Remove genes with unlisted sites- check robustness
Briana Mittleman
6/4/2020
Last updated: 2020-06-09
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/TrialFiltersMeta.txt.sb-9845453e-R58Y0Q/
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-284518db-RGf0kd/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/AREstabilityScores.Rmd
Untracked: analysis/AllLoc_effectSizeCor.Rmd
Untracked: analysis/Conservation_bydAPAset.Rmd
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/HandC.TvN
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/ResultsNoUnlifted.md
Untracked: analysis/annotationBias.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: code/._AlignmentScores.sh
Untracked: code/._BothFCMM.sh
Untracked: code/._BothFCMMPrim.sh
Untracked: code/._BothFCnewOInclusive.sh
Untracked: code/._ChimpStarMM2.sh
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._ClosestorthoEx.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._Filter255MM.sh
Untracked: code/._FilterPrimSec.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._MismatchNumbers.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._SAF2Bed.py
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._StarMM2.sh
Untracked: code/._TestFC.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2Bedbothstrand.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chimpMultiCov.sh
Untracked: code/._chimpMultiCov255.sh
Untracked: code/._chimpMultiCovInclusive.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopGeneral.ph
Untracked: code/._extractPhylopGeneral.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._filterPrimaryread.py
Untracked: code/._filterSecondaryread.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixFCheadforExp.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._humanMultiCov.sh
Untracked: code/._humanMultiCov255.sh
Untracked: code/._humanMultiCov_inclusive.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapMMandOrthoexon.sh
Untracked: code/._overlapPASandOrthoexon.sh
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantLiftedPASPrimary.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ALLPAS_sequenceDF.err
Untracked: code/ALLPAS_sequenceDF.out
Untracked: code/AlignmentScores.err
Untracked: code/AlignmentScores.out
Untracked: code/AlignmentScores.sh
Untracked: code/BothFCMM.err
Untracked: code/BothFCMM.out
Untracked: code/BothFCMM.sh
Untracked: code/BothFCMMPrim.err
Untracked: code/BothFCMMPrim.out
Untracked: code/BothFCMMPrim.sh
Untracked: code/BothFCnewOInclusive.sh
Untracked: code/BothFCnewOInclusive.sh.err
Untracked: code/BothFCnewOInclusive.sh.out
Untracked: code/ChimpStarMM2.err
Untracked: code/ChimpStarMM2.out
Untracked: code/ChimpStarMM2.sh
Untracked: code/ClassifyLeafviz.sh
Untracked: code/ClosestorthoEx.err
Untracked: code/ClosestorthoEx.out
Untracked: code/ClosestorthoEx.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DTUTR.sh
Untracked: code/DiffDom_RNAmotif_4.err
Untracked: code/DiffDom_RNAmotif_4.out
Untracked: code/DiffDom_RNAmotif_4.sh
Untracked: code/DiffDom_RNAmotif_4_splitDE.err
Untracked: code/DiffDom_RNAmotif_4_splitDE.out
Untracked: code/DiffDom_RNAmotif_4_splitDE.sh
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/Filter255.err
Untracked: code/Filter255.out
Untracked: code/Filter255MM.sh
Untracked: code/FilterPrimSec.err
Untracked: code/FilterPrimSec.out
Untracked: code/FilterPrimSec.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindDomXCutoff.py
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/GetTopminus2Usage.py
Untracked: code/H3K36me3DTplot.err
Untracked: code/H3K36me3DTplot.out
Untracked: code/H3K36me3DTplot.sh
Untracked: code/H3K36me3DTplot_DiffIso.err
Untracked: code/H3K36me3DTplot_DiffIso.out
Untracked: code/H3K36me3DTplot_DiffIso.sh
Untracked: code/H3K36me3DTplot_Specific.err
Untracked: code/H3K36me3DTplot_Specific.out
Untracked: code/H3K36me3DTplot_Specific.sh
Untracked: code/H3K36me3DTplot_distalPAS.err
Untracked: code/H3K36me3DTplot_distalPAS.out
Untracked: code/H3K36me3DTplot_distalPAS.sh
Untracked: code/H3K36me3DTplot_transcript.err
Untracked: code/H3K36me3DTplot_transcript.out
Untracked: code/H3K36me3DTplot_transcript.sh
Untracked: code/H3K36me3DTplotwide.err
Untracked: code/H3K36me3DTplotwide.out
Untracked: code/H3K36me3DTplotwide.sh
Untracked: code/H3K9me3DTplot_transcript.err
Untracked: code/H3K9me3DTplot_transcript.out
Untracked: code/H3K9me3DTplot_transcript.sh
Untracked: code/H3K9me3_processandDT.sh
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/InfoContentShannon.py
Untracked: code/InfoContentbyInd.py
Untracked: code/IntersectMMandOrtho.err
Untracked: code/IntersectMMandOrtho.out
Untracked: code/IntersectPASandOrtho.err
Untracked: code/IntersectPASandOrtho.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/MismatchNumbers.err
Untracked: code/MismatchNumbers.out
Untracked: code/MismatchNumbers.sh
Untracked: code/NuclearDTUTR.err
Untracked: code/NuclearDTUTRt.out
Untracked: code/NuclearPlotsDEandDiffDom_4.err
Untracked: code/NuclearPlotsDEandDiffDom_4.out
Untracked: code/NuclearPlotsDEandDiffDom_4.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/PlotNuclearUsagebySpecies_DF_4DIC.R
Untracked: code/PlotNuclearUsagebySpecies_DF_DEout.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/RNAmotif_PAS.err
Untracked: code/RNAmotif_PAS.out
Untracked: code/RNAmotif_PAS.sh
Untracked: code/RNAmotif_PAS_chimp.err
Untracked: code/RNAmotif_PAS_chimp.out
Untracked: code/RNAmotif_PAS_chimp.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunNewDom.err
Untracked: code/RunNewDom.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/SAF2Bed.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/StarMM2.err
Untracked: code/StarMM2.out
Untracked: code/StarMM2.sh
Untracked: code/TestFC.err
Untracked: code/TestFC.out
Untracked: code/TestFC.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/UTR2FASTA.py
Untracked: code/Upstream10Bases_general.py
Untracked: code/allPASSeq_df.sh
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/apaQTLsnakefiltPAS.err
Untracked: code/apaQTLsnakefiltPAS.out
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2Bedbothstrand.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chimpMultiCov.err
Untracked: code/chimpMultiCov.out
Untracked: code/chimpMultiCov.sh
Untracked: code/chimpMultiCov255.sh
Untracked: code/chimpMultiCovInclusive.err
Untracked: code/chimpMultiCovInclusive.out
Untracked: code/chimpMultiCovInclusive.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhaastConGeneral.py
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopGeneral.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterPrimaryread.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSecondaryread.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFCheadforExp.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getAlloverlap.py
