Categories from Khan
Briana Mittleman
3/5/2020
Last updated: 2020-03-06
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ClassifyLeafviz.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OverlapBenchmark/
Untracked: data/PAS/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalHvC/
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/ExploredAPA_DF.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/SpliceSiteStrength.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/dAPA_Conservation.Rmd
Modified: analysis/dAPAandapaQTL_DF.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/speciesSpecific.Rmd
Modified: analysis/speciesSpecific_DF.Rmd
Modified: analysis/upsetter_DF.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | be1ed38 | brimittleman | 2020-03-06 | add exampels |
| html | 8d7a76e | brimittleman | 2020-03-06 | Build site. |
| Rmd | 8400ab6 | brimittleman | 2020-03-06 | add enrichment results |
| html | a1fd883 | brimittleman | 2020-03-05 | Build site. |
| Rmd | a71e15c | brimittleman | 2020-03-05 | wflow_publish(c(“analysis/index.Rmd”, “analysis/DirSelectionKhan.Rmd”)) |
I will use the directional selection categories from Khan et al.
library(tidyverse)── Attaching packages ─────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()Model from Khan et al.
model.num.rna: : 1 = mRNA expression level pattern consistent with directional selection along human lineage, 2 = mRNA expression level pattern consistent with directional selection along chimpanzee lineage, 3 = undetermined pattern, 4 = patterns with no significant difference between mean expression levels; 5 = evidence for relaxation of constraint along human lineage, 6 = evidence of relaxation of constraint along chimpanzee lineage
model.num.protein: 1 = protein expression level pattern consistent with directional selection along human lineage, 2 = protein expression level pattern consistent with directional selection along chimpanzee lineage, 3 = undetermined pattern, 4 = patterns with no significant difference between mean expression levels; 5 = evidence for relaxation of constraint along human lineage, 6 = evidence of relaxation of constraint along chimpanzee lineage
KhanData=read.csv("../data/Khan_prot/Khan_TableS4.csv",stringsAsFactors = F) %>% select(gene.symbol,contains("model") ) %>% rename("gene"=gene.symbol, "Protein"=model.num.protein, "RNA"=model.num.rna)
