Differential Expression Remove Bad Individuals
Briana Mittleman
11/19/2019
Last updated: 2019-11-20
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.5.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq/
Ignored: data/RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx/
Ignored: data/metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF/
Ignored: data/metadata_HCpanel.txt.sb-f4823d1e-qihGek/
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/assessReadQual.Rmd
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._filter5percPAS.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/.snakemake/
Untracked: code/Config_chimp.yaml
Untracked: code/Config_human.yaml
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bed215upbed.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/converBam2Junc.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: data/._.DS_Store
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata.xlsx
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/CompapaQTLpas/
Untracked: data/DTmatrix/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffSplice/
Untracked: data/MapStats/
Untracked: data/NuclearHvC/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_total/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata.xlsx
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/liftover_files/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Unstaged changes:
Modified: analysis/CorrbetweenInd.Rmd
Modified: analysis/InvestigateBadSamples.Rmd
Modified: analysis/MapRNAhg19.Rmd
Modified: analysis/PASnumperSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/diffSplicing.Rmd
Modified: analysis/verifyBAM.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | db0484c | brimittleman | 2019-11-21 | add PC corr |
| html | 9ecd769 | brimittleman | 2019-11-20 | Build site. |
| Rmd | db85b26 | brimittleman | 2019-11-20 | add remove 2 analysis |
This is exactly the diffExpression analysis but I will remove 2 individuals that may be hurting the analysis.
These are:
- Chimp 4973
- Human 18498
library(workflowr)This is workflowr version 1.5.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library("scales")
Attaching package: 'scales'The following object is masked from 'package:purrr':
discardThe following object is masked from 'package:readr':
col_factorlibrary("gplots")
Attaching package: 'gplots'The following object is masked from 'package:stats':
lowesslibrary("edgeR")Loading required package: limmalibrary("R.utils")Loading required package: R.ooLoading required package: R.methodsS3R.methodsS3 v1.7.1 (2016-02-15) successfully loaded. See ?R.methodsS3 for help.R.oo v1.22.0 (2018-04-21) successfully loaded. See ?R.oo for help.
Attaching package: 'R.oo'The following objects are masked from 'package:methods':
getClasses, getMethodsThe following objects are masked from 'package:base':
attach, detach, gc, load, saveR.utils v2.7.0 successfully loaded. See ?R.utils for help.
Attaching package: 'R.utils'The following object is masked from 'package:tidyr':
extractThe following object is masked from 'package:utils':
timestampThe following objects are masked from 'package:base':
cat, commandArgs, getOption, inherits, isOpen, parse, warningslibrary("limma")
library("VennDiagram")Loading required package: gridLoading required package: futile.loggerlibrary("RColorBrewer")
library(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsFor this analysis I do preprocessing with the Snakemake pipeline. The snakemake will map the RNA seq and quantify orthologous exons.
From FastQC:
Does not look like there is adapter contamination
No reads tagged as bad quality
Assess mapping:
metaData=read.table("../data/RNASEQ_metadata_2Removed.txt", header = T, stringsAsFactors = F)
metaData$Species=as.factor(metaData$Species)
metaData$Collection=as.factor(metaData$Collection)readInfo=metaData %>% mutate(AAUnMapped= Reads-Mapped, ABNotOrtho= Mapped-AssignedOrtho) %>% select(Line, Species, AAUnMapped, ABNotOrtho, AssignedOrtho) %>% gather(key="Category", value="Number", -Line, -Species)
ggplot(readInfo, aes(x=Line,y=Number, fill=Category)) + geom_bar(stat="identity") + scale_fill_brewer(palette = "Dark2",name = "Type", labels = c("Unmapped", "Mapped not ortho", "Assigned Ortho Exon"))+theme(axis.text.x = element_text( hjust = 0,vjust = 1, size = 6, angle = 90)) + labs(y="Reads", title="Human and chimp read statistics") 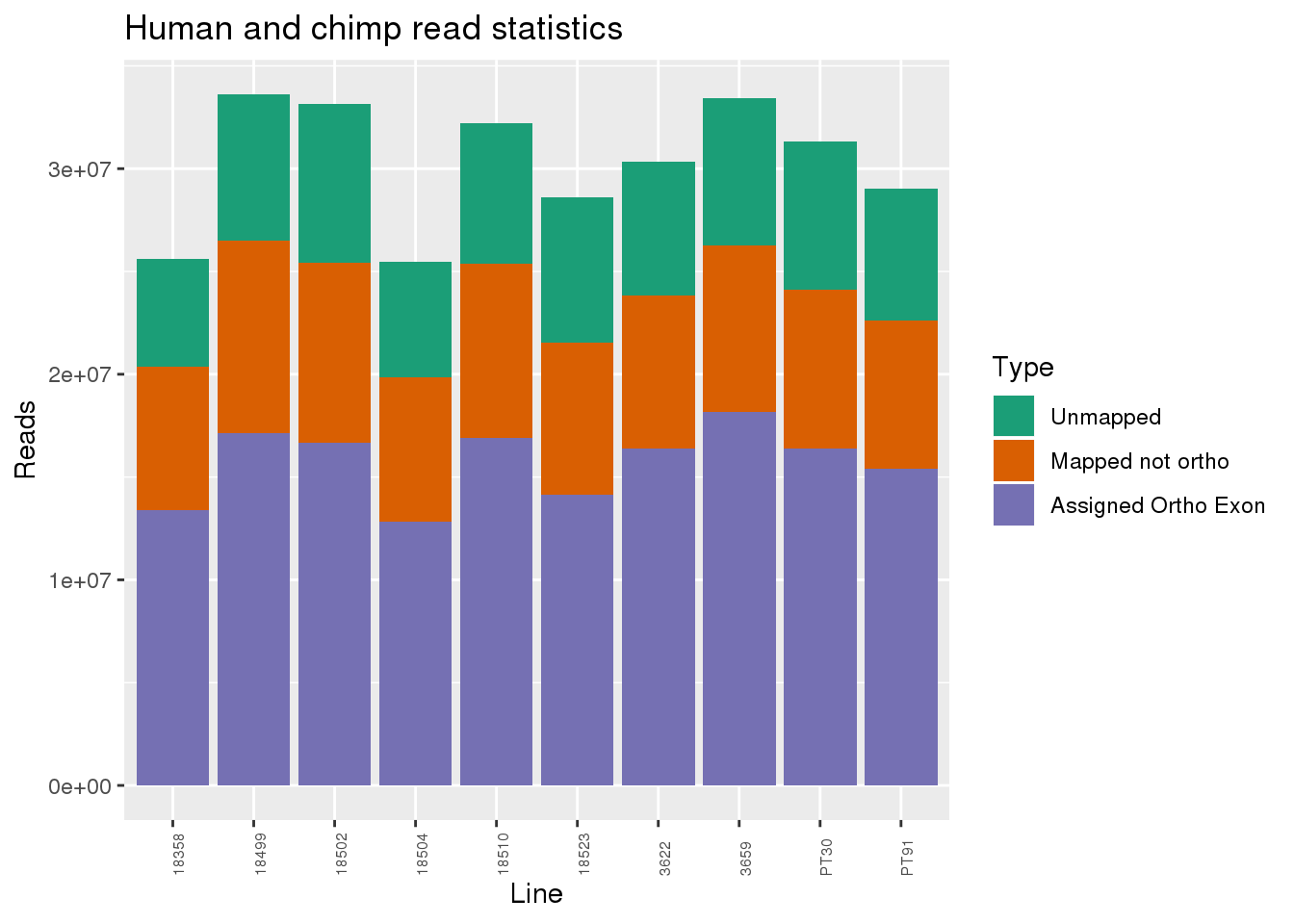
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Proportion of reads.
