Difference in protein and APA not expression
Briana Mittleman
3/7/2020
Last updated: 2020-04-23
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/TrialFiltersMeta.txt.sb-9845453e-R58Y0Q/
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-284518db-RGf0kd/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/AREstabilityScores.Rmd
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/HandC.TvN
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/annotationBias.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: code/._AlignmentScores.sh
Untracked: code/._BothFCMM.sh
Untracked: code/._BothFCMMPrim.sh
Untracked: code/._BothFCnewOInclusive.sh
Untracked: code/._ChimpStarMM2.sh
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._ClosestorthoEx.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._Filter255MM.sh
Untracked: code/._FilterPrimSec.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._MismatchNumbers.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._SAF2Bed.py
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._StarMM2.sh
Untracked: code/._TestFC.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2Bedbothstrand.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chimpMultiCov.sh
Untracked: code/._chimpMultiCov255.sh
Untracked: code/._chimpMultiCovInclusive.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopGeneral.ph
Untracked: code/._extractPhylopGeneral.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._filterPrimaryread.py
Untracked: code/._filterSecondaryread.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixFCheadforExp.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._humanMultiCov.sh
Untracked: code/._humanMultiCov255.sh
Untracked: code/._humanMultiCov_inclusive.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapMMandOrthoexon.sh
Untracked: code/._overlapPASandOrthoexon.sh
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantLiftedPASPrimary.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ALLPAS_sequenceDF.err
Untracked: code/ALLPAS_sequenceDF.out
Untracked: code/AlignmentScores.err
Untracked: code/AlignmentScores.out
Untracked: code/AlignmentScores.sh
Untracked: code/BothFCMM.err
Untracked: code/BothFCMM.out
Untracked: code/BothFCMM.sh
Untracked: code/BothFCMMPrim.err
Untracked: code/BothFCMMPrim.out
Untracked: code/BothFCMMPrim.sh
Untracked: code/BothFCnewOInclusive.sh
Untracked: code/BothFCnewOInclusive.sh.err
Untracked: code/BothFCnewOInclusive.sh.out
Untracked: code/ChimpStarMM2.err
Untracked: code/ChimpStarMM2.out
Untracked: code/ChimpStarMM2.sh
Untracked: code/ClassifyLeafviz.sh
Untracked: code/ClosestorthoEx.err
Untracked: code/ClosestorthoEx.out
Untracked: code/ClosestorthoEx.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DTUTR.sh
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/Filter255.err
Untracked: code/Filter255.out
Untracked: code/Filter255MM.sh
Untracked: code/FilterPrimSec.err
Untracked: code/FilterPrimSec.out
Untracked: code/FilterPrimSec.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindDomXCutoff.py
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/GetTopminus2Usage.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/IntersectMMandOrtho.err
Untracked: code/IntersectMMandOrtho.out
Untracked: code/IntersectPASandOrtho.err
Untracked: code/IntersectPASandOrtho.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/MismatchNumbers.err
Untracked: code/MismatchNumbers.out
Untracked: code/MismatchNumbers.sh
Untracked: code/NuclearDTUTR.err
Untracked: code/NuclearDTUTRt.out
Untracked: code/NuclearPlotsDEandDiffDom_4.err
Untracked: code/NuclearPlotsDEandDiffDom_4.out
Untracked: code/NuclearPlotsDEandDiffDom_4.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/PlotNuclearUsagebySpecies_DF_DEout.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunNewDom.err
Untracked: code/RunNewDom.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/SAF2Bed.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/StarMM2.err
Untracked: code/StarMM2.out
Untracked: code/StarMM2.sh
Untracked: code/TestFC.err
Untracked: code/TestFC.out
Untracked: code/TestFC.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/UTR2FASTA.py
Untracked: code/Upstream10Bases_general.py
Untracked: code/allPASSeq_df.sh
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/apaQTLsnakefiltPAS.err
Untracked: code/apaQTLsnakefiltPAS.out
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2Bedbothstrand.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chimpMultiCov.err
Untracked: code/chimpMultiCov.out
Untracked: code/chimpMultiCov.sh
Untracked: code/chimpMultiCov255.sh
Untracked: code/chimpMultiCovInclusive.err
Untracked: code/chimpMultiCovInclusive.out
Untracked: code/chimpMultiCovInclusive.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopGeneral.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterPrimaryread.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSecondaryread.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFCheadforExp.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getAlloverlap.py
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/humanMultiCov.err
Untracked: code/humanMultiCov.out
Untracked: code/humanMultiCov.sh
Untracked: code/humanMultiCov255.err
Untracked: code/humanMultiCov255.out
Untracked: code/humanMultiCov255.sh