KhanData_g=KhanData %>% gather("Set", "Model", -gene)
KhanData_g$Model= as.factor(KhanData_g$Model)ggplot(KhanData_g, aes(x=Model, by=Set, fill=Set)) +geom_bar(stat="count",position = "dodge")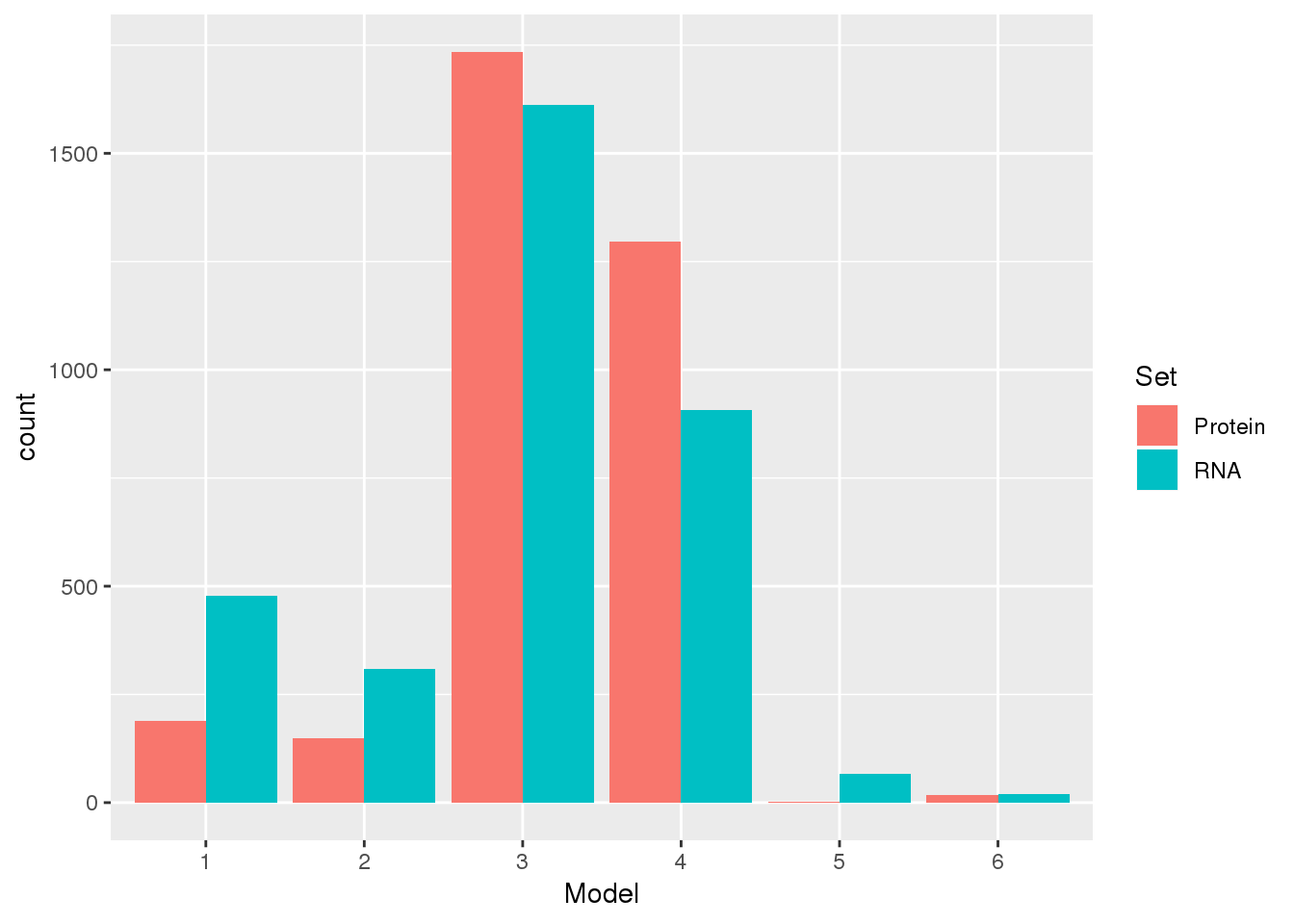
| Version | Author | Date |
|---|---|---|
| a1fd883 | brimittleman | 2020-03-05 |
- directional human
- directional in chimp 3, undetermined
- no mean difference
- relaxed in human
- related in chimp
Proportion
KhanData_group=KhanData_g %>% group_by(Set, Model) %>% summarise(Nset=n(), Prortion=Nset/nrow(KhanData))
ggplot(KhanData_group, aes(x=Model, by=Set, fill=Set, y=Prortion)) +geom_bar(stat="identity",position = "dodge")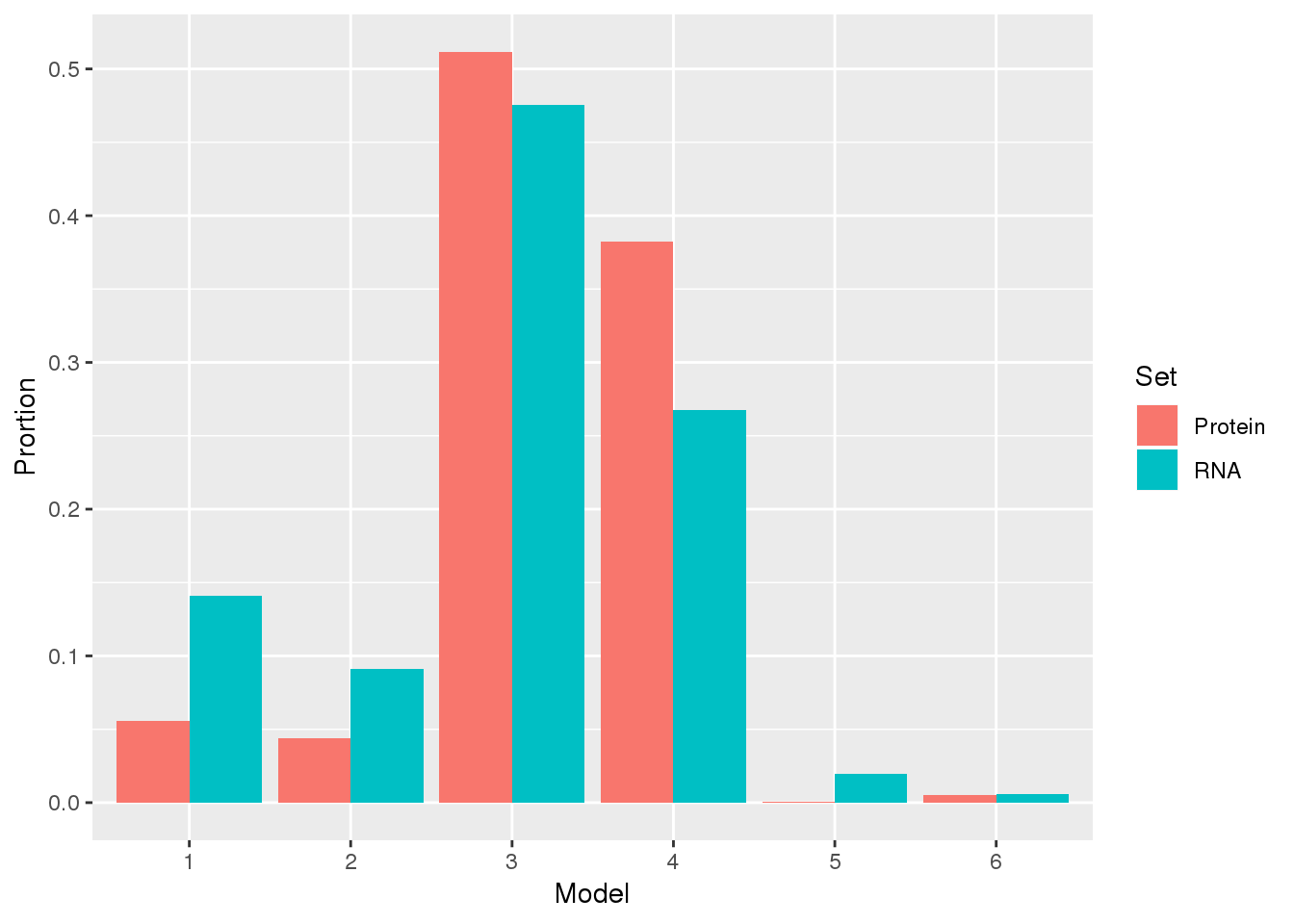
| Version | Author | Date |
|---|---|---|
| a1fd883 | brimittleman | 2020-03-05 |
This is their results. I will overlap this with the genes I found differences in.
DiffIso=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T, stringsAsFactors = F) %>% inner_join(KhanData, by="gene")
nrow(DiffIso)[1] 11245nrow(DiffIso %>% select(gene) %>% unique())[1] 2495We have data for 11,203 PAS in 2,485 genes.