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/humanMultiCov.err
Untracked: code/humanMultiCov.out
Untracked: code/humanMultiCov.sh
Untracked: code/humanMultiCov255.err
Untracked: code/humanMultiCov255.out
Untracked: code/humanMultiCov255.sh
Untracked: code/humanMultiCovInclusive.err
Untracked: code/humanMultiCovInclusive.out
Untracked: code/humanMultiCov_inclusive.sh
Untracked: code/infoContentSimpson.py
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftVCF.out
Untracked: code/liftVCF.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/lliftVCF.err
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeDIC.err
Untracked: code/makeDIC.out
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/makedICPlots_DF.sh
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/mergeChimpRNA.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergeandsort_H3K9me3
Untracked: code/mergeandsort_h3k36me3
Untracked: code/mergeandsorth3k36me3.sh
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapMMandOrthoexon.sh
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapPASandOrthoexon.sh
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quantLiftedPASPrimary.err
Untracked: code/quantLiftedPASPrimary.out
Untracked: code/quantLiftedPASPrimary.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runChimpDiffIsoDF.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runHumanDiffIsoDF.sh
Untracked: code/runNewDom.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_dF.err
Untracked: code/run_Humanleafcutter_dF.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePAS_Human.batch
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.batch
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Chimp_tvN_DF.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN_DF.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/test.txt
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/tripseq-analysis/
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._TrialFiltersMeta.txt
Untracked: data/._TrialFiltersMeta.txt.sb-9845453e-R58Y0Q
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/AREelements/
Untracked: data/BaseComp/
Untracked: data/CleanLiftedPeaks_FC_primary/
Untracked: data/CompapaQTLpas/
Untracked: data/DIC_Viz/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffDomandDE_example/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DistTwoDom/
Untracked: data/DomDefGreaterX/
Untracked: data/DomStructure_4/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/DoubleFilterUsageNumeric/
Untracked: data/EvalPantro5/
Untracked: data/H3K36me3/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/HumanMolPheno/
Untracked: data/HumanUTR/
Untracked: data/IndInfoContent/
Untracked: data/InfoContent/
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OrthoExonBed/
Untracked: data/OverlapBenchmark/
Untracked: data/OverlappingPAS/
Untracked: data/PAS/
Untracked: data/PAS_SAF/
Untracked: data/PAS_doubleFilter/
Untracked: data/PTM/
Untracked: data/Peaks_5perc/
Untracked: data/PhastCon/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/Pol2Chip/
Untracked: data/QTLPASoverlap/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/TestAnnoBiasOE/
Untracked: data/TestMM2/
Untracked: data/TestMM2_AS/
Untracked: data/TestMM2_PrimaryRead/
Untracked: data/TestMM2_SeondaryRead/
Untracked: data/TestMM2_mismatch/
Untracked: data/TestMM2_quality/
Untracked: data/TestWithinMergePAS/
Untracked: data/Test_FC_methods/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalFractionPAS/
Untracked: data/TotalHvC/
Untracked: data/TrialFiltersMeta.txt
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/UnliftedSites/
Untracked: data/UrichElements/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/bioGRID/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_extra.txt
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/miRNA/
Untracked: data/multimap/
Untracked: data/orthoUTR/
Untracked: data/paiDecay/
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/testQuant/
Untracked: data/utrDB/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/DEandAPA.txt
Untracked: output/DEandAPA_sig.txt
Untracked: output/DEandTEeffectsize
Untracked: output/DEandTEeffectsize.pdf
Untracked: output/DEeffectsize
Untracked: output/DEeffectsize.pdf
Untracked: output/FigureDF/
Untracked: output/PropSamesdom
Untracked: output/PropSamesdom.pdf
Untracked: output/TEeffectsize
Untracked: output/TEeffectsize.pdf
Untracked: output/Total_DEeffectsize
Untracked: output/Total_DEeffectsize.pdf
Untracked: output/Total_DEeffectsizeNotJusttop.pdf
Untracked: output/Total_DErelationship.pdf
Untracked: output/Total_TEeffectsize
Untracked: output/Total_TEeffectsize.pdf
Untracked: output/Ubiqplot
Untracked: output/Ubiqplot.pdf
Untracked: output/dAPAandDomEnrich.png
Untracked: output/dEandDomEnrich.png
Untracked: output/dediffdom.pdf
Untracked: output/dpnotDE
Untracked: output/dtPlots/
Untracked: output/exandte
Untracked: output/fig1.pdf
Untracked: output/fig2.pdf
Untracked: output/fig3.pdf
Untracked: output/fig4.pdf
Untracked: output/fig5.pdf
Untracked: output/fig6.pdf
Untracked: output/piecharts
Untracked: output/piecharts.pdf
Untracked: output/removeUnilift_fig6.pdf
Untracked: output/removeUnlift_fig3.pdf
Untracked: output/removeUnlift_fig4.pdf
Untracked: output/removeUnlift_fig5.pdf
Untracked: output/simpson.pdf
Untracked: output/whichSiteplot.pdf
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/DeandNumPAS.Rmd
Modified: analysis/DirSelectionKhan.Rmd
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/MMExpreiment.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/PTM_analysis.Rmd
Modified: analysis/SetsdAPADIC.Rmd
Modified: analysis/TotalDomStructure.Rmd
Modified: analysis/TotalVNuclearBothSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/changeMisprimcut.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/dInforContent.Rmd
Modified: analysis/diffExpression.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/incorporateQTLsAncestral.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/mRNADecay.Rmd
Modified: analysis/miRNAanalysis.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/phastCon.Rmd
Modified: analysis/pol2.Rmd
Modified: analysis/speciesSpecific.Rmd
Modified: analysis/unliftedsites.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 1f3d41d | brimittleman | 2020-06-09 | add figure out |
| html | bb8d220 | brimittleman | 2020-06-04 | Build site. |
| Rmd | a276deb | brimittleman | 2020-06-04 | add robust to unlift |
In a previous analysis I found 300 genes that may be affected by unlifted PAS. I will use this analysis to check if my results are consistent if we are conservative and remove these genes.