readProp=metaData %>% mutate(Aunmapped=1-percentMapped, MappednotOrtho=percentMapped-percentOrtho) %>% select(Line,Species, percentOrtho, MappednotOrtho, Aunmapped) %>% gather(key="Category", value="Proportion", -Line, -Species)
ggplot(readProp, aes(x=Line,y=Proportion, fill=Category)) + geom_bar(stat="identity") + scale_fill_brewer(palette = "Dark2", name="", labels = c("Unmapped", "Mapped not ortho", "Assigned Ortho Exon"))+theme(axis.text.x = element_text( hjust = 0,vjust = 1, size = 6, angle = 90)) + labs(y="Reads", title="Human and chimp read proportions") 
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
By species:
ggplot(readInfo,aes(x=Category, y=Number, by=Species, fill=Species)) + geom_boxplot() +scale_x_discrete( breaks=c("AAUnMapped","ABNotOrtho","AssignedOrtho"),labels=c("Unmapped", "Not in OrthoExon", "Assigned to OrthoExon")) + scale_fill_brewer(palette = "Dark2") + labs(title="Mapped reads by Species", y="Reads", x="")
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
ggplot(readProp,aes(x=Category, y=Proportion, by=Species, fill=Species)) + geom_boxplot() + scale_fill_brewer(palette = "Dark2") + labs(title="Map Proportion by Species", y="Proportion", x="") + scale_x_discrete( breaks=c("Aunmapped","MappedNotOrtho","percentOrtho"),labels=c("Unmapped", "Not in OrthoExon", "Assigned to OrthoExon"))
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Diffferential Expression
Code originally from Lauren Blake (http://lauren-blake.github.io/Reg_Evo_Primates/analysis/Normalization_plots.html)
Raw Counts
HumanCounts=read.table("../Human/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fc", header = T, stringsAsFactors = F) %>% select(-Chr,-Start,-End,-Strand, -Length, -NA18498)
ChimpCounts=read.table("../Chimp/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fc", header = T, stringsAsFactors = F) %>% select(-Chr,-Start,-End,-Strand, -Length, -NA4973)
counts_genes=HumanCounts %>% inner_join(ChimpCounts,by="Geneid") %>% column_to_rownames(var="Geneid")
head(counts_genes) NA18504 NA18510 NA18523 NA18499 NA18502 NAPT30 NAPT91
ENSG00000188976 24 50 31 34 58 2 1
ENSG00000188157 106 65 106 128 39 5 7
ENSG00000273443 40 43 54 48 11 21 2
ENSG00000217801 60 36 164 61 19 34 8
ENSG00000237330 0 1 0 1 1 0 0
ENSG00000223823 0 0 0 0 0 0 0
NA3622 NA3659 NA18358
ENSG00000188976 1 1 0
ENSG00000188157 7 8 6
ENSG00000273443 3 78 16
ENSG00000217801 19 139 31
ENSG00000237330 0 2 0
ENSG00000223823 0 0 0# Load colors
colors <- colorRampPalette(c(brewer.pal(9, "Blues")[1],brewer.pal(9, "Blues")[9]))(100)
pal <- c(brewer.pal(9, "Set1"), brewer.pal(8, "Set2"), brewer.pal(12, "Set3"))
labels <- paste(metaData$Species,metaData$Line, sep=" ")#PCA function (original code from Julien Roux)
#Load in the plot_scores function
plot_scores <- function(pca, scores, n, m, cols, points=F, pchs =20, legend=F){
xmin <- min(scores[,n]) - (max(scores[,n]) - min(scores[,n]))*0.05
if (legend == T){ ## let some room (35%) for a legend
