOpp_Human, ShannonEH=human_EH)
BothInfoTypes_eh_c=BothResBothInfoDomEH %>% select(gene,numPAS, simpOpp_Chimp, chimp_EH) %>% mutate(species="Chimp")%>% rename(Simpson= simpOpp_Chimp, ShannonEH=chimp_EH)
BothInfoTypes_bothEH=BothInfoTypes_eh_h %>% bind_rows(BothInfoTypes_eh_c)
ggplot(BothInfoTypes_bothEH,aes(x=Simpson, y=ShannonEH, by=species, col=species)) +geom_point(alpha=.4) +geom_density2d(col="black") + stat_cor(label.x=0) + geom_smooth(col="black",method = "lm") + facet_grid(~species) + labs(title="Correlation between Indicies") +theme(legend.position = "none")+scale_color_brewer(palette = "Set1")Warning: Removed 1 rows containing non-finite values (stat_density2d).Warning: Removed 1 rows containing non-finite values (stat_cor).Warning: Removed 1 rows containing non-finite values (stat_smooth).Warning: Removed 1 rows containing missing values (geom_point).
Look at it with dominance:
ggplot(BothResBothInfoDomEH,aes(x=human_EH,y= chimp_EH, col=Dom )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + scale_color_brewer(palette = "Set2") + labs(x="Human equitability", y="Chimp equitability", title="Shannon equitability colored by whether gene has a dominant PAS")Warning: Removed 1 rows containing missing values (geom_point).