library(ggpubr)Loading required package: ggplot2Loading required package: magrittrlibrary(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ tibble 2.1.1 ✔ purrr 0.3.2
✔ tidyr 0.8.3 ✔ dplyr 0.8.0.1
✔ readr 1.3.1 ✔ stringr 1.3.1
✔ tibble 2.1.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ tidyr::extract() masks magrittr::extract()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
✖ purrr::set_names() masks magrittr::set_names()library(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggpubr':
get_legendThe following object is masked from 'package:ggplot2':
ggsaveUnfliftGenes=read.table("../data/UnliftedSites/GeneswUnliftedandPassingPAS.txt", header = T,stringsAsFactors = F)-Effect size relationships
-enrichment for DE/TE
-same diff dominant
- p not e
Effect size relationships
Meta=read.table("../data/PAS_doubleFilter/PAS_5perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F) %>% dplyr::select(PAS, chr, start,end, loc)
DiffIso= read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T,stringsAsFactors = F) %>% inner_join(Meta, by=c("chr", 'start','end')) %>% filter(loc %in% c("intron","utr3"))
DiffIsoTop=DiffIso %>% mutate(AvgUsageBoth=(Human+Chimp)/2) %>% group_by(gene) %>% arrange(p.adjust,desc(AvgUsageBoth)) %>% slice(1) %>% ungroup()
nameID=read.table("../../genome_anotation_data/ensemble_to_genename.txt",sep="\t", header = T, stringsAsFactors = F) %>% dplyr::select(Gene_stable_ID, Gene.name)
DE=read.table("../data/DiffExpression/DEtested_allres.txt",stringsAsFactors = F,header = F, col.names = c("Gene_stable_ID" ,"logFC" ,"AveExpr" , "t" , "P.Value" , "adj.P.Val", "B" )) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::rename('gene'=Gene.name) %>% dplyr::select(-Gene_stable_ID) %>% mutate(CorrectedlogFC=-1*logFC)DeandAPA= DiffIsoTop %>% inner_join(DE, by="gene") %>%anti_join(UnfliftGenes,by="gene")DE_all=ggplot(DeandAPA,aes(y=deltaPAU, x=CorrectedlogFC)) + geom_point(alpha=.3) + geom_smooth(method="lm") + labs(title="APA v DE \n Remove 353 unlifted", x="DE log effect size", y="Difference in PAS Usage") + scale_color_brewer(palette = "Set1",name="", labels=c("Intronic", "3' UTR"))+ stat_cor(label.x = -8,label.y = -1) +theme_classic(base_size = 12)
DE_all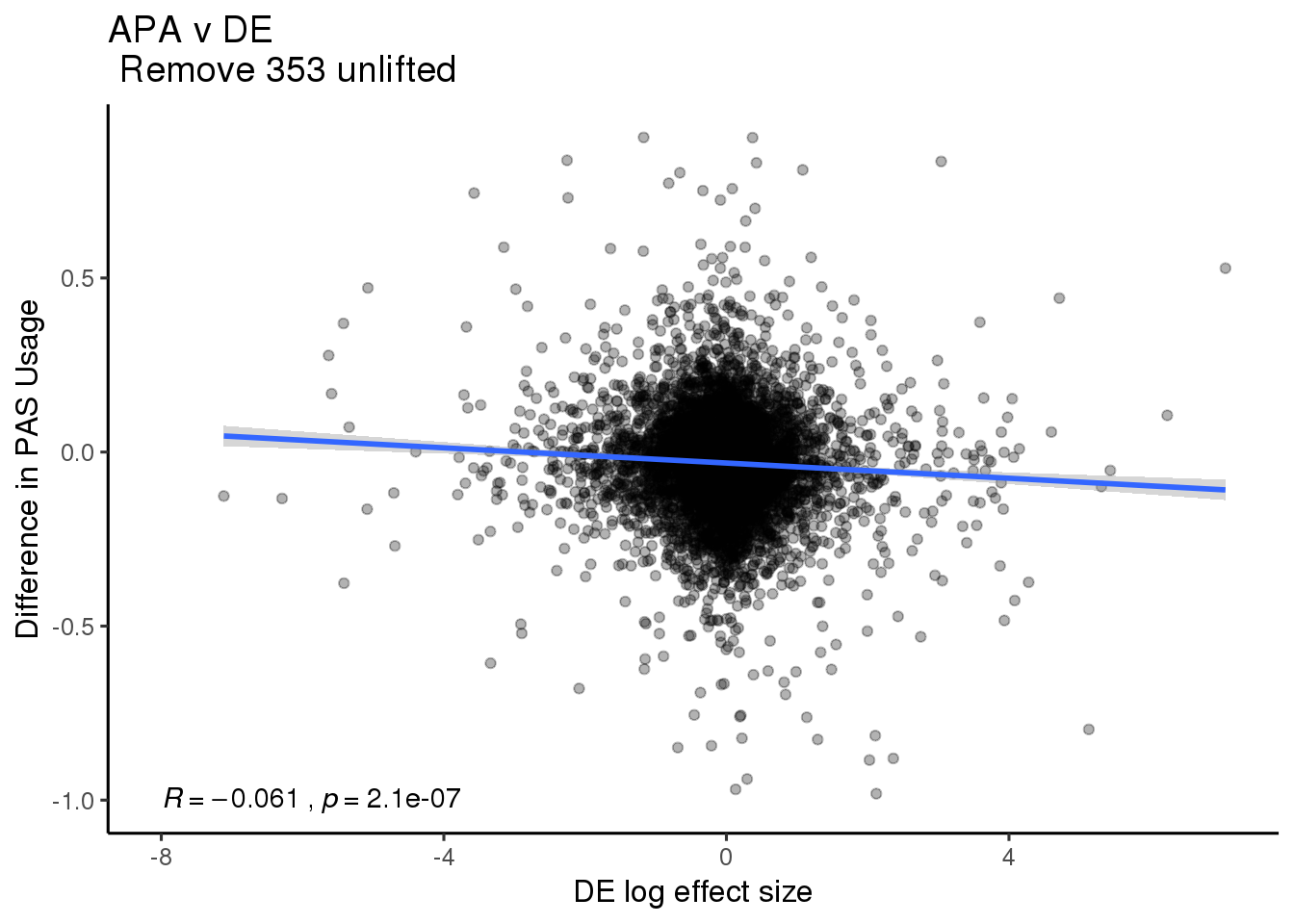
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
DE_split=ggplot(DeandAPA,aes(y=deltaPAU, x=CorrectedlogFC, col=loc)) + geom_point(alpha=.3) + geom_smooth(aes(col=loc),method="lm") + labs(title="APA v DE \n Remove 353 unlifted", x="DE log effect size", y="Difference in PAS Usage") + scale_color_brewer(palette = "Set1",name="", labels=c("Intronic", "3' UTR"))+ stat_cor(aes(col=loc),label.x = c(-8,0),label.y = -1) +theme_classic(base_size = 12)
DE_split
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
DeandAPASig= DeandAPA %>% filter(SigPAU2=="Yes", adj.P.Val<=0.05)
nrow(DeandAPASig)[1] 393DE_sig_all=ggplot(DeandAPASig,aes(y=deltaPAU, x=CorrectedlogFC)) + geom_point(alpha=.3) + geom_smooth(method="lm") + labs(title="Significant differences in APA and expression\n Remove 353 unlifted", x="DE log effect size", y="Difference in PAS Usage") + scale_color_brewer(palette = "Set1",name="", labels=c("Intronic", "3' UTR"))+ stat_cor(label.x = -8,label.y = -1) +theme_classic(base_size = 12)
DE_sig_all
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
DE_sig_split=ggplot(DeandAPASig,aes(y=deltaPAU, x=CorrectedlogFC, col=loc)) + geom_point(alpha=.3) + geom_smooth(aes(col=loc),method="lm") + labs(title="Significant differences in APA and expression\n Remove 353 unlifted", x="DE log effect size", y="Difference in PAS Usage") + scale_color_brewer(palette = "Set1",name="", labels=c("Intronic", "3' UTR"))+ stat_cor(aes(col=loc),label.x = c(-8,0),label.y = -1) +theme_classic(base_size = 12)
DE_sig_split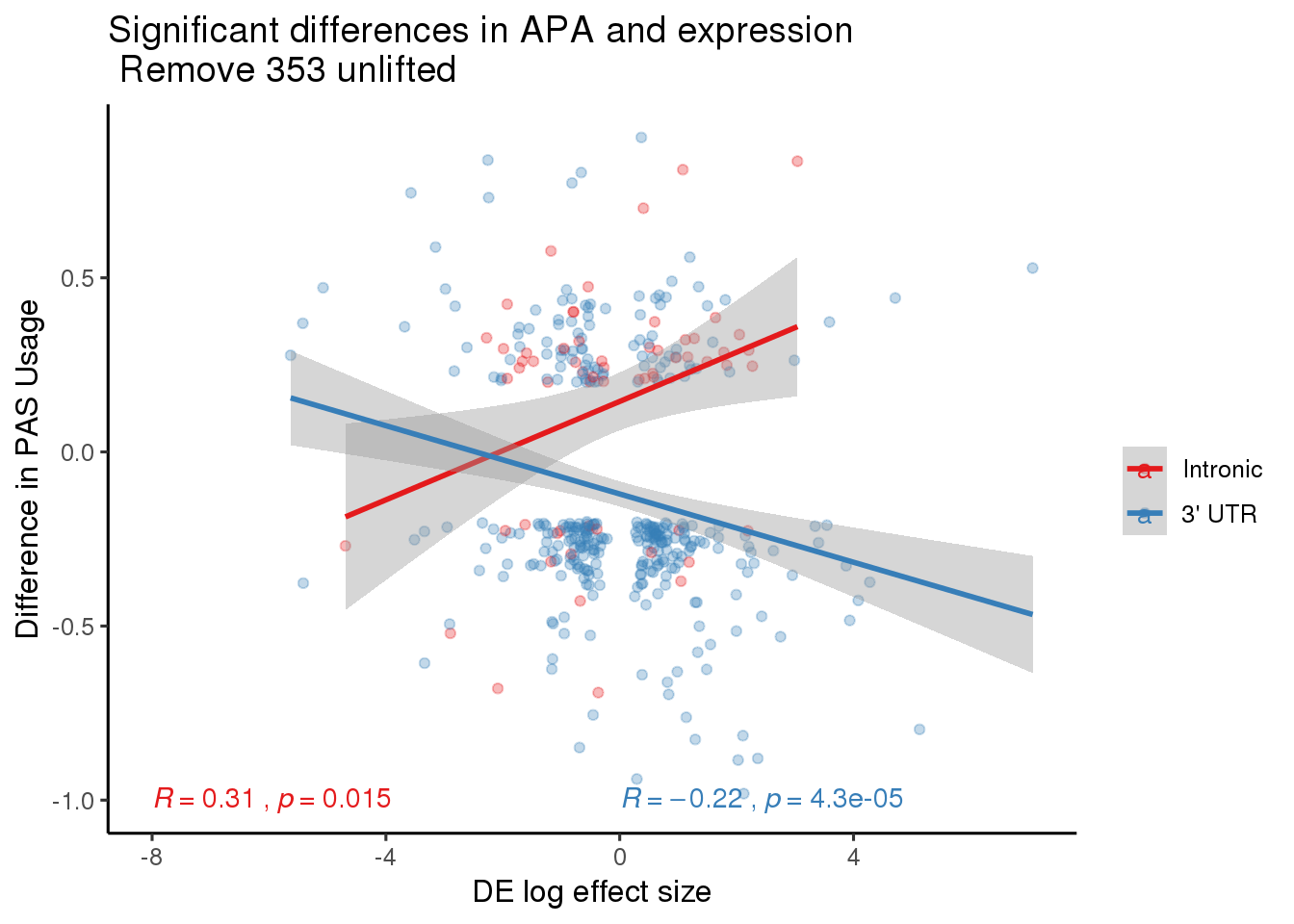
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
DE v TE
Meta=read.table("../data/PAS_doubleFilter/PAS_5perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F)
Meta_genes= Meta %>% select(gene) %>% unique()
Meta_PAS=Meta %>% select(PAS,gene)