xmax <- max(scores[,n]) + (max(scores[,n]) - min(scores[,n]))*0.50
}
else {
xmax <- max(scores[,n]) + (max(scores[,n]) - min(scores[,n]))*0.05
}
ymin <- min(scores[,m]) - (max(scores[,m]) - min(scores[,m]))*0.05
ymax <- max(scores[,m]) + (max(scores[,m]) - min(scores[,m]))*0.05
plot(scores[,n], scores[,m], xlab=paste("PC", n, ": ", round(summary(pca)$importance[2,n],3)*100, "% variance explained", sep=""), ylab=paste("PC", m, ": ", round(summary(pca)$importance[2,m],3)*100, "% variance explained", sep=""), xlim=c(xmin, xmax), ylim=c(ymin, ymax), type="n")
if (points == F){
text(scores[,n],scores[,m], rownames(scores), col=cols, cex=1)
}
else {
points(scores[,n],scores[,m], col=cols, pch=pchs, cex=1.3)
}
}# Clustering (original code from Julien Roux)
cors <- cor(counts_genes, method="spearman", use="pairwise.complete.obs")
heatmap.2( cors, scale="none", col = colors, margins = c(12, 12), trace='none', denscol="white", labCol=labels, ColSideColors=pal[as.integer(as.factor(metaData$Species))], RowSideColors=pal[as.integer(as.factor(metaData$Collection))+9], cexCol = 0.2 + 1/log10(15), cexRow = 0.2 + 1/log10(15))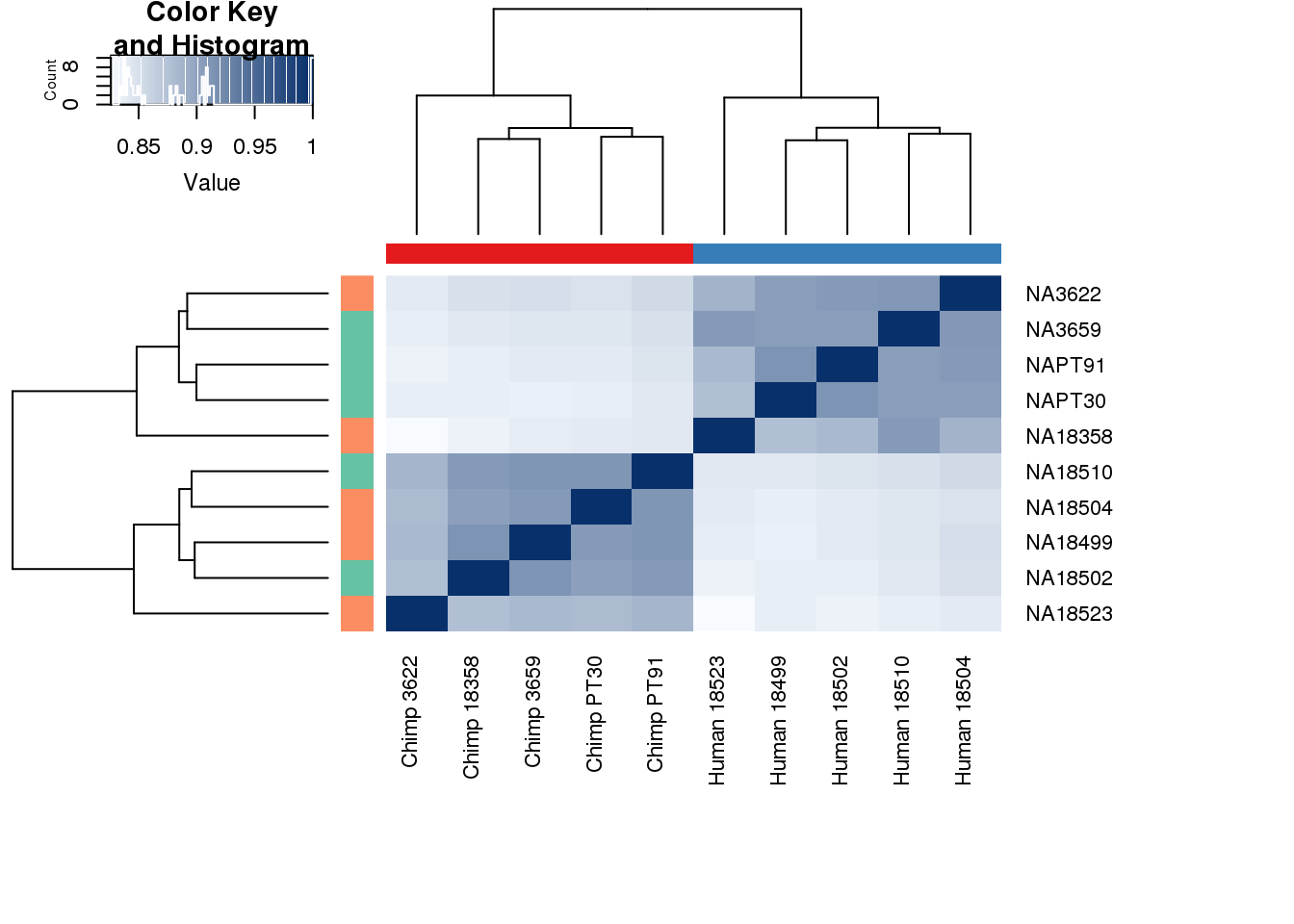
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
select <- counts_genes
summary(apply(select, 1, var) == 0) Mode FALSE TRUE
logical 31883 12242 # Perform PCA
pca_genes <- prcomp(t(counts_genes), scale = F)
scores <- pca_genes$x
#Make PCA plots with the factors colored by species
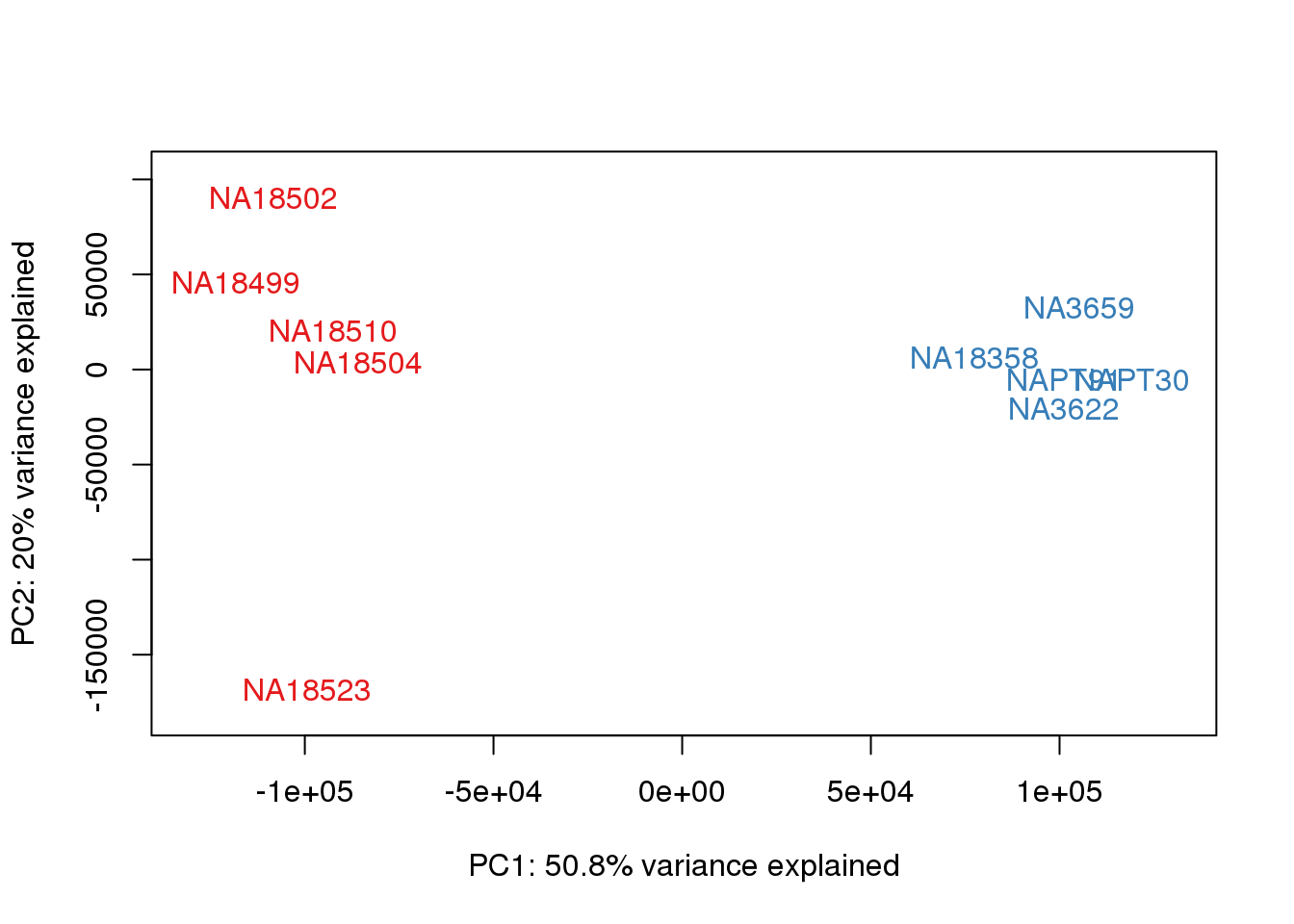
### PCs 1 and 2 Raw Data
for (n in 1:1){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
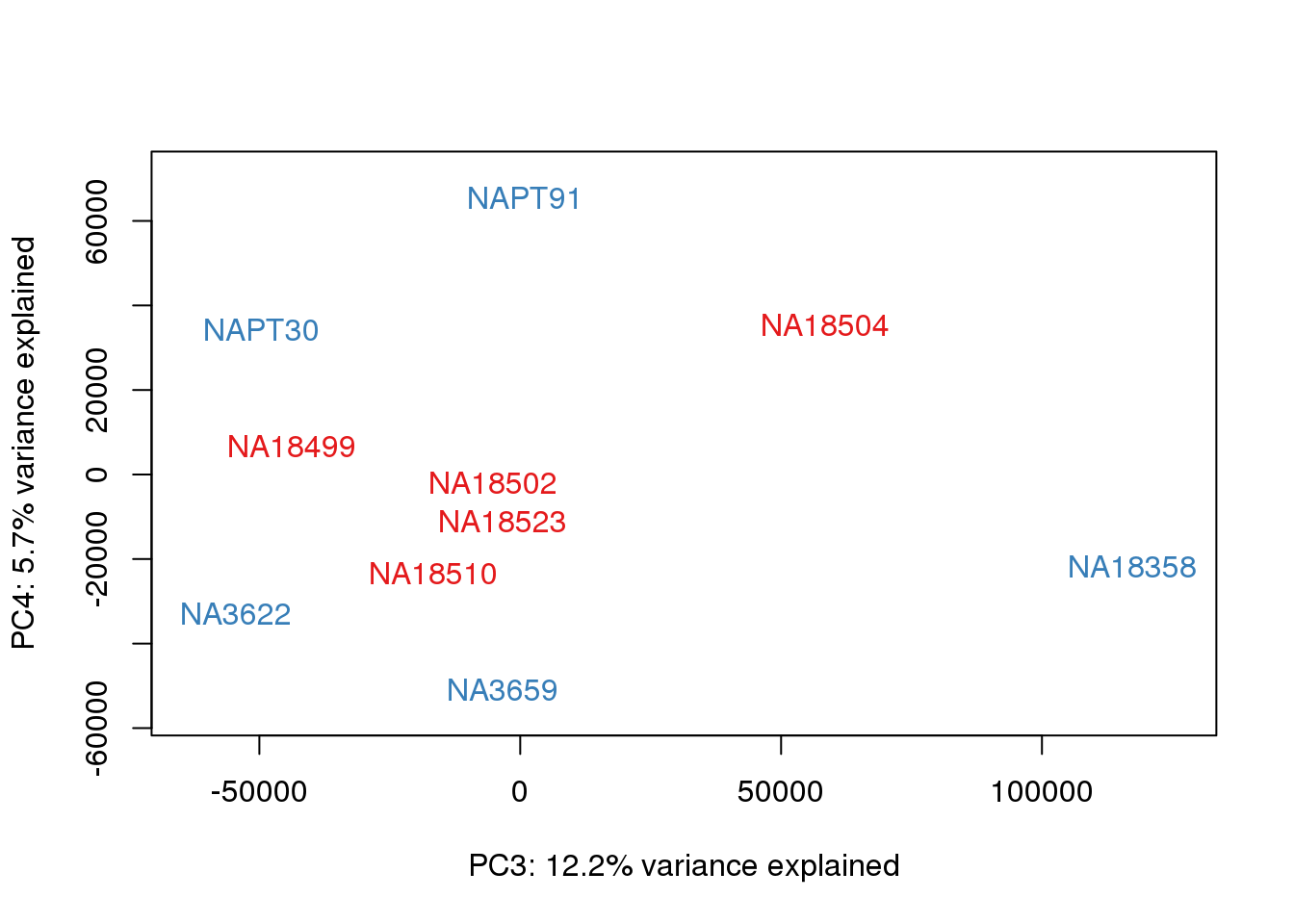
### PCs 3 and 4 Raw Data
for (n in 3:3){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Plot density for raw data:
density_plot_18504 <- ggplot(counts_genes, aes(x = NA18504)) + geom_density() + labs(title = "Density plot of raw gene counts of NA18504") + labs(x = "Raw counts for each gene")
density_plot_18504
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Convert to log2
log_counts_genes <- as.data.frame(log2(counts_genes))
head(log_counts_genes) NA18504 NA18510 NA18523 NA18499 NA18502 NAPT30
ENSG00000188976 4.584963 5.643856 4.954196 5.087463 5.857981 1.000000
ENSG00000188157 6.727920 6.022368 6.727920 7.000000 5.285402 2.321928
ENSG00000273443 5.321928 5.426265 5.754888 5.584963 3.459432 4.392317
ENSG00000217801 5.906891 5.169925 7.357552 5.930737 4.247928 5.087463
ENSG00000237330 -Inf 0.000000 -Inf 0.000000 0.000000 -Inf
ENSG00000223823 -Inf -Inf -Inf -Inf -Inf -Inf
NAPT91 NA3622 NA3659 NA18358
ENSG00000188976 0.000000 0.000000 0.000000 -Inf
ENSG00000188157 2.807355 2.807355 3.000000 2.584963
ENSG00000273443 1.000000 1.584963 6.285402 4.000000
ENSG00000217801 3.000000 4.247928 7.118941 4.954196
ENSG00000237330 -Inf -Inf 1.000000 -Inf
ENSG00000223823 -Inf -Inf -Inf -Infdensity_plot_18504 <- ggplot(log_counts_genes, aes(x = 18504)) + geom_density()
density_plot_18504 + labs(title = "Density plot of log2 counts of 18504") + labs(x = "Log2 counts for each gene") + geom_vline(xintercept = 1) 