I want to get this down to gene level
GenewDiffIso= DiffIso %>% group_by(gene,SigPAU2) %>% summarise(nEach=n()) %>% filter(SigPAU2=="Yes")
KhanData_withAPAinfo= KhanData %>% mutate(dAPA=ifelse(gene %in%GenewDiffIso$gene, "Yes", "No" ))Plot the proportion with a dAPA in each
KhanData_gwAPA=KhanData_withAPAinfo %>% gather("Set", "Model", -gene, -dAPA)
KhanData_gwAPA$Model= as.factor(KhanData_gwAPA$Model)
KhanData_gwAPA$Set=factor(KhanData_gwAPA$Set, levels=c("RNA", "Protein"))
ggplot(KhanData_gwAPA, aes(x=Model, by=dAPA, fill=dAPA)) +geom_bar(stat="count", position = "stack") + facet_grid(~Set) + scale_fill_brewer(palette = "Dark2") + labs(y="Number of Genes") + scale_x_discrete( labels=c("Selection Human","Selection Chimp","Undetermined","No mean difference","Relaxation in Human","Relaxation in Chimp"))+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16))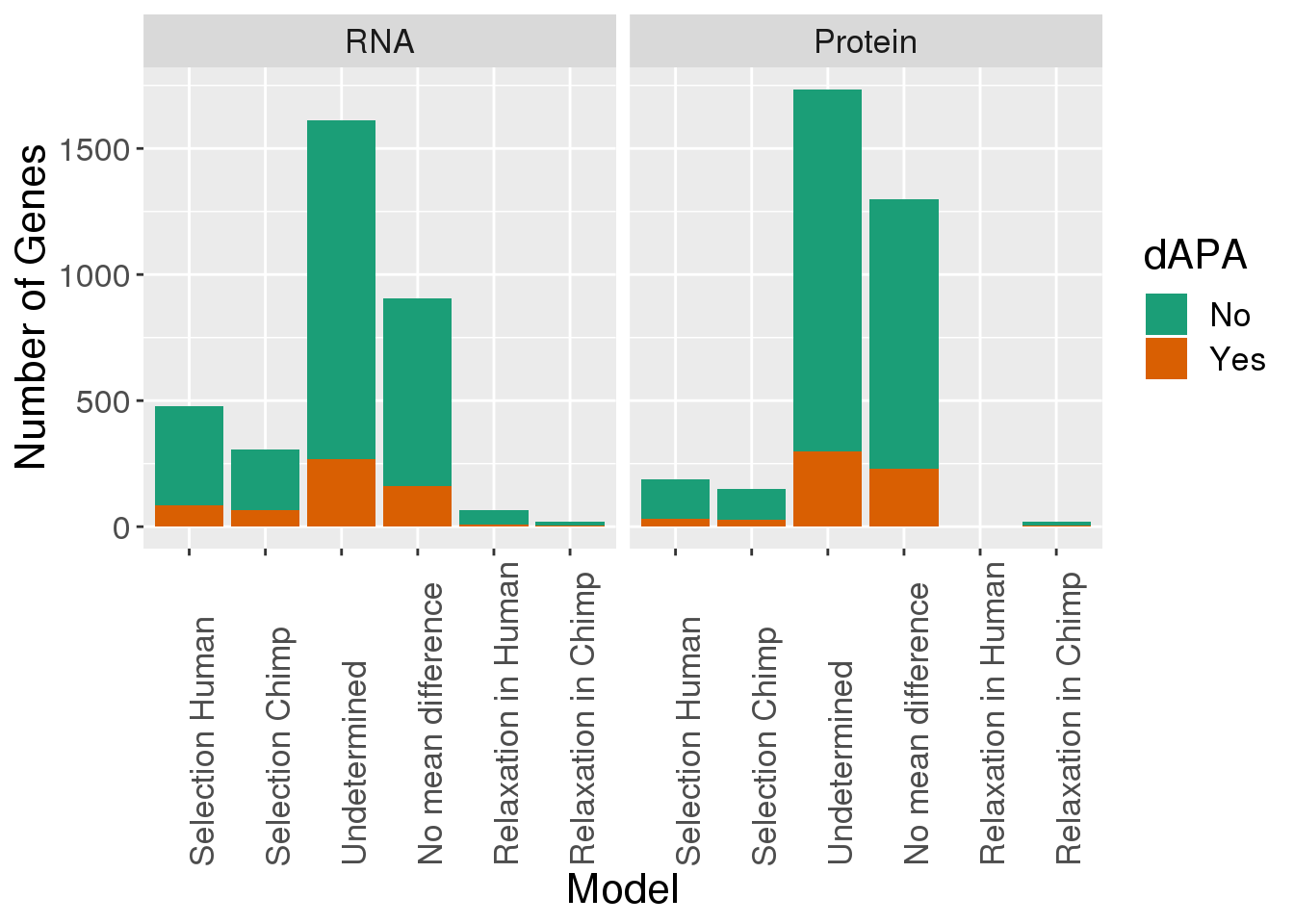
Look at the genes with directional selection:
DirSelectionRNA=KhanData_gwAPA %>% filter(Set=="RNA", Model %in% c(1,2))
DirSelectionRNA %>% group_by(Model,dAPA) %>% summarise(n())# A tibble: 4 x 3
# Groups: Model [2]
Model dAPA `n()`
<fct> <chr> <int>
1 1 No 394
2 1 Yes 84
3 2 No 243
4 2 Yes 65DirSelectionProt=KhanData_gwAPA %>% filter(Set=="Protein", Model %in% c(1,2))
DirSelectionProt %>% group_by(Model,dAPA) %>% summarise(n())# A tibble: 4 x 3
# Groups: Model [2]
Model dAPA `n()`
<fct> <chr> <int>
1 1 No 157
2 1 Yes 32
3 2 No 121
4 2 Yes 28RNA -84 genes directional to human
-64 genes directional to chimp
Protein
-32 genes directional in human
-27 genes directional in chimp
Relaxed selection:
RelSelectionRNA=KhanData_gwAPA %>% filter(Set=="RNA", Model %in% c(5,6))
RelSelectionRNA %>% group_by(Model,dAPA) %>% summarise(n())# A tibble: 4 x 3
# Groups: Model [2]
Model dAPA `n()`
<fct> <chr> <int>
1 5 No 57
2 5 Yes 9
3 6 No 14
4 6 Yes 5RelSelectionProt=KhanData_gwAPA %>% filter(Set=="Protein", Model %in% c(5,6))
RelSelectionProt %>% group_by(Model,dAPA) %>% summarise(n())# A tibble: 3 x 3
# Groups: Model [2]
Model dAPA `n()`
<fct> <chr> <int>
1 5 No 2
2 6 No 12
3 6 Yes 69/57, 5/13
RNA -9 genes relax to human (9/66) 13% -5 genes relax to chimp (5/18) 27%
Protein
-0 genes relax in human
-6 genes relax in chimp
Enrichement:
Write a loop that gets the pvalue and enrichment for each of these:
Model=seq(1,6)
EnrichmentRNA=c()
PvalueRNA=c()
for (i in seq(1:6)){
x=nrow(KhanData_gwAPA %>% filter(Set=="RNA", dAPA=="Yes", Model==i))
m=nrow(KhanData_gwAPA %>% filter(Set=="RNA", Model==i))
n=nrow(KhanData_gwAPA %>% filter(Set=="RNA", Model!=i))
k=nrow(KhanData_gwAPA %>% filter(Set=="RNA", dAPA=="Yes"))
N=nrow(KhanData_gwAPA %>% filter(Set=="RNA"))
PvalueRNA=c(PvalueRNA, phyper(x,m,n,k,lower.tail=F))
enrich=(x/k)/(m/N)
EnrichmentRNA=c(EnrichmentRNA, enrich)
}
EnrichProt=c()
PvalueProt=c()
for (i in seq(1:6)){
x=nrow(KhanData_gwAPA %>% filter(Set=="Protein", dAPA=="Yes", Model==i))
m=nrow(KhanData_gwAPA %>% filter(Set=="Protein", Model==i))
n=nrow(KhanData_gwAPA %>% filter(Set=="Protein", Model!=i))
k=nrow(KhanData_gwAPA %>% filter(Set=="Protein", dAPA=="Yes"))
N=nrow(KhanData_gwAPA %>% filter(Set=="Protein"))
PvalueProt=c(PvalueProt, phyper(x,m,n,k,lower.tail=F))
enrich=(x/k)/(m/N)
EnrichProt=c(EnrichProt, enrich)