Untracked: code/humanMultiCovInclusive.err
Untracked: code/humanMultiCovInclusive.out
Untracked: code/humanMultiCov_inclusive.sh
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/mergeChimpRNA.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapMMandOrthoexon.sh
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapPASandOrthoexon.sh
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quantLiftedPASPrimary.err
Untracked: code/quantLiftedPASPrimary.out
Untracked: code/quantLiftedPASPrimary.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runChimpDiffIsoDF.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runHumanDiffIsoDF.sh
Untracked: code/runNewDom.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_dF.err
Untracked: code/run_Humanleafcutter_dF.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePAS_Human.batch
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.batch
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Chimp_tvN_DF.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN_DF.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/test.txt
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._TrialFiltersMeta.txt
Untracked: data/._TrialFiltersMeta.txt.sb-9845453e-R58Y0Q
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/AREelements/
Untracked: data/BaseComp/
Untracked: data/CleanLiftedPeaks_FC_primary/
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffDomandDE_example/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DomDefGreaterX/
Untracked: data/DomStructure_4/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OrthoExonBed/
Untracked: data/OverlapBenchmark/
Untracked: data/OverlappingPAS/
Untracked: data/PAS/
Untracked: data/PAS_SAF/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/Pol2Chip/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/TestAnnoBiasOE/
Untracked: data/TestMM2/
Untracked: data/TestMM2_AS/
Untracked: data/TestMM2_PrimaryRead/
Untracked: data/TestMM2_SeondaryRead/
Untracked: data/TestMM2_mismatch/
Untracked: data/TestMM2_quality/
Untracked: data/TestWithinMergePAS/
Untracked: data/Test_FC_methods/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalFractionPAS/
Untracked: data/TotalHvC/
Untracked: data/TrialFiltersMeta.txt
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/bioGRID/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_extra.txt
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/miRNA/
Untracked: data/multimap/
Untracked: data/orthoUTR/
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/testQuant/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dAPAandDomEnrich.png
Untracked: output/dEandDomEnrich.png
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/DiffTop2SecondDom.Rmd
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/ExploredAPA_DF.Rmd
Modified: analysis/MMExpreiment.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/PTM_analysis.Rmd
Modified: analysis/TotalDomStructure.Rmd
Modified: analysis/TotalVNuclearBothSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/changeMisprimcut.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/pol2.Rmd
Modified: analysis/signalsites_doublefilter.Rmd
Modified: analysis/speciesSpecific.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | c1bc496 | brimittleman | 2020-04-23 | add interaction density, dapa and e and order proble |
| html | d969183 | brimittleman | 2020-04-23 | Build site. |
| Rmd | 3770355 | brimittleman | 2020-04-23 | add overlap diff |
| html | 3790efa | brimittleman | 2020-04-23 | Build site. |
| Rmd | e513e9f | brimittleman | 2020-04-23 | add dot chart |
| html | 5d82297 | brimittleman | 2020-04-22 | Build site. |
| Rmd | 1e814ec | brimittleman | 2020-04-22 | change colors |
| html | 6d8725a | brimittleman | 2020-04-10 | Build site. |
| Rmd | fbabfb7 | brimittleman | 2020-04-10 | remove 18499 |
| html | c2a0778 | brimittleman | 2020-04-06 | Build site. |
| Rmd | 9676d72 | brimittleman | 2020-04-06 | updated anno |
| html | 30bfaaa | brimittleman | 2020-03-16 | Build site. |
| Rmd | 94211e8 | brimittleman | 2020-03-16 | add change misprime analysis and dapa qtls |
| html | f33ef5a | brimittleman | 2020-03-08 | Build site. |
| Rmd | 6146c33 | brimittleman | 2020-03-08 | add effect corr |
| html | 25a4790 | brimittleman | 2020-03-07 | Build site. |
| Rmd | c6a11f0 | brimittleman | 2020-03-07 | add expression indep |
library(tidyverse)── Attaching packages ────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(UpSetR)
library(ggpubr)Loading required package: magrittr
Attaching package: 'magrittr'The following object is masked from 'package:purrr':
set_namesThe following object is masked from 'package:tidyr':
extractUpload:
Protein=read.table("../data/Khan_prot/HC_SigProtein.txt", header = T, stringsAsFactors = F)%>% dplyr::rename("gene"=gene.symbol)
nameID=read.table("../../genome_anotation_data/ensemble_to_genename.txt",sep="\t", header = T, stringsAsFactors = F)
DEgenes=read.table("../data/DiffExpression/DE_genes.txt", header = F,col.names = c("Gene_stable_ID"),stringsAsFactors = F) %>% inner_join(nameID, by="Gene_stable_ID") %>% dplyr::select(Gene.name) %>% dplyr::rename("gene"=Gene.name)
NucAPA=read.table("../data/DiffIso_Nuclear_DF/SignifianceEitherGENES_Nuclear.txt",header = T,stringsAsFactors = F)I will do this first with these then I can start to look at it by significance.