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
plotDensities(log_counts_genes, col=pal[as.numeric(metaData$Species)], legend="topright")
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Convert to CPM
cpm <- cpm(counts_genes, log=TRUE)
head(cpm) NA18504 NA18510 NA18523 NA18499 NA18502
ENSG00000188976 0.9894703 1.620342 1.209959 1.068977 1.8425919
ENSG00000188157 3.0594442 1.985078 2.926705 2.916115 1.2946137
ENSG00000273443 1.6891590 1.412390 1.976621 1.540857 -0.3532791
ENSG00000217801 2.2551156 1.169332 3.547816 1.873190 0.3334631
ENSG00000237330 -2.9815417 -2.429988 -2.981542 -2.437603 -2.4245831
ENSG00000223823 -2.9815417 -2.981542 -2.981542 -2.981542 -2.9815417
NAPT30 NAPT91 NA3622 NA3659 NA18358
ENSG00000188976 -2.008556 -2.3855698 -2.4153553 -2.4615104 -2.9815417
ENSG00000188157 -1.212936 -0.7860576 -0.8558140 -0.8206567 -0.8020982
ENSG00000273443 0.492301 -1.9650589 -1.6935886 2.1415838 0.3987400
ENSG00000217801 1.136923 -0.6333308 0.3592322 2.9568407 1.2842881
ENSG00000237330 -2.981542 -2.9815417 -2.9815417 -2.0800683 -2.9815417
ENSG00000223823 -2.981542 -2.9815417 -2.9815417 -2.9815417 -2.9815417plotDensities(cpm, col=pal[as.numeric(metaData$Species)], legend="topright")
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Log2 CPM
TMM/log2(CPM)
## Create edgeR object (dge) to calculate TMM normalization
dge_original <- DGEList(counts=as.matrix(counts_genes), genes=rownames(counts_genes), group = as.character(t(labels)))
dge_original <- calcNormFactors(dge_original)
tmm_cpm <- cpm(dge_original, normalized.lib.sizes=TRUE, log=TRUE, prior.count = 0.25)
head(cpm) NA18504 NA18510 NA18523 NA18499 NA18502
ENSG00000188976 0.9894703 1.620342 1.209959 1.068977 1.8425919
ENSG00000188157 3.0594442 1.985078 2.926705 2.916115 1.2946137
ENSG00000273443 1.6891590 1.412390 1.976621 1.540857 -0.3532791
ENSG00000217801 2.2551156 1.169332 3.547816 1.873190 0.3334631
ENSG00000237330 -2.9815417 -2.429988 -2.981542 -2.437603 -2.4245831
ENSG00000223823 -2.9815417 -2.981542 -2.981542 -2.981542 -2.9815417
NAPT30 NAPT91 NA3622 NA3659 NA18358
ENSG00000188976 -2.008556 -2.3855698 -2.4153553 -2.4615104 -2.9815417
ENSG00000188157 -1.212936 -0.7860576 -0.8558140 -0.8206567 -0.8020982
ENSG00000273443 0.492301 -1.9650589 -1.6935886 2.1415838 0.3987400
ENSG00000217801 1.136923 -0.6333308 0.3592322 2.9568407 1.2842881
ENSG00000237330 -2.981542 -2.9815417 -2.9815417 -2.0800683 -2.9815417
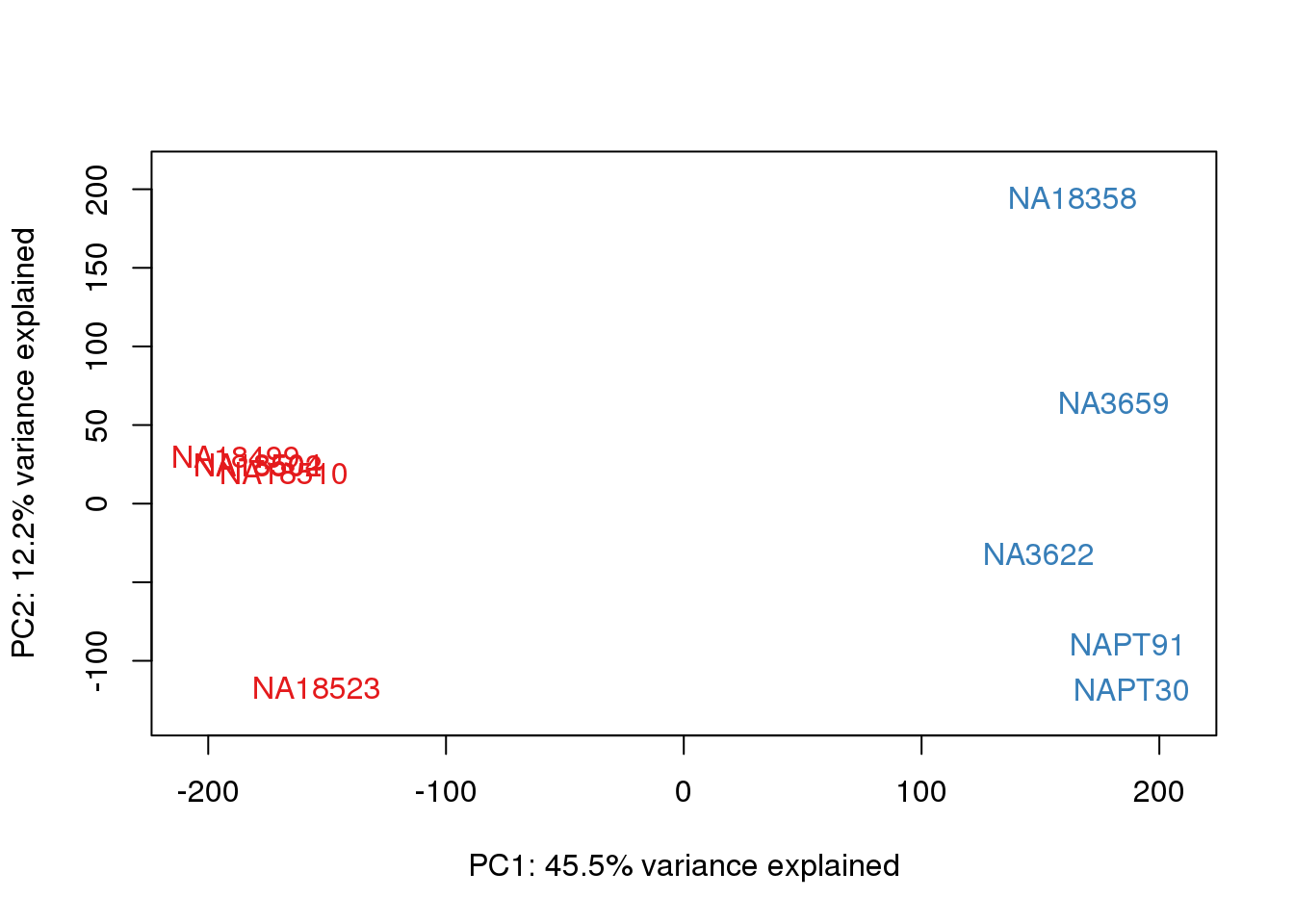
ENSG00000223823 -2.981542 -2.9815417 -2.9815417 -2.9815417 -2.9815417pca_genes <- prcomp(t(tmm_cpm), scale = F)
scores <- pca_genes$x
for (n in 1:2){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |

| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
# Plot library size
boxplot_library_size <- ggplot(dge_original$samples, aes(x=metaData$Species, y = dge_original$samples$lib.size, fill = metaData$Species)) + geom_boxplot()