}EnrichmentResults=as.data.frame(cbind(Model, EnrichmentRNA,EnrichProt,PvalueRNA, PvalueProt))
EnrichmentG= EnrichmentResults %>% select(Model, EnrichmentRNA, EnrichProt) %>% rename("RNA"=EnrichmentRNA, "Protein"=EnrichProt) %>% gather("Set", "Enrichment", -Model)
PvalG= EnrichmentResults %>% select(Model, PvalueRNA, PvalueProt) %>% rename("RNA"=PvalueRNA, "Protein"=PvalueProt) %>% gather("Set", "Pvalue", -Model)
Alldata=EnrichmentG %>% inner_join(PvalG, by=c("Model","Set"))
Alldata$Set=factor(Alldata$Set, levels=c("RNA", "Protein"))
Alldata$Model=factor(Alldata$Model)
ggplot(Alldata,aes(x=Model, y=Enrichment,fill=Set)) + geom_bar(stat = "identity") + geom_hline(yintercept = 1) + geom_text(aes(label=round(Pvalue,2), vjust=0))+ facet_grid(~Set) + scale_fill_brewer(palette = "Dark2") + scale_x_discrete( labels=c("Selection Human","Selection Chimp","Undetermined","No mean difference","Relaxation in Human","Relaxation in Chimp"))+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16)) +labs(title="Enrichment of dAPA genes in directional selection sets")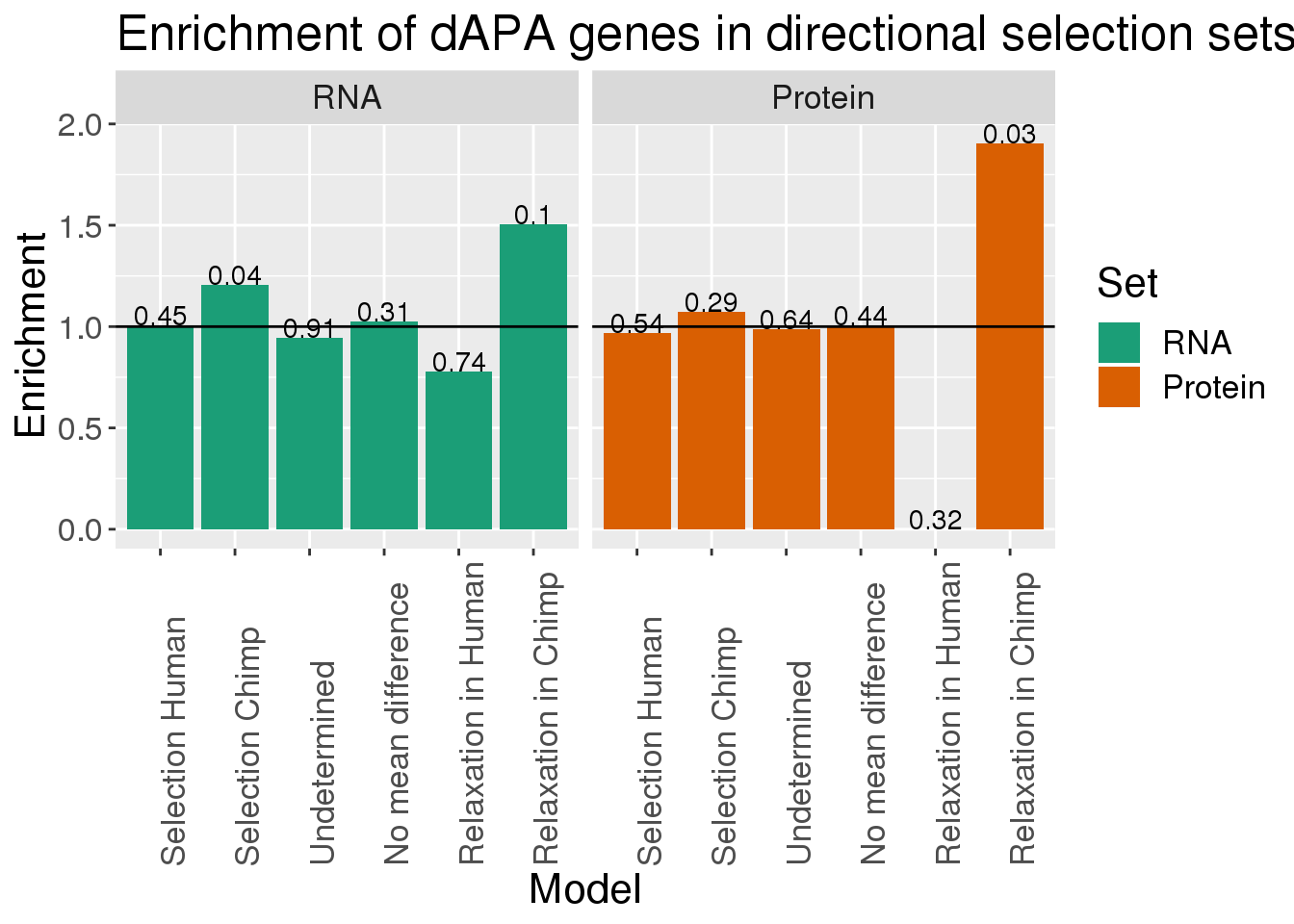
| Version | Author | Date |
|---|---|---|
| 8d7a76e | brimittleman | 2020-03-06 |
I need a way to relate this to something about APA
Examples:
Look at all of the relaxed
RelSelectionRNA %>% filter(dAPA=="Yes", Model=="6") gene dAPA Set Model
1 LIMA1 Yes RNA 6
2 CUL1 Yes RNA 6
3 APBB1IP Yes RNA 6
4 PTCD3 Yes RNA 6
5 STOM Yes RNA 6RelSelectionProt%>% filter(dAPA=="Yes", Model=="6") gene dAPA Set Model
1 PTBP3 Yes Protein 6
2 IRF5 Yes Protein 6
3 FABP5 Yes Protein 6
4 OTUB1 Yes Protein 6
5 ECI1 Yes Protein 6
6 LYRM7 Yes Protein 6IRF5 OTUB1