APAandPnotE= NucAPA %>% inner_join(Protein, by="gene") %>% anti_join(DEgenes,by="gene")listInput_nucOnly <- list(DE=DEgenes$gene, DAPA=NucAPA$gene, DP=Protein$gene)
#upset(fromList(listInput_nosplice), queries = list(list(query=intersects, params=list("DAPA", "DT", "DP"), color="red", active=T,query.name="APA, Ribo, Protein"),list(query=intersects, params=list("DE", "DT", "DP"), color="orange", active=T, query.name="Expression,Ribo, Protein"),list(query=intersects, params=list("DAPA", "DT"), color="blue", active=T, query.name="APA,Ribo") ,list(query=intersects, params=list("DAPA", "DP"), color="purple", active=T, query.name="APA, Protein"),list(query=intersects, params=list("DAPA", "DE"), color="green", active=T, query.name="APA, Expression")), order.by = "freq", query.legend = "bottom")
upset(fromList(listInput_nucOnly), order.by = "freq", keep.order = T,empty.intersections = "on", queries = list(list(query=intersects, params=list("DAPA", "DP"), color="darkorchid4", active=T,query.name="APA, Protein")))
90 of these genes.
Learn about these genes.
Selection:
model.num.rna: : 1 = mRNA expression level pattern consistent with directional selection along human lineage, 2 = mRNA expression level pattern consistent with directional selection along chimpanzee lineage, 3 = undetermined pattern, 4 = patterns with no significant difference between mean expression levels; 5 = evidence for relaxation of constraint along human lineage, 6 = evidence of relaxation of constraint along chimpanzee lineage
model.num.protein: 1 = protein expression level pattern consistent with directional selection along human lineage, 2 = protein expression level pattern consistent with directional selection along chimpanzee lineage, 3 = undetermined pattern, 4 = patterns with no significant difference between mean expression levels; 5 = evidence for relaxation of constraint along human lineage, 6 = evidence of relaxation of constraint along chimpanzee lineage
KhanData=read.csv("../data/Khan_prot/Khan_TableS4.csv",stringsAsFactors = F) %>% dplyr::select(gene.symbol,contains("model") ) %>% dplyr::rename("gene"=gene.symbol, "Protein"=model.num.protein, "RNA"=model.num.rna)
APAandPnotE_sel= APAandPnotE %>% inner_join(KhanData,by="gene")Plot the information about the RNA and protein for these:
APAandPnotE_sel_g=APAandPnotE_sel %>% dplyr::select(gene, Protein, RNA) %>% gather("Set", "Model", -gene)
APAandPnotE_sel_g$Model= as.factor(APAandPnotE_sel_g$Model)
ggplot(APAandPnotE_sel_g,aes(x=Model, by=Set, fill=Set)) + geom_bar(stat="count", position="dodge") + scale_fill_brewer(palette = "RdYlBu")
Plot protein only:
APAandPnotE_sel_gOnlyP= APAandPnotE_sel_g %>% filter(Set=="Protein")
APAandPnotE_sel_gOnlyP$Model= as.factor(APAandPnotE_sel_gOnlyP$Model)
ggplot(APAandPnotE_sel_gOnlyP,aes(x=Model)) + geom_bar(stat="count", position="dodge", fill="darkorchid4") + labs(y="Number of Genes", x="Protein Selection Model", title="Protein and APA differences\n no difference in Expression") + scale_x_discrete( labels=c("Selection Human","Selection Chimp","Undetermined","No mean difference","Relaxation in Chimp"))+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16)) 
The genes in 1,2,5,6 are interesting.
APAandPnotE_selCalled= APAandPnotE_sel_g %>% filter(Set=="Protein", Model %in% c(1,2,5,6))There are 20 of these genes:
APAandPnotE_selCalled gene