boxplot_library_size + labs(title = "Library size by Species") + labs(y = "Library size") + labs(x = "Species") + guides(fill=guide_legend(title="Species"))
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
plotDensities(tmm_cpm, col=pal[as.numeric(metaData$Species)], legend="topright")
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Filter low expressed gene
Filter based on log2 cpm
filter log2(cpm >1) in at least 10 of the samples (2/3)
#filter counts
keep.exprs=rowSums(tmm_cpm>1) >8
counts_filtered= counts_genes[keep.exprs,]
plotDensities(counts_filtered, col=pal[as.numeric(metaData$Species)], legend="topright")
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
labels <- paste(metaData$Species, metaData$Line, sep=" ")
dge_in_cutoff <- DGEList(counts=as.matrix(counts_filtered), genes=rownames(counts_filtered), group = as.character(t(labels)))
dge_in_cutoff <- calcNormFactors(dge_in_cutoff)
cpm_in_cutoff <- cpm(dge_in_cutoff, normalized.lib.sizes=TRUE, log=TRUE, prior.count = 0.25)
head(cpm_in_cutoff) NA18504 NA18510 NA18523 NA18499 NA18502 NAPT30
ENSG00000186891 5.019760 5.041382 4.900641 5.711124 3.366537 4.651440
ENSG00000078808 6.869945 7.037620 7.477403 6.629313 6.741815 6.705657
ENSG00000176022 4.763855 4.687804 4.803574 4.617006 4.705504 4.849646
ENSG00000160087 4.849052 5.381048 5.119697 4.965717 5.086619 5.207984
ENSG00000131584 6.069280 4.920816 4.539908 4.737501 5.356339 5.133882
ENSG00000169972 3.357225 3.394890 3.555318 3.773273 3.315091 3.621735
NAPT91 NA3622 NA3659 NA18358
ENSG00000186891 3.175319 4.739154 6.460823 6.498061
ENSG00000078808 6.781323 6.788155 6.796852 7.076976
ENSG00000176022 5.566032 4.857458 5.289102 5.577640
ENSG00000160087 5.451318 5.542696 5.337421 5.414395
ENSG00000131584 5.709962 5.380906 5.258878 5.565531
ENSG00000169972 3.552346 4.077282 3.072501 3.321316hist(cpm_in_cutoff, xlab = "Log2(CPM)", main = "Log2(CPM) values for genes meeting the filtering criteria", breaks = 100 )
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Voom transformation:
Species <- factor(metaData$Species)
design <- model.matrix(~ 0 + Species)
head(design) SpeciesChimp SpeciesHuman
1 1 0
2 1 0
3 1 0
4 1 0
5 1 0
6 0 1colnames(design) <- gsub("Species", "", dput(colnames(design)))c("SpeciesChimp", "SpeciesHuman")Voom creates a random effect.
# Voom with individual as a random variable
cpm.voom<- voom(counts_filtered, design, normalize.method="quantile", plot=T)
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
boxplot(cpm.voom$E, col = pal[as.numeric(metaData$Species)],las=2)
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
plotDensities(cpm.voom, col = pal[as.numeric(metaData$Species)], legend = "topleft") 
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
Looks like i still have a skew on the lower side of the distribution.
# PCA
pca_genes <- prcomp(t(cpm.voom$E), scale = T)
scores <- pca_genes$x
eigsGene <- pca_genes$sdev^2
proportionG = eigsGene/sum(eigsGene)
plot(proportionG)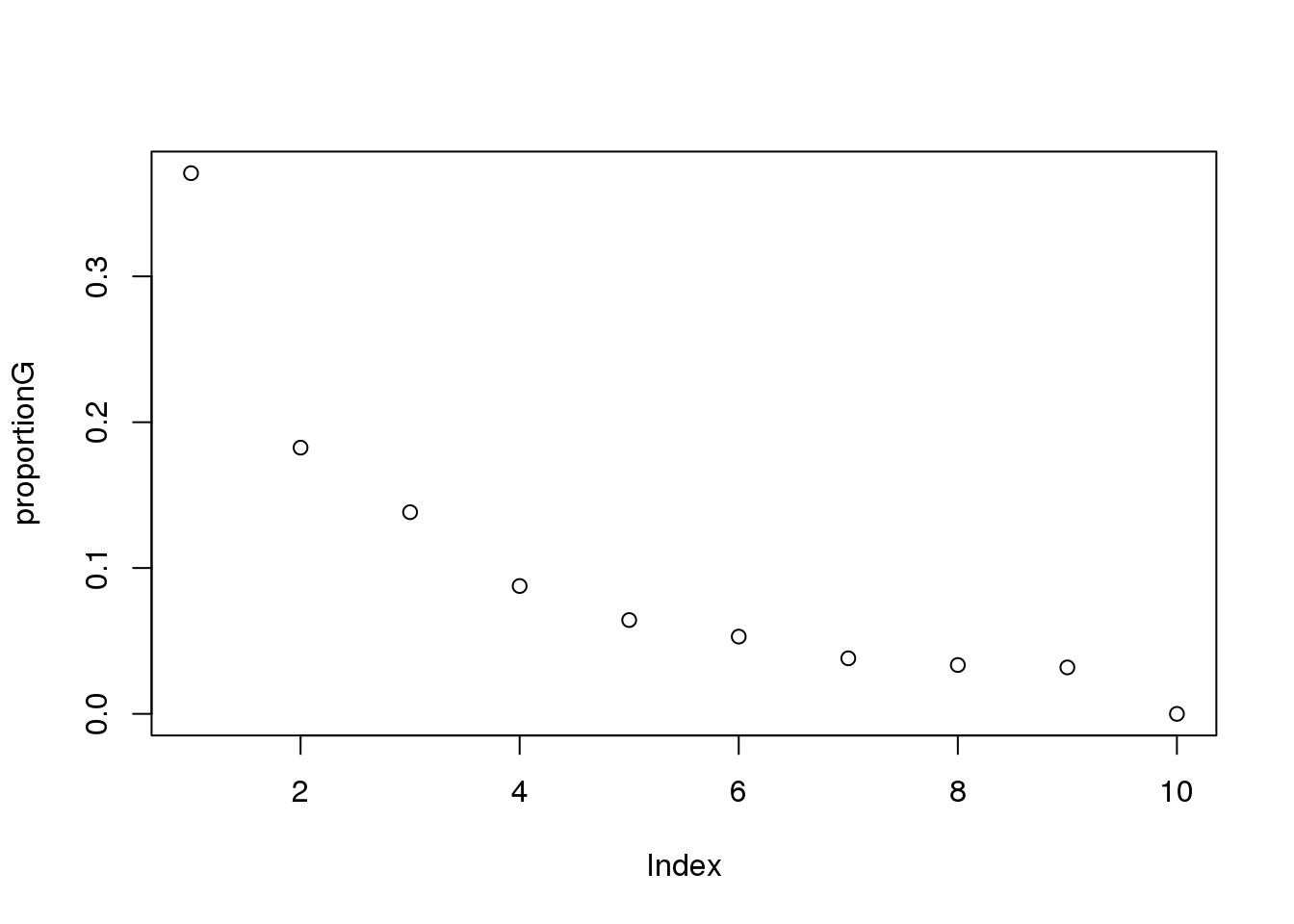
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