ggplot(BothResBothInfoDomEH,aes(x=human_EH,y= chimp_EH, col=Set )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + scale_color_brewer(palette = "Set2") +geom_density2d()+ labs(x="Human equitability", y="Chimp equitability", title="Shannon equitability colored by Dominance Structure")Warning: Removed 1 rows containing non-finite values (stat_density2d).
Warning: Removed 1 rows containing missing values (geom_point).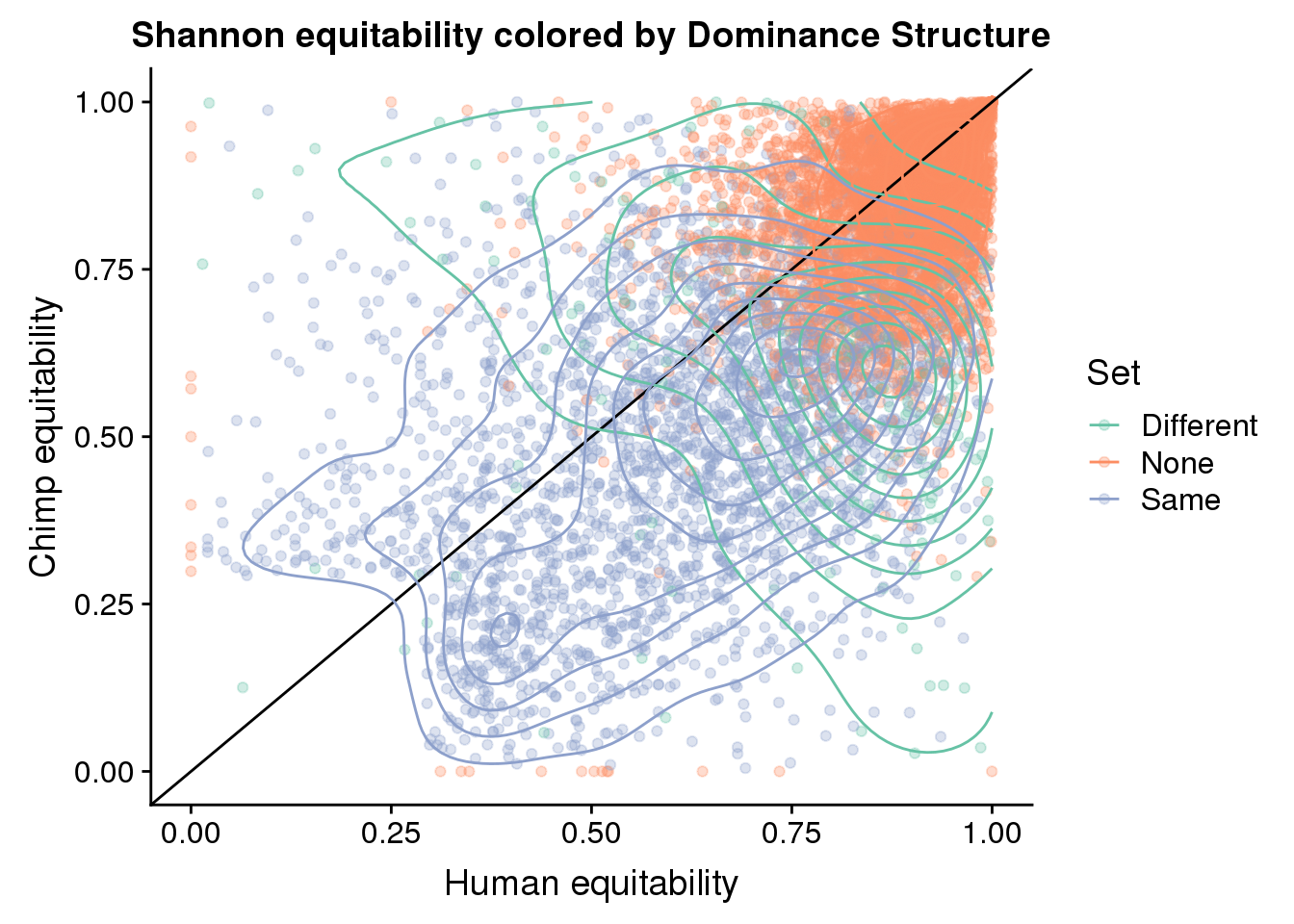
BothResBothInfoDomEH$numPAS=as.factor(BothResBothInfoDomEH$numPAS)
ggplot(BothResBothInfoDomEH,aes(x=human_EH,y= chimp_EH, col=numPAS )) + geom_point(alpha=.3) + geom_abline(slope=1, intercept = 0) + labs(x="Human equitability", y="Chimp equitability", title="Shannon Equitability colored by number of PAS ") + facet_grid(~Dom)Warning: Removed 1 rows containing missing values (geom_point).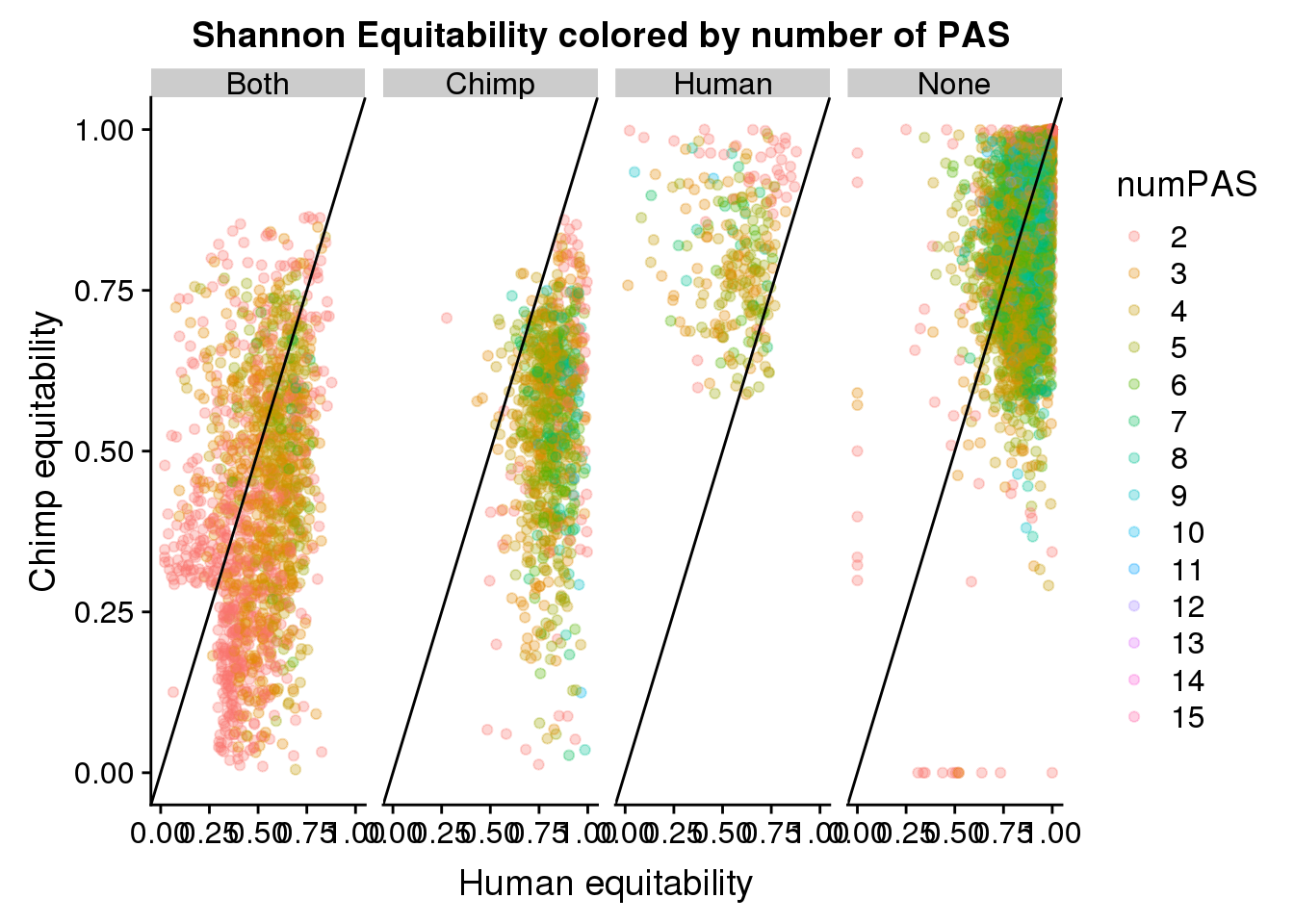
plot simpson h/c colors:
simpsonind=ggplot(BothOppSimp_g, aes(x=SimpsonOpp, fill=species)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Dark2",labels=c("Chimp", "Human"))+labs(title="Simpson Index to measure isoform diversity", x="Simpson Index")+ theme_classic2()
simpsonind
| Version | Author | Date |
|---|---|---|
| cb0a024 | brimittleman | 2020-04-30 |
Plot only 1 color to demonstrate:
ggplot(BothOppSimp_g, aes(x=SimpsonOpp )) + geom_density(fill="grey") +labs(title="Simpson Index", x="Simpson Index")+ theme_classic2()
| Version | Author | Date |
|---|---|---|
| 10590ea | brimittleman | 2020-05-07 |
Plot number of PAS and info content to use:
ggplot(BothInfoTypes,aes(x=simpOpp_Human,y= simpOpp_Chimp ,col=numPAS)) + geom_point(alpha=.4) + geom_abline(slope=1, intercept = 0)+labs(title="Simpson Index Colored by number of PAS", x="Human", y="Chimp") + theme_classic()
| Version | Author | Date |
|---|---|---|
| 33d6feb | brimittleman | 2020-05-07 |
Plot shannon with HC colors
ggplot(BothResInfo_2, aes(x=base2, fill=species)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Dark2",labels=c("Chimp", "Human"))+labs(title="Shannon Index to measure isoform diversity", x="Shannon Index")+ theme_classic2()Warning: Removed 1 rows containing non-finite values (stat_density).
| Version | Author | Date |
|---|---|---|
| 6a947a5 | brimittleman | 2020-05-12 |
shannonPlot=ggplot(BothResInfo_2, aes(x=base2, fill=species)) + geom_density(alpha=.5) + scale_fill_brewer(palette = "Dark2",labels=c("Chimp", "Human"))+labs(title="Shannon Index to measure isoform diversity", x="Shannon Index")+ theme_classic2()
shannonPlotWarning: Removed 1 rows containing non-finite values (stat_density).
| Version | Author | Date |
|---|---|---|
| 6a947a5 | brimittleman | 2020-05-12 |
pdf of figures
ggplot(BothInfoTypes,aes(x=Human_Base2,y= Chimp_Base2 ,col=numPAS)) + geom_point(alpha=.4) + geom_abline(slope=1, intercept = 0)+labs(title="Shannon Index Colored by number of PAS", x="Human", y="Chimp") + theme_classic()Warning: Removed 1 rows containing missing values (geom_point).