dAPAGenes=read.table("../data/DiffIso_Nuclear_DF/SignifianceEitherGENES_Nuclear.txt", header = T, stringsAsFactors = F)
dAPAPAS=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T, stringsAsFactors = F) %>% inner_join(Meta, by=c("chr","start", "end","gene")) %>% select(PAS,gene,SigPAU2 )
dAPAPAS_genes= dAPAPAS %>% select(gene) %>% unique()
dAPATestedGenes= dAPAPAS %>% select(gene) %>% unique() %>% mutate(dAPA=ifelse(gene %in% dAPAGenes$gene,"Yes", "No"))
dICdata= read.table("../data/IndInfoContent/SimpsonMedianSignificance.txt", header = T, stringsAsFactors = F)%>% select(sIC,gene)
dICdata_sig= dICdata %>% filter(sIC=="Yes")
dAPAandDic= dICdata %>% inner_join(dAPATestedGenes,by="gene") %>% mutate(Both=ifelse(sIC=="Yes" & dAPA=="Yes", "Yes","No"),OnlyIC=ifelse(sIC=="Yes" & dAPA=="No", "Yes","No"),OnlyAPA=ifelse(sIC=="No" & dAPA=="Yes", "Yes","No"))
DiffExp=read.table("../data/DiffExpression/DEtested_allres.txt",stringsAsFactors = F,header = F, col.names = c("Gene_stable_ID" ,"logFC" ,"AveExpr" , "t" , "P.Value" , "adj.P.Val", "B" )) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::rename('gene'=Gene.name) %>% dplyr::select(-Gene_stable_ID) %>% mutate(DE=ifelse(adj.P.Val<.05, "Yes", "No")) %>% select(gene,DE)Join DE:
DEandAPA=DiffExp %>% inner_join(dAPAandDic,by="gene") %>% anti_join(UnfliftGenes,by="gene") Expression enrichments:
sets=c("OnlyAPA", "OnlyIC", "Both")
DE_pval=c()
DE_enrich=c()
x=nrow(DEandAPA %>% filter(OnlyAPA=="Yes", DE=="Yes"))
m=nrow(DEandAPA %>% filter(DE=="Yes"))
n=nrow(DEandAPA %>% filter(DE=="No"))
k=nrow(DEandAPA %>% filter(OnlyAPA=="Yes"))
N=nrow(DEandAPA)
phyper(x-1,m,n,k,lower.tail=F)[1] 0.0006534149DE_pval=c(DE_pval, phyper(x-1,m,n,k,lower.tail=F))
DE_enrich=c(DE_enrich, (x/k)/(m/N))
x=nrow(DEandAPA %>% filter(OnlyIC=="Yes", DE=="Yes"))
m=nrow(DEandAPA %>% filter(DE=="Yes"))
n=nrow(DEandAPA %>% filter(DE=="No"))
k=nrow(DEandAPA %>% filter(OnlyIC=="Yes"))
N=nrow(DEandAPA)
phyper(x-1,m,n,k,lower.tail=F)[1] 0.2234779DE_pval=c(DE_pval, phyper(x-1,m,n,k,lower.tail=F))
DE_enrich=c(DE_enrich, (x/k)/(m/N))
x=nrow(DEandAPA %>% filter(Both=="Yes", DE=="Yes"))
m=nrow(DEandAPA %>% filter(DE=="Yes"))
n=nrow(DEandAPA %>% filter(DE=="No"))
k=nrow(DEandAPA %>% filter(Both=="Yes"))
N=nrow(DEandAPA)
phyper(x-1,m,n,k,lower.tail=F)[1] 0.0162844DE_pval=c(DE_pval, phyper(x-1,m,n,k,lower.tail=F))
DE_enrich=c(DE_enrich, (x/k)/(m/N))
DEdf=as.data.frame(cbind(sets,DE_pval, DE_enrich)) %>% rename(Pval=DE_pval, Enrichment=DE_enrich) %>% mutate(Pheno="Expression")
DEdf sets Pval Enrichment Pheno
1 OnlyAPA 0.000653414923024692 1.12256754451574 Expression
2 OnlyIC 0.223477911524423 1.05270145258434 Expression
3 Both 0.0162844040325435 1.14539262950798 ExpressionRibo=read.table("../data/Wang_ribo/Additionaltable5_translationComparisons.txt",header = T, stringsAsFactors = F) %>% rename("Gene_stable_ID"= ENSG) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::select(Gene.name, HvC.beta, HvC.pvalue, HvC.FDR) %>% rename("gene"=Gene.name) %>% mutate(dTE=ifelse(HvC.FDR <0.05, "Yes","No"))
RiboSmall= Ribo %>% select(gene,dTE)
DTandAPA=Ribo %>% inner_join(dAPAandDic,by="gene") %>% anti_join(UnfliftGenes,by="gene") DT_pval=c()
DT_enrich=c()
x=nrow(DTandAPA %>% filter(OnlyAPA=="Yes", dTE=="Yes"))
m=nrow(DTandAPA %>% filter(dTE=="Yes"))
n=nrow(DTandAPA %>% filter(dTE=="No"))
k=nrow(DTandAPA %>% filter(OnlyAPA=="Yes"))
N=nrow(DTandAPA)
phyper(x-1,m,n,k,lower.tail=F)[1] 0.04848155DT_pval=c(DT_pval, phyper(x-1,m,n,k,lower.tail=F))
DT_enrich=c(DT_enrich, (x/k)/(m/N))
x=nrow(DTandAPA %>% filter(OnlyIC=="Yes", dTE=="Yes"))
m=nrow(DTandAPA %>% filter(dTE=="Yes"))
n=nrow(DTandAPA %>% filter(dTE=="No"))
k=nrow(DTandAPA %>% filter(OnlyIC=="Yes"))
N=nrow(DTandAPA)
phyper(x-1,m,n,k,lower.tail=F)[1] 0.02402838DT_pval=c(DT_pval, phyper(x-1,m,n,k,lower.tail=F))
DT_enrich=c(DT_enrich, (x/k)/(m/N))
x=nrow(DTandAPA %>% filter(Both=="Yes", dTE=="Yes"))
m=nrow(DTandAPA %>% filter(dTE=="Yes"))
n=nrow(DTandAPA %>% filter(dTE=="No"))
k=nrow(DTandAPA %>% filter(Both=="Yes"))
N=nrow(DTandAPA)
phyper(x-1,m,n,k,lower.tail=F)[1] 0.02586374DT_pval=c(DT_pval, phyper(x-1,m,n,k,lower.tail=F))
DT_enrich=c(DT_enrich, (x/k)/(m/N))