for (n in 1:2){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |

| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
#Clustering (original code from Julien Roux)
cors <- cor(cpm.voom$E, method="spearman", use="pairwise.complete.obs")
heatmap.2( cors, scale="none", col = colors, margins = c(12, 12), trace='none', denscol="white", labCol=labels, ColSideColors=pal[as.integer(as.factor(metaData$Species))], RowSideColors=pal[as.integer(as.factor(metaData$Species))+9], cexCol = 0.2 + 1/log10(15), cexRow = 0.2 + 1/log10(15))
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
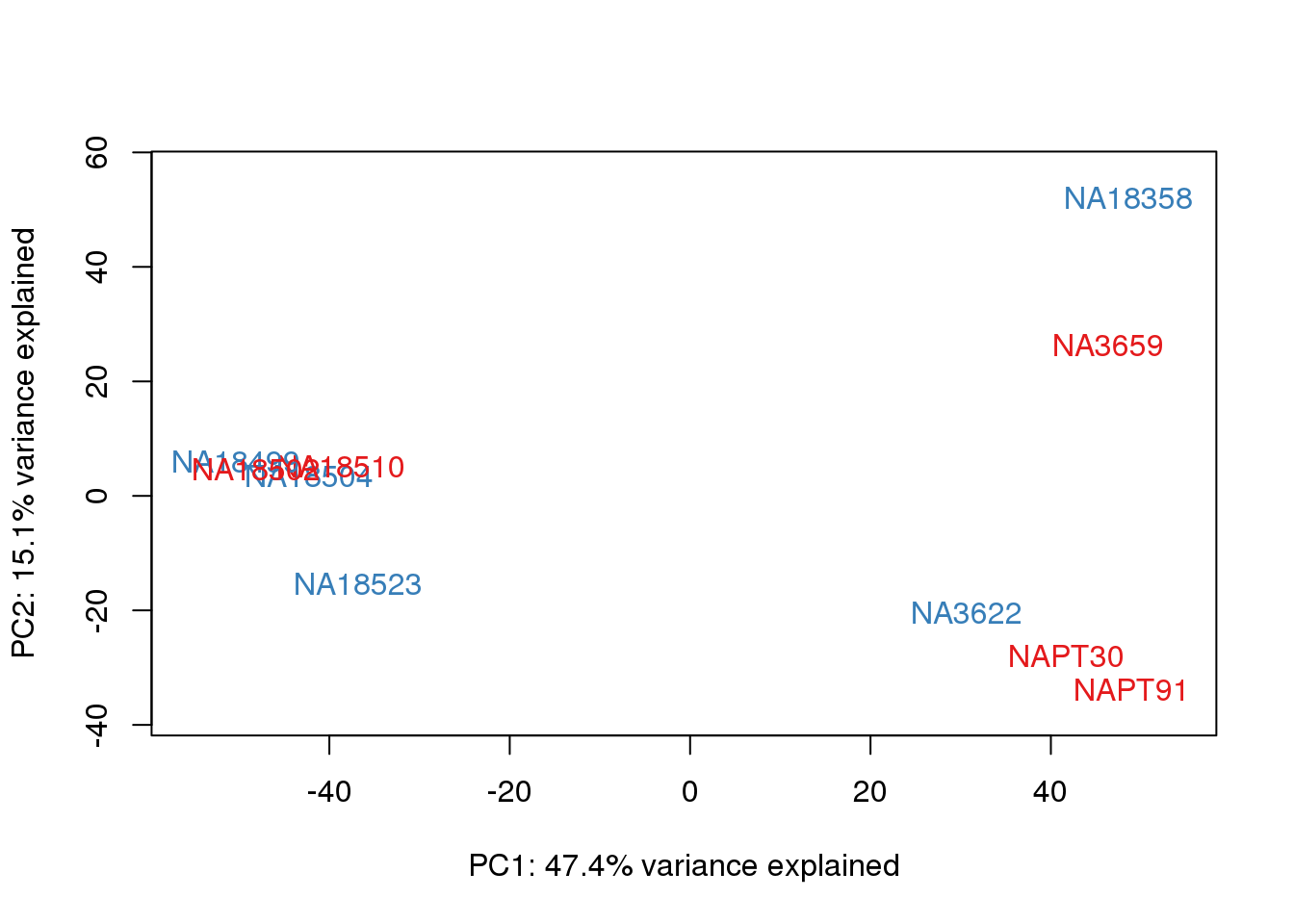
This is wierd. Normalization moves 2 samples to opposite species clusters but the samples that separate in the correlation are not those samples. 4973 and 18498 are the samples that looked funny on the original 3’ data. This may be a sample swap at the RNA stage. These samples were in the same extraction batch. It could have happened then. I will look into this more.
One thing I can do is look at the correlation between the PCs and other factors in the data.
# PCA
pca_genes <- prcomp(t(cpm.voom$E), scale = F)
scores <- pca_genes$x
for (n in 1:2){
col.v <- pal[as.integer(metaData$Collection)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |

| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
metaData$Extraction=as.factor(metaData$Extraction)
for (n in 1:2){
col.v <- pal[as.integer(metaData$Extraction)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |

| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
It does not look like batch (who collected or extraction date batch)
cols = brewer.pal(9, "Blues")
palC = colorRampPalette(cols)
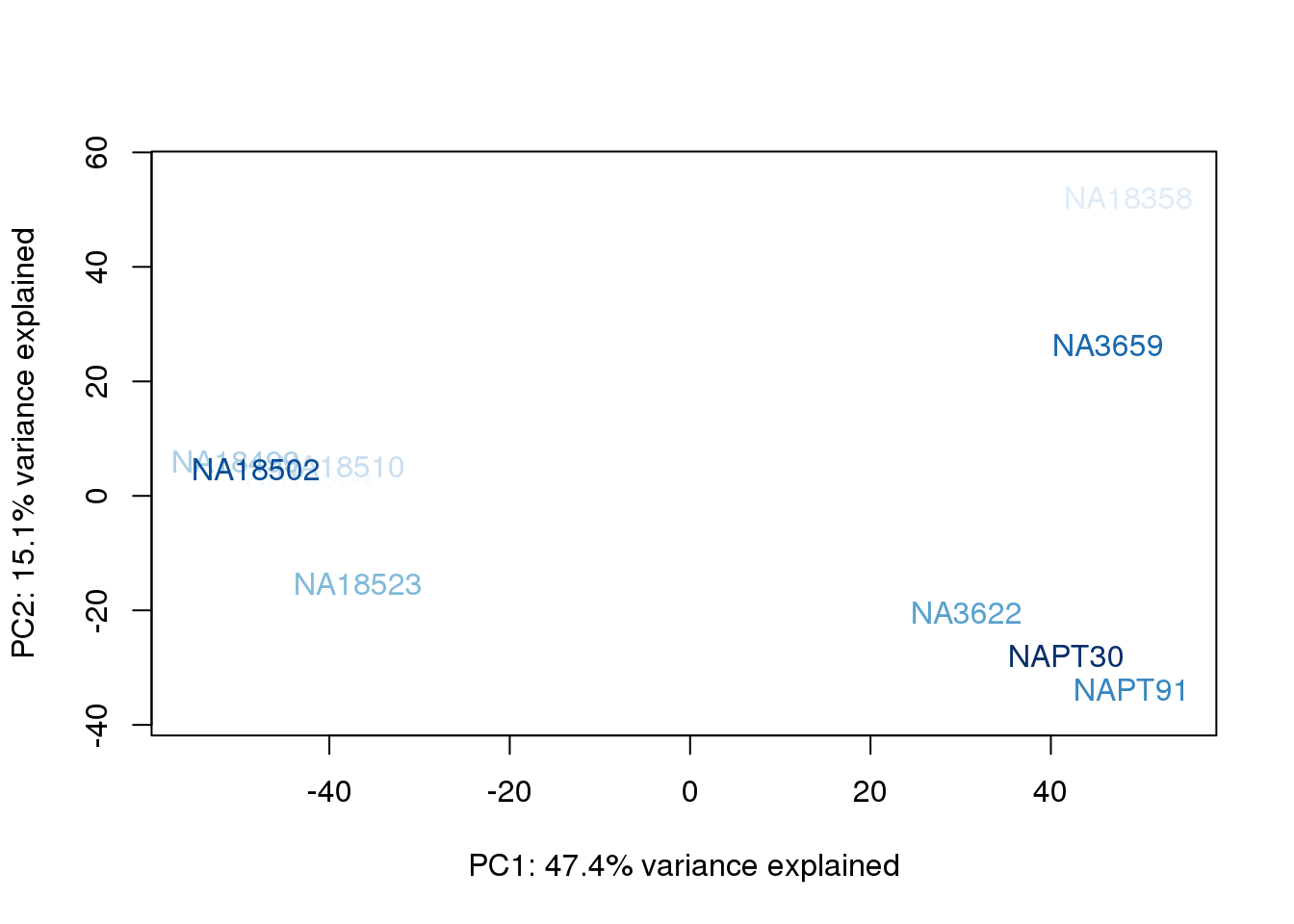
metaData$UndilutedAverageorder = findInterval(metaData$UndilutedAverage, sort(metaData$UndilutedAverage))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$UndilutedAverageorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |

| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
metaData$BioAConcorder = findInterval(metaData$BioAConc, sort(metaData$BioAConc))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$BioAConcorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |

| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
metaData$RinConcorder = findInterval(metaData$Rin, sort(metaData$Rin))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$RinConcorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
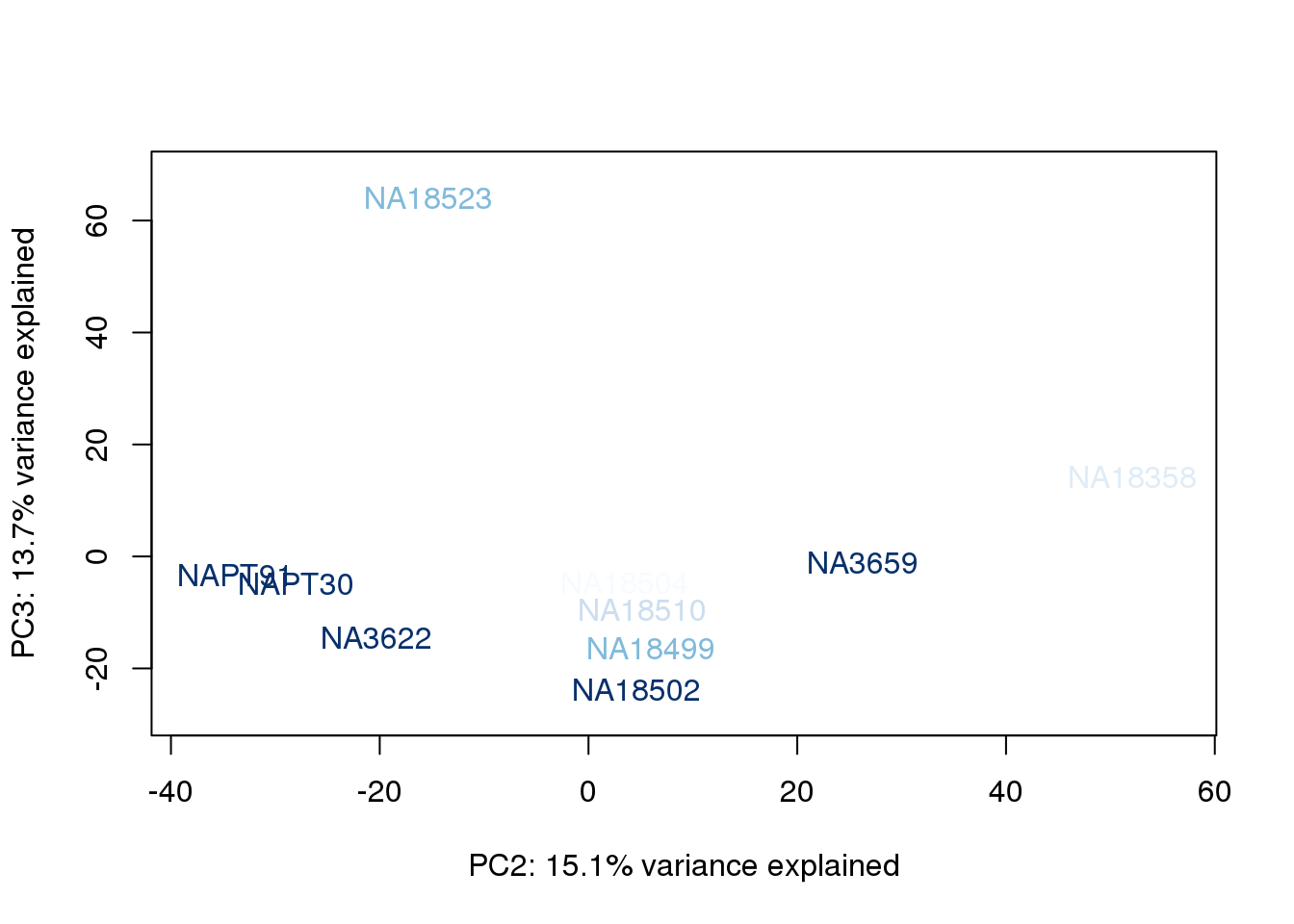
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
The samples do not cluster by collection concentration, RNA rin score or RNA concentration.
metaData$AssignedOrthoorder = findInterval(metaData$AssignedOrtho, sort(metaData$AssignedOrtho))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$AssignedOrthoorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
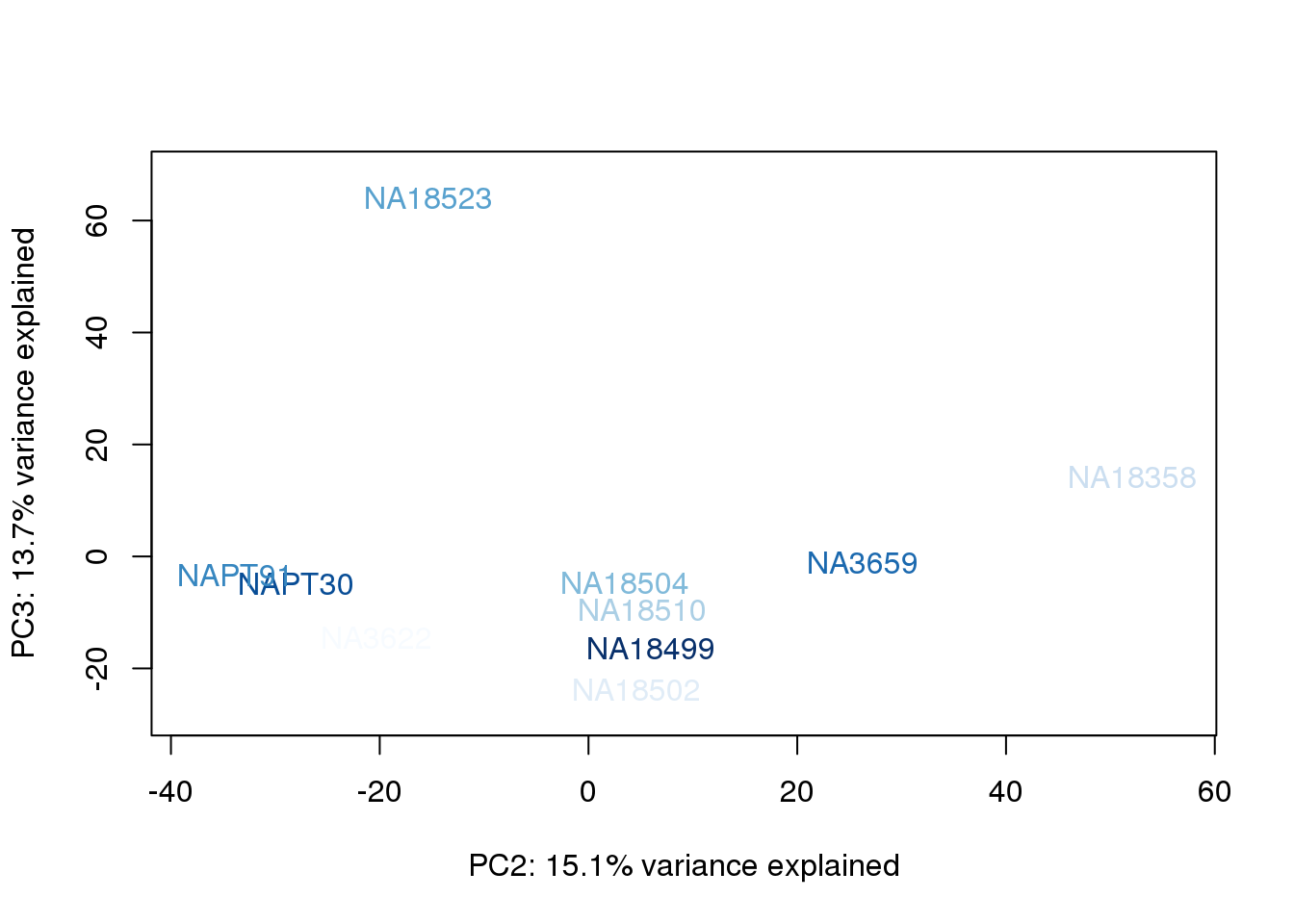
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