| Version | Author | Date |
|---|---|---|
| 6a947a5 | brimittleman | 2020-05-12 |
Plot number of PAS by index:
ggplot(BothInfoTypes,aes(x=numPAS,y= Chimp_Base2 )) + geom_point(alpha=.4) + stat_cor()+ theme_classic()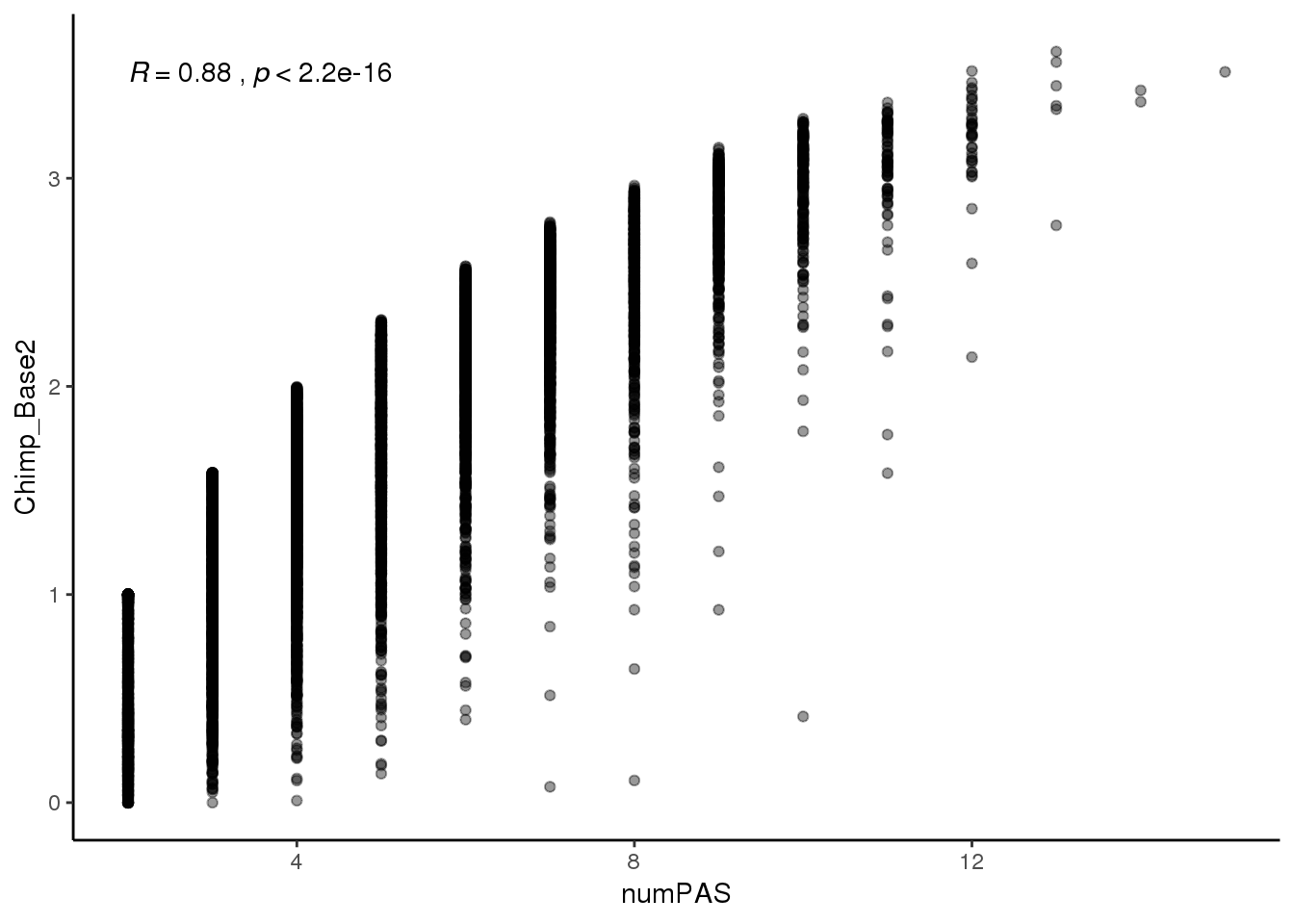
| Version | Author | Date |
|---|---|---|
| 6a947a5 | brimittleman | 2020-05-12 |
ggplot(BothInfoTypes,aes(x=numPAS,y= Human_Base2 )) + geom_point(alpha=.4) + stat_cor()+ theme_classic()Warning: Removed 1 rows containing non-finite values (stat_cor).Warning: Removed 1 rows containing missing values (geom_point).