DTdf=as.data.frame(cbind(sets,DT_pval, DT_enrich)) %>% rename(Pval=DT_pval, Enrichment=DT_enrich) %>% mutate(Pheno="Translation")
DTdf sets Pval Enrichment Pheno
1 OnlyAPA 0.0484815452167291 1.10358865436524 Translation
2 OnlyIC 0.0240283752892615 1.20480789240106 Translation
3 Both 0.0258637425077943 1.21130122254139 TranslationAllDF= DEdf %>% bind_rows(DTdf)Warning in bind_rows_(x, .id): Unequal factor levels: coercing to characterWarning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vectorWarning in bind_rows_(x, .id): Unequal factor levels: coercing to characterWarning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vectorAllDF$Pval=as.numeric(AllDF$Pval)
AllDF$Enrichment=as.numeric(AllDF$Enrichment)
AllDF$Pheno=factor(AllDF$Pheno, levels=c("Expression", "Translation", "Protein"))
useCOl <- c("#d73027", "#4575b4","#fee090")
enrichpoint=ggplot(AllDF,aes(x=sets,col=sets,y=Enrichment,label = round(Enrichment,3)))+ geom_bar(stat="identity",color="grey",aes(y=AllDF$Enrichment),width=.01)+geom_point(size=10) + coord_flip() + geom_hline(yintercept = 1) + facet_grid(~Pheno)+scale_color_manual(values=useCOl)+ labs( title="Enrichment for APA phenotype differences in other regulatory phenotypes \n Remove 353 unlifted",x="Set", y="Enrichment")+geom_text(color = "black", size = 3) + theme(legend.position = "none")
pvalplot=ggplot(AllDF,aes(x=Pheno, by=sets, y=-log10(Pval),fill=sets)) +geom_bar(stat = "identity",position = "dodge") +geom_hline(yintercept =1.3)+ scale_fill_manual(values=useCOl)+ theme(legend.position = "bottom")
plot_grid(enrichpoint,pvalplot, nrow=2)
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
dominant PAS
HumanRes=read.table("../data/DomDefGreaterX/Human_AllGenes_DiffTop.txt", col.names = c("Human_PAS", "gene","Human_DiffDom"),stringsAsFactors = F)
ChimpRes=read.table("../data/DomDefGreaterX/Chimp_AllGenes_DiffTop.txt", col.names = c("Chimp_PAS", "gene","Chimp_DiffDom"),stringsAsFactors = F)
BothRes=HumanRes %>% inner_join(ChimpRes,by="gene")%>% anti_join(UnfliftGenes,by="gene")
BothRes_10=BothRes %>% filter(Chimp_DiffDom >=0.1 | Human_DiffDom>=0.1) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=10)
BothRes_20=BothRes %>% filter(Chimp_DiffDom >=0.2 | Human_DiffDom>=0.2) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=20)
BothRes_30=BothRes %>% filter(Chimp_DiffDom >=0.3 | Human_DiffDom>=0.3) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=30)
BothRes_40=BothRes %>% filter(Chimp_DiffDom >=0.4 | Human_DiffDom>=0.4) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=40)
BothRes_50=BothRes %>% filter(Chimp_DiffDom >=0.5 | Human_DiffDom>=0.5) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=50)
BothRes_60=BothRes %>% filter(Chimp_DiffDom >=0.6 | Human_DiffDom>=0.6) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=60)
BothRes_70=BothRes %>% filter(Chimp_DiffDom >=0.7 | Human_DiffDom>=0.7) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=70)
BothRes_80=BothRes %>% filter(Chimp_DiffDom >=0.8 | Human_DiffDom>=0.8) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=80)
BothRes_90=BothRes %>% filter(Chimp_DiffDom >=0.9 | Human_DiffDom>=0.9) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=90)
BothResAll=BothRes_10 %>% bind_rows(BothRes_20) %>% bind_rows(BothRes_30) %>% bind_rows(BothRes_40) %>% bind_rows(BothRes_50) %>% bind_rows(BothRes_60) %>% bind_rows(BothRes_70) %>% bind_rows(BothRes_80) %>% bind_rows(BothRes_90)
Pval=c()
Enrich=c()
set=c(10,20,30,40,50,60,70,80,90)
expected=c()
actual=c()
nameID=read.table("../../genome_anotation_data/ensemble_to_genename.txt",sep="\t", header = T, stringsAsFactors = F)
DE= read.table("../data/DiffExpression/DEtested_allres.txt",header=F, stringsAsFactors = F,col.names = c('Gene_stable_ID', 'logFC' ,'AveExpr', 't', 'P.Value', 'adj.P.Val', 'B')) %>% inner_join(nameID, by="Gene_stable_ID") %>% dplyr::select(-Gene_stable_ID, -Source_of_gene_name) %>% rename("gene"=Gene.name) %>% mutate(DE=ifelse(adj.P.Val<=.05, "Yes","No")) %>% select(gene,DE)
DE_yes= DE %>% filter(DE=="Yes")
All4= BothResAll %>% select(gene,cut,Set) %>% inner_join(DE, by="gene")
for (i in set){
x=nrow(All4 %>% filter(cut==i, Set=="Different", DE=="Yes"))