RelSelectionRNA %>% filter(dAPA=="Yes", Model=="5") gene dAPA Set Model
1 PYGB Yes RNA 5
2 EIF4H Yes RNA 5
3 MTR Yes RNA 5
4 PTBP3 Yes RNA 5
5 SNRNP27 Yes RNA 5
6 STX17 Yes RNA 5
7 ADPGK Yes RNA 5
8 LIMS1 Yes RNA 5
9 IARS Yes RNA 5DirSelectionRNA %>% filter(dAPA=="Yes", Model=="2") gene dAPA Set Model
1 RECQL Yes RNA 2
2 PPP5C Yes RNA 2
3 SMARCD1 Yes RNA 2
4 SEL1L Yes RNA 2
5 DYNC1I2 Yes RNA 2
6 EPS15 Yes RNA 2
7 TMED2 Yes RNA 2
8 GOLGA3 Yes RNA 2
9 GLG1 Yes RNA 2
10 CDC23 Yes RNA 2
11 ASCC2 Yes RNA 2
12 PSMC1 Yes RNA 2
13 ADNP Yes RNA 2
14 INTS6 Yes RNA 2
15 ZFAND1 Yes RNA 2
16 GOSR2 Yes RNA 2
17 DDX6 Yes RNA 2
18 RAB5B Yes RNA 2
19 HDDC2 Yes RNA 2
20 RPL22 Yes RNA 2
21 CAPZA1 Yes RNA 2
22 ARHGEF2 Yes RNA 2
23 CBX3 Yes RNA 2
24 PLCG1 Yes RNA 2
25 TUBGCP3 Yes RNA 2
26 IRF3 Yes RNA 2
27 HERC2 Yes RNA 2
28 DUT Yes RNA 2
29 ELP3 Yes RNA 2
30 SYNCRIP Yes RNA 2
31 SCYL2 Yes RNA 2
32 FLNB Yes RNA 2
33 RAC1 Yes RNA 2
34 RB1 Yes RNA 2
35 SORD Yes RNA 2
36 IRF8 Yes RNA 2
37 SERBP1 Yes RNA 2
38 TPM3 Yes RNA 2
39 RPL7L1 Yes RNA 2
40 ARHGAP18 Yes RNA 2
41 DGKE Yes RNA 2
42 CC2D1B Yes RNA 2
43 SAMSN1 Yes RNA 2
44 UBE2Z Yes RNA 2
45 NCSTN Yes RNA 2
46 UTP15 Yes RNA 2
47 GFM2 Yes RNA 2
48 RPL13 Yes RNA 2
49 STX18 Yes RNA 2
50 POLR1C Yes RNA 2
51 BCL2L1 Yes RNA 2
52 DCAKD Yes RNA 2
53 DENND4A Yes RNA 2
54 TMEM70 Yes RNA 2
55 FAM91A1 Yes RNA 2
56 AP3S1 Yes RNA 2
57 SNRPE Yes RNA 2
58 ADI1 Yes RNA 2
59 UBE2L3 Yes RNA 2
60 MORF4L1 Yes RNA 2
61 MYO6 Yes RNA 2
62 MRPL42 Yes RNA 2
63 ANKRD28 Yes RNA 2
64 HEXA Yes RNA 2
65 SEC22B Yes RNA 2DirSelectionProt %>% filter(dAPA=="Yes", Model=="2") gene dAPA Set Model
1 CUL1 Yes Protein 2
2 GNAI3 Yes Protein 2
3 DYNC1I2 Yes Protein 2