BothInfoTypesShanG= BothInfoTypes %>% select(gene, numPAS,Human_Base2,Chimp_Base2 ) %>% rename(Human=Human_Base2, Chimp=Chimp_Base2) %>% gather("Species", "value", -gene, -numPAS)
shanoNum=ggplot(BothInfoTypesShanG,aes(x=numPAS,y= value ,col=Species)) + geom_point(alpha=.4) + stat_cor(col="black",label.y.npc="bottom")+ theme_classic() + facet_grid(~Species) + scale_color_brewer(palette = "Dark2") + labs(y="Shannon Information Content", title="Shannon Information Content and PAS number", x= "number of PAS in gene")
shanoNumWarning: Removed 1 rows containing non-finite values (stat_cor).Warning: Removed 1 rows containing missing values (geom_point).
| Version | Author | Date |
|---|---|---|
| 6a947a5 | brimittleman | 2020-05-12 |
BothInfoTypesSimpG= BothInfoTypes %>% select(gene, numPAS,simpOpp_Human,simpOpp_Chimp ) %>% rename(Human=simpOpp_Human, Chimp=simpOpp_Chimp) %>% gather("Species", "value", -gene, -numPAS)
simpnum=ggplot(BothInfoTypesSimpG,aes(x=numPAS,y= value ,col=Species)) + geom_point(alpha=.4) + stat_cor(col="black",label.y.npc="bottom")+ theme_classic() + facet_grid(~Species) + scale_color_brewer(palette = "Dark2")+ labs(y="Simpson Diversity", title="Simpson Diversity and PAS number", x= "number of PAS in gene")
simpnum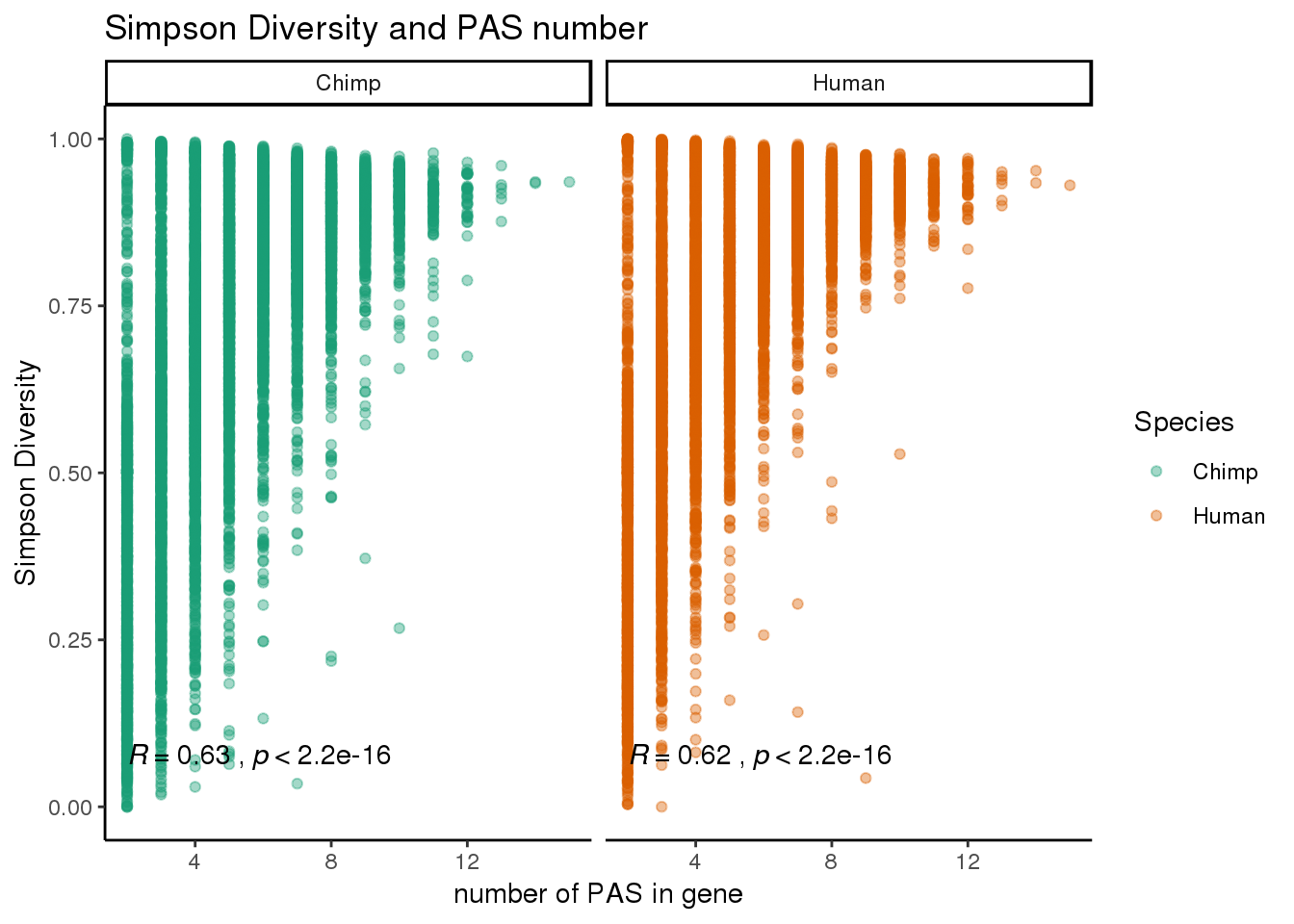
| Version | Author | Date |
|---|---|---|
| 6a947a5 | brimittleman | 2020-05-12 |