PCA heatmap: Code from Michelle Ward:
x.pca <- pca_genes
tech_factors <- metaData
tech_factors_sum <- tech_factors[,c(2:14)] %>% select(-CollectionDate)
p_comps <- 1:6
pc_cov_cor <- matrix(nrow = ncol(tech_factors_sum), ncol = length(p_comps),
dimnames = list(colnames(tech_factors_sum), colnames(x.pca$x)[p_comps]))
for (pc in p_comps) {
for (covariate in 1:ncol(tech_factors_sum)) {
lm_result <- lm(x.pca$x[, pc] ~ tech_factors_sum[, covariate])
r2 <- summary(lm_result)$r.squared
pc_cov_cor[covariate, pc] <- r2
}
}
pc_cov_pval <- matrix(nrow = ncol(tech_factors_sum), ncol = length(p_comps),
dimnames = list(colnames(tech_factors_sum), colnames(x.pca$x)[p_comps]))
for (pc in p_comps) {
for (covariate_2 in 1:ncol(tech_factors_sum)) {
lm_result_2 <- lm(x.pca$x[, pc] ~ tech_factors_sum[, covariate_2])
pval <- anova(lm_result_2)$'Pr(>F)'[1]
pc_cov_pval[covariate_2, pc] <- pval
}
}
PCs <- c("PC1", "PC2", "PC3", "PC4", "PC5", "PC6")
Tech_fac <- colnames(tech_factors_sum)
#Tech_fac <- c("Species", "Individual", "O2.", "Condition" , "Sex", "RIN" , "CO2", "Purity_high", "Purity_med" ,
#"Expt_Batch", "RNA_Batch", "Library_Batch", "Seq_pool", "Episomal_integration" )
heatmap.2(as.matrix(pc_cov_cor[Tech_fac,PCs]),col=brewer.pal(4, "Greens"), trace="none",
Rowv=FALSE, Colv=FALSE, key=T, main="Cor. PCs & tech factors", dendrogram="none",
key.title=NA, cexRow=0.9, cexCol=0.9)
| Version | Author | Date |
|---|---|---|
| 9ecd769 | brimittleman | 2019-11-20 |
log10_pc_cov_pval <- -log(pc_cov_pval)
heatmap.2(as.matrix(log10_pc_cov_pval[Tech_fac,PCs]), col=brewer.pal(9, "Greens"), trace="none",
Rowv=FALSE, Colv=FALSE, key=T, main="-log10 pval of cor. PCs & tech factors", dendrogram="none",
key.title=NA, cexRow=0.9, cexCol=0.9)
Test for DE
fit.cpm.voom = lmFit(cpm.voom, design, plot=T)
head(coef(fit.cpm.voom)) Chimp Human
ENSG00000186891 4.795816 5.095375
ENSG00000078808 6.947449 6.828201
ENSG00000176022 4.693342 5.230150
ENSG00000160087 5.073666 5.402331
ENSG00000131584 5.103075 5.410320
ENSG00000169972 3.468275 3.544200contr <- makeContrasts(Chimp - Human, levels = colnames(coef(fit.cpm.voom)))
contr Contrasts
Levels Chimp - Human
Chimp 1
Human -1tmp <- contrasts.fit(fit.cpm.voom, contr)
tmp <- eBayes(tmp)
top.table <- topTable(tmp, sort.by = "P", n = Inf)
head(top.table, 20) logFC AveExpr t P.Value adj.P.Val
ENSG00000105372 4.885120 8.015919 49.50888 1.051297e-13 4.138512e-10
ENSG00000204463 -6.291373 5.272106 -49.50052 1.053142e-13 4.138512e-10
ENSG00000205531 -5.980720 5.127592 -47.39996 1.651394e-13 4.138512e-10
ENSG00000145741 6.272278 5.704197 47.26164 1.702216e-13 4.138512e-10
ENSG00000133112 6.037437 8.611334 45.96615 2.270894e-13 4.416889e-10
ENSG00000142937 5.708662 8.279388 44.22833 3.386317e-13 5.488655e-10
ENSG00000142541 6.366791 6.767260 39.58302 1.069006e-12 1.485155e-09
ENSG00000100316 6.010986 8.347096 37.36455 1.941895e-12 2.360616e-09
ENSG00000147604 -6.194041 5.570670 -34.97357 3.847768e-12 4.157728e-09
ENSG00000071082 5.845714 6.590811 31.97632 9.707041e-12 9.440097e-09