actual=c(actual, x)
m=nrow(All4 %>% filter(cut==i, DE=="Yes"))
n=nrow(All4 %>% filter(cut==i, DE=="No"))
k=nrow(All4 %>% filter(cut==i, Set=="Different"))
N=nrow(All4 %>% filter(cut==i))
val=phyper(x-1,m,n,k,lower.tail=F)
Pval= c(Pval, val)
en=(x/k)/(m/N)
Enrich=c(Enrich, en)
#ex=which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))
ex=k*(m/N)
expected=c(expected,ex)
}
ResDF=as.data.frame(cbind(set,Pval,Enrich, actual, expected))
ResDF$set=as.factor(ResDF$set)
ResDF$Pval=as.numeric(as.character(ResDF$Pval))
ResDF$Enrich=as.numeric(as.character(ResDF$Enrich))
diffP=ggplot(ResDF,aes(x=set, y=-log10(Pval),fill=set)) + geom_bar(stat="identity") +labs(title="Enrichment pvalues for DE and different dominant \n Remove 353 unlifted",x="Dominance Cutoff")+ scale_fill_brewer(palette = "RdYlBu") + theme(legend.position = "none")+ geom_hline(yintercept = 1.30103)
diffE=ggplot(ResDF,aes(x=set, y=Enrich,fill=set)) + geom_bar(stat="identity") + geom_hline(yintercept = 1)+labs(title="Enrichment for DE and different dominant \n Remove 353 unlifted",x="Dominance Cutoff")+ scale_fill_brewer(palette = "RdYlBu") + theme(legend.position = "none")
PvalSame=c()
EnrichSame=c()
expectedSame=c()
actualSame=c()
for (i in set){
x=nrow(All4 %>% filter(cut==i, Set=="Same", DE=="Yes"))
actualSame=c(actualSame, x)
m=nrow(All4 %>% filter(cut==i, DE=="Yes"))
n=nrow(All4 %>% filter(cut==i, DE=="No"))
k=nrow(All4 %>% filter(cut==i, Set=="Same"))
N=nrow(All4 %>% filter(cut==i))
val=phyper(x-1,m,n,k,lower.tail=F)
PvalSame= c(PvalSame, val)
en=(x/k)/(m/N)
EnrichSame=c(EnrichSame, en)
#ex=which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))
ex=k*(m/N)
expectedSame=c(expectedSame,ex)
}
ResDFSame=as.data.frame(cbind(set,PvalSame,EnrichSame, actualSame,expectedSame))
ResDFSame$set=as.factor(ResDFSame$set)
ResDFSame$PvalSame=as.numeric(as.character(ResDFSame$PvalSame))
ResDFSame$EnrichSame=as.numeric(as.character(ResDFSame$EnrichSame))
Samep=ggplot(ResDFSame,aes(x=set, y=-log10(PvalSame),fill=set)) + geom_bar(stat="identity") +labs(title="Enrichment pvalues for DE and same dominant \n Remove 353 unlifted",x="Dominance Cutoff")+ scale_fill_brewer(palette = "RdYlBu") + theme(legend.position = "none")+ geom_hline(yintercept = 1.30103)
SameE=ggplot(ResDFSame,aes(x=set, y=EnrichSame,fill=set)) + geom_bar(stat="identity") + geom_hline(yintercept = 1)+labs(title="Enrichment for DE and same dominant \n Remove 353 unlifted",x="Dominance Cutoff")+ scale_fill_brewer(palette = "RdYlBu") + theme(legend.position = "none")
plot_grid(diffE, SameE, diffP, Samep)
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
plot_grid( diffP, Samep)
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
p not e
Meta=read.table("../data/PAS_doubleFilter/PAS_5perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F)
Meta_genes= Meta %>% select(gene) %>% unique()
Meta_PAS=Meta %>% select(PAS,gene)
dAPAGenes=read.table("../data/DiffIso_Nuclear_DF/SignifianceEitherGENES_Nuclear.txt", header = T, stringsAsFactors = F) %>% anti_join(UnfliftGenes,by="gene")
dAPAPAS=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T, stringsAsFactors = F) %>% inner_join(Meta, by=c("chr","start", "end","gene")) %>% select(PAS,gene,SigPAU2 ) %>% anti_join(UnfliftGenes,by="gene")
dAPAPAS_genes= dAPAPAS %>% select(gene) %>% unique()
dAPATestedGenes= dAPAPAS %>% select(gene) %>% unique() %>% mutate(dAPA=ifelse(gene %in% dAPAGenes$gene,"Yes", "No"))
dICdata= read.table("../data/IndInfoContent/SimpsonMedianSignificance.txt", header = T, stringsAsFactors = F)%>% select(sIC,gene)%>% anti_join(UnfliftGenes,by="gene")
dAPAandDic= dICdata %>% inner_join(dAPATestedGenes,by="gene") %>% mutate(Both=ifelse(sIC=="Yes" & dAPA=="Yes", "Yes","No"),OnlyIC=ifelse(sIC=="Yes" & dAPA=="No", "Yes","No"),OnlyAPA=ifelse(sIC=="No" & dAPA=="Yes", "Yes","No"))
dIConly=dAPAandDic %>%filter(OnlyIC=="Yes")
Both=dAPAandDic %>%filter(Both=="Yes")
dAPAonly=dAPAandDic %>%filter(OnlyAPA=="Yes")nameID=read.table("../../genome_anotation_data/ensemble_to_genename.txt",sep="\t", header = T, stringsAsFactors = F) %>% dplyr::select(Gene_stable_ID, Gene.name)