plot_grid(shanoNum,simpnum, nrow=2)Warning: Removed 1 rows containing non-finite values (stat_cor).Warning: Removed 1 rows containing missing values (geom_point).
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] vegan_2.5-3 lattice_0.20-38 permute_0.9-4 cowplot_0.9.4
[5] workflowr_1.6.0 ggpubr_0.2 magrittr_1.5 forcats_0.3.0
[9] stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1
[13] tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.4.6 lubridate_1.7.4 assertthat_0.2.0
[4] rprojroot_1.3-2 digest_0.6.18 utf8_1.1.4
[7] R6_2.3.0 cellranger_1.1.0 plyr_1.8.4
[10] backports_1.1.2 evaluate_0.12 httr_1.3.1
[13] pillar_1.3.1 rlang_0.4.0 lazyeval_0.2.1
[16] readxl_1.1.0 rstudioapi_0.10 whisker_0.3-2
[19] Matrix_1.2-15 reticulate_1.10 rmarkdown_1.10
[22] labeling_0.3 munsell_0.5.0 broom_0.5.1
[25] compiler_3.5.1 httpuv_1.4.5 modelr_0.1.2
[28] pkgconfig_2.0.2 mgcv_1.8-25 htmltools_0.3.6
[31] tidyselect_0.2.5 fansi_0.4.0 crayon_1.3.4
[34] withr_2.1.2 later_0.7.5 MASS_7.3-51.1
[37] grid_3.5.1 nlme_3.1-137 jsonlite_1.6
[40] gtable_0.2.0 git2r_0.26.1 scales_1.0.0
[43] cli_1.1.0 stringi_1.2.4 reshape2_1.4.3
[46] fs_1.3.1 promises_1.0.1 xml2_1.2.0
[49] generics_0.0.2 RColorBrewer_1.1-2 tools_3.5.1
[52] glue_1.3.0 hms_0.4.2 parallel_3.5.1
[55] yaml_2.2.0 colorspace_1.3-2 cluster_2.0.7-1
[58] rvest_0.3.2 knitr_1.20 haven_1.1.2