ENSG00000183020 -4.817991 4.189720 -30.95079 1.358744e-11 1.201253e-08
ENSG00000186298 3.182017 6.870342 30.14030 1.786324e-11 1.447667e-08
ENSG00000198242 4.240871 5.024421 25.20195 1.125488e-10 8.042832e-08
ENSG00000165672 2.443572 5.535737 25.13245 1.157837e-10 8.042832e-08
ENSG00000153395 -4.940243 5.261343 -24.69814 1.384552e-10 8.829558e-08
ENSG00000116478 3.404755 5.871950 24.58273 1.452678e-10 8.829558e-08
ENSG00000089009 2.275400 7.530109 24.11044 1.772287e-10 1.013852e-07
ENSG00000171863 3.179473 6.726062 22.92492 2.969928e-10 1.554700e-07
ENSG00000105640 -2.873676 8.228281 -22.87459 3.037460e-10 1.554700e-07
ENSG00000198034 6.351635 7.259074 22.55804 3.502709e-10 1.689663e-07
B
ENSG00000105372 21.10198
ENSG00000204463 19.68397
ENSG00000205531 19.45798
ENSG00000145741 19.60711
ENSG00000133112 20.53890
ENSG00000142937 20.21086
ENSG00000142541 18.80122
ENSG00000100316 18.80876
ENSG00000147604 17.57624
ENSG00000071082 17.14047
ENSG00000183020 16.48622
ENSG00000186298 16.88711
ENSG00000198242 14.96025
ENSG00000165672 15.09170
ENSG00000153395 14.76334
ENSG00000116478 14.86424
ENSG00000089009 14.74940
ENSG00000171863 14.23110
ENSG00000105640 14.21959
ENSG00000198034 14.02234length(which(top.table$adj.P.Val < 0.05))[1] 3200Make a table to plot:
-log10(bh adjusted pval) vs logFC (log3 fold change)
top.table=top.table %>% mutate(Species=ifelse(logFC > 1 & adj.P.Val<.05, "Chimp", ifelse(logFC < -1 & adj.P.Val< .05, "Human", "Neither")))
ggplot(top.table, aes(x=logFC, y= -log10(adj.P.Val))) + geom_point(aes(col=Species), alpha=.3)
summary(decideTests(tmp)) Chimp - Human
Down 1584
NotSig 6525
Up 1616
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] reshape2_1.4.3 RColorBrewer_1.1-2 VennDiagram_1.6.20
[4] futile.logger_1.4.3 R.utils_2.7.0 R.oo_1.22.0
[7] R.methodsS3_1.7.1 edgeR_3.24.0 limma_3.38.2
[10] gplots_3.0.1 scales_1.0.0 forcats_0.3.0
[13] stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2
[16] readr_1.3.1 tidyr_0.8.3 tibble_2.1.1
[19] ggplot2_3.1.1 tidyverse_1.2.1 workflowr_1.5.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.2 locfit_1.5-9.1 lubridate_1.7.4
[4] lattice_0.20-38 gtools_3.8.1 assertthat_0.2.0
[7] rprojroot_1.3-2 digest_0.6.18 R6_2.3.0
[10] cellranger_1.1.0 plyr_1.8.4 futile.options_1.0.1
[13] backports_1.1.2 evaluate_0.12 httr_1.3.1
[16] pillar_1.3.1 rlang_0.4.0 lazyeval_0.2.1
[19] readxl_1.1.0 rstudioapi_0.10 gdata_2.18.0
[22] whisker_0.3-2 rmarkdown_1.10 labeling_0.3
[25] munsell_0.5.0 broom_0.5.1 compiler_3.5.1
[28] httpuv_1.4.5 modelr_0.1.2 pkgconfig_2.0.2
[31] htmltools_0.3.6 tidyselect_0.2.5 crayon_1.3.4
[34] withr_2.1.2 later_0.7.5 bitops_1.0-6
[37] nlme_3.1-137 jsonlite_1.6 gtable_0.2.0
[40] formatR_1.5 git2r_0.26.1 magrittr_1.5
[43] KernSmooth_2.23-15 cli_1.1.0 stringi_1.2.4
[46] fs_1.3.1 promises_1.0.1 xml2_1.2.0
[49] generics_0.0.2 lambda.r_1.2.3 tools_3.5.1
[52] glue_1.3.0 hms_0.4.2 yaml_2.2.0
[55] colorspace_1.3-2 caTools_1.17.1.1 rvest_0.3.2
[58] knitr_1.20 haven_1.1.2