DiffExp=read.table("../data/DiffExpression/DEtested_allres.txt",stringsAsFactors = F,header = F, col.names = c("Gene_stable_ID" ,"logFC" ,"AveExpr" , "t" , "P.Value" , "adj.P.Val", "B" )) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::rename('gene'=Gene.name) %>% dplyr::select(-Gene_stable_ID) %>% mutate(DE=ifelse(adj.P.Val<.05, "Yes", "No")) %>% select(gene,DE) %>% filter(DE=="Yes")
Prot= read.table("../data/Khan_prot/ProtData_effectSize.txt",header = T,stringsAsFactors = F) %>% mutate(dP=ifelse(pval<0.05, "Yes", "No")) %>% filter(dP=="Yes")
Ribo=read.table("../data/Wang_ribo/Additionaltable5_translationComparisons.txt",header = T, stringsAsFactors = F) %>% rename("Gene_stable_ID"= ENSG) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::select(Gene.name, HvC.beta, HvC.pvalue, HvC.FDR) %>% rename("gene"=Gene.name) %>% mutate(dTE=ifelse(HvC.FDR <0.05, "Yes","No")) %>% filter(dTE=="Yes")
RiboAll=read.table("../data/Wang_ribo/Additionaltable5_translationComparisons.txt",header = T, stringsAsFactors = F) %>% rename("Gene_stable_ID"= ENSG) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::select(Gene.name, HvC.beta, HvC.pvalue, HvC.FDR) %>% rename("gene"=Gene.name) %>% mutate(dTE=ifelse(HvC.FDR <0.05, "Yes","No"))
Set=c("OnlydIC", "Both", "OnlyAPA")
Number=c(40, 33,76)
SetSize=c(nrow(dIConly),nrow(Both),nrow(dAPAonly) )
useCOl <- c("#d73027", "#4575b4","#fee090")
DFres=data.frame(cbind(Set,Number,SetSize))
DFres$Number=as.numeric(as.character(DFres$Number))
DFres$SetSize=as.numeric(as.character(DFres$SetSize))
DFres_prop=DFres %>% mutate(Prop=Number/SetSize)
numberPlot=ggplot(DFres_prop,aes(x=Set, fill=Set, y=Number))+ geom_bar(stat="identity")+ scale_fill_manual(values=useCOl)+geom_text(aes(label=Number), position=position_dodge(width=0.9), vjust=2)+ theme(legend.position = "none") + labs(title="Number of dP not DE genes", y="Number of Genes",x="")+scale_x_discrete(labels=c(Both="Both", OnlyAPA="PAS\nLevel",OnlydIC= "Isoform\n Diversity"))
propPlot=ggplot(DFres_prop,aes(x=Set, fill=Set, y=Prop))+ geom_bar(stat="identity")+ scale_fill_manual(values=useCOl) + labs(title="Proportion of APA set that are\n dP not DE", y="Proportion of APA set",x="")+geom_text(aes(label=round(Prop,3)), position=position_dodge(width=0.9), vjust=2) + theme(legend.position = "none")+scale_x_discrete(labels=c(Both="Both", OnlyAPA="PAS\nLevel",OnlydIC= "Isoform\n Diversity"))numberprop=plot_grid(numberPlot,propPlot)
numberprop
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
ProtInfo=read.table("../data/PTM/ProtLength.txt", sep = "\t",stringsAsFactors = F,header = T,col.names = c("entry","organism", "nAA", "gene")) %>% select(nAA, gene)
Interactions=read.table("../data/bioGRID/GeneswInteractions.txt",stringsAsFactors = F, header = T) %>% inner_join(ProtInfo, by="gene")%>% mutate(NormInter=nInt/nAA)
#DiffExp$gene, DT=Ribo$gene, DP=Prot$gene
dAPAandDic_wP=dAPAandDic %>% mutate(dE=ifelse(gene %in%DiffExp$gene, "Yes", "No" ), dP=ifelse(gene %in%Prot$gene,"Yes","No" ), dPnotDE=ifelse(dE=="No"&dP=="Yes", "Yes","No")) %>% inner_join(Interactions, by="gene")
dAPAandDic_wP_dAPA= dAPAandDic_wP %>% filter(dAPA=="Yes")
dAPAandDic_wP_both= dAPAandDic_wP %>% filter(Both=="Yes")
dAPAandDic_wP_IC= dAPAandDic_wP %>% filter(OnlyIC=="Yes")dapaProt=ggplot(dAPAandDic_wP_dAPA,aes(x=dPnotDE, y=log10(NormInter),fill=dPnotDE)) + geom_boxplot(notch = T) + stat_compare_means() + scale_fill_manual(values = c("grey", "#4575b4"))+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=10), legend.position = "false") + labs(x="Expression independent set", y="log10(Normalized Interaction)", title="")
bothProt=ggplot(dAPAandDic_wP_both,aes(x=dPnotDE, y=log10(NormInter),fill=dPnotDE)) + geom_boxplot(notch = T) + stat_compare_means() + scale_fill_manual(values = c("grey", "#d73027"))+ theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=10), legend.position = "false") + labs(x="Expression independent set", y="log10(Normalized Interaction)", title="")