4 CPOX Yes Protein 2
5 SAMM50 Yes Protein 2
6 PYGB Yes Protein 2
7 EIF4H Yes Protein 2
8 RAB5B Yes Protein 2
9 TRIM38 Yes Protein 2
10 KYNU Yes Protein 2
11 WDR77 Yes Protein 2
12 DUT Yes Protein 2
13 YARS2 Yes Protein 2
14 GALNT2 Yes Protein 2
15 CCT6A Yes Protein 2
16 CCT5 Yes Protein 2
17 DGKE Yes Protein 2
18 CC2D1B Yes Protein 2
19 TTC9C Yes Protein 2
20 UBLCP1 Yes Protein 2
21 NFU1 Yes Protein 2
22 MFN1 Yes Protein 2
23 PGAM1 Yes Protein 2
24 BCL2L1 Yes Protein 2
25 DCAKD Yes Protein 2
26 MYO6 Yes Protein 2
27 PRIM1 Yes Protein 2
28 SEC22B Yes Protein 2
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.3.0 stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2
[5] readr_1.3.1 tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1
[9] tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 reshape2_1.4.3 haven_1.1.2
[4] lattice_0.20-38 colorspace_1.3-2 generics_0.0.2
[7] htmltools_0.3.6 yaml_2.2.0 utf8_1.1.4
[10] rlang_0.4.0 later_0.7.5 pillar_1.3.1
[13] glue_1.3.0 withr_2.1.2 RColorBrewer_1.1-2
[16] modelr_0.1.2 readxl_1.1.0 plyr_1.8.4
[19] munsell_0.5.0 gtable_0.2.0 workflowr_1.6.0
[22] cellranger_1.1.0 rvest_0.3.2 evaluate_0.12
[25] labeling_0.3 knitr_1.20 httpuv_1.4.5
[28] fansi_0.4.0 broom_0.5.1 Rcpp_1.0.2
[31] promises_1.0.1 scales_1.0.0 backports_1.1.2
[34] jsonlite_1.6 fs_1.3.1 hms_0.4.2
[37] digest_0.6.18 stringi_1.2.4 grid_3.5.1
[40] rprojroot_1.3-2 cli_1.1.0 tools_3.5.1
[43] magrittr_1.5 lazyeval_0.2.1 crayon_1.3.4
[46] whisker_0.3-2 pkgconfig_2.0.2 xml2_1.2.0
[49] lubridate_1.7.4 assertthat_0.2.0 rmarkdown_1.10
[52] httr_1.3.1 rstudioapi_0.10 R6_2.3.0
[55] nlme_3.1-137 git2r_0.26.1 compiler_3.5.1