dicProt=ggplot(dAPAandDic_wP_IC,aes(x=dPnotDE, y=log10(NormInter),fill=dPnotDE)) + geom_boxplot(notch = T) + stat_compare_means() + scale_fill_manual(values = c("grey", "#fee090"))+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=10), legend.position = "false") + labs(x="Expression independent set", y="log10(Normalized Interaction)", title="")
interactionplots=plot_grid(bothProt,dapaProt,dicProt, nrow=1)dAPAandDic_wP_trans= dAPAandDic_wP %>% inner_join(RiboAll,by="gene")
dAPAandDic_wP_trans_dpnote= dAPAandDic_wP_trans %>% filter(dPnotDE=="Yes") %>% select(gene,Both, OnlyIC, OnlyAPA,dTE)
dAPAandDic_wP_trans_dpnoteOnlyAPA=dAPAandDic_wP_trans_dpnote %>% filter(OnlyAPA=="Yes") %>% group_by(dTE) %>% summarise(ndTE=n()) %>% mutate(set="OnlyAPA")
dAPAandDic_wP_trans_dpnoteBoth=dAPAandDic_wP_trans_dpnote %>% filter(Both=="Yes") %>% group_by(dTE) %>% summarise(ndTE=n()) %>% mutate(set="Both")
dAPAandDic_wP_trans_dpnoteIC=dAPAandDic_wP_trans_dpnote %>% filter(OnlyIC=="Yes") %>% group_by(dTE) %>% summarise(ndTE=n()) %>% mutate(set="OnlyIC")
AllTenum= dAPAandDic_wP_trans_dpnoteOnlyAPA %>% bind_rows(dAPAandDic_wP_trans_dpnoteBoth) %>% bind_rows(dAPAandDic_wP_trans_dpnoteIC)numTE=ggplot(AllTenum, aes(x=set,by=dTE, y=ndTE,fill=set, alpha=dTE)) +geom_bar(stat="identity", position = "dodge") + labs(title="Most dP not dE genes are not dTE", y="Number of Genes",x="") + scale_fill_manual(values = useCOl ) + scale_alpha_manual(values=c(.6, 1)) + theme(legend.position = "bottom") + geom_text(aes(label=ndTE), position=position_dodge(width=0.9), vjust=1)+guides(fill = FALSE)+scale_x_discrete(labels=c(Both="Both", OnlyAPA="PAS\nLevel",OnlyIC= "Isoform\n Diversity"))
numTE
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
propTe=ggplot(AllTenum, aes(x=set,by=dTE, y=ndTE,fill=set, alpha=dTE)) +geom_bar(stat="identity", position = "fill") + labs(title="Most dP not dE genes \nare not dTE", y="Proportion",x="") + scale_fill_manual(values = useCOl ) + scale_alpha_manual(values=c(.4, 1)) + theme(legend.position = "bottom")+guides(fill = FALSE)+scale_x_discrete(labels=c(Both="Both", OnlyAPA="PAS\nLevel",OnlyIC= "Isoform\n Diversity"))
propTe
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
teplots=plot_grid(numTE, propTe, nrow =1)
teplots
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
fullplot=plot_grid(numberprop, teplots,interactionplots, nrow = 3)
fullplot
| Version | Author | Date |
|---|---|---|
| bb8d220 | brimittleman | 2020-06-04 |
Plots:
fig3:
plot_grid(DE_all,DE_split,DE_sig_all,DE_sig_split, scale = c(.9,.9,.9,.9), labels=c("a","b","c","d"),rel_widths=c(1,1.3,1,1.3))
pdf("../output/removeUnlift_fig3.pdf", height=8, width=12)
plot_grid(DE_all,DE_split,DE_sig_all,DE_sig_split, scale = c(.9,.9,.9,.9), labels=c("a","b","c","d"),rel_widths=c(1,1.3,1,1.3))
dev.off()png
2 fig4
pdf("../output/removeUnlift_fig4.pdf", height=6, width=8,useKerning=F)
plot_grid(enrichpoint,pvalplot, nrow=2)
dev.off()png
2 fig 5:
pdf("../output/removeUnlift_fig5.pdf", height=6, width=10,useKerning=F)
plot_grid(diffE, SameE, diffP, Samep)
dev.off()png
2 fig 6:
pdf("../output/removeUnilift_fig6.pdf", height=9, width=12,useKerning=F)
fullplot
dev.off()png
2
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cowplot_0.9.4 forcats_0.3.0 stringr_1.3.1 dplyr_0.8.0.1
[5] purrr_0.3.2 readr_1.3.1 tidyr_0.8.3 tibble_2.1.1
[9] tidyverse_1.2.1 workflowr_1.6.0 ggpubr_0.2 magrittr_1.5
[13] ggplot2_3.1.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 reshape2_1.4.3 haven_1.1.2
[4] lattice_0.20-38 colorspace_1.3-2 generics_0.0.2
[7] htmltools_0.3.6 yaml_2.2.0 rlang_0.4.0
[10] later_0.7.5 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 RColorBrewer_1.1-2 modelr_0.1.2
[16] readxl_1.1.0 plyr_1.8.4 munsell_0.5.0
[19] gtable_0.2.0 cellranger_1.1.0 rvest_0.3.2
[22] evaluate_0.12 labeling_0.3 knitr_1.20
[25] httpuv_1.4.5 broom_0.5.1 Rcpp_1.0.4.6
[28] promises_1.0.1 scales_1.0.0 backports_1.1.2
[31] jsonlite_1.6 fs_1.3.1 hms_0.4.2
[34] digest_0.6.18 stringi_1.2.4 grid_3.5.1
[37] rprojroot_1.3-2 cli_1.1.0 tools_3.5.1
[40] lazyeval_0.2.1 crayon_1.3.4 whisker_0.3-2
[43] pkgconfig_2.0.2 xml2_1.2.0 lubridate_1.7.4
[46] assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[49] rstudioapi_0.10 R6_2.3.0 nlme_3.1-137
[52] git2r_0.26.1 compiler_3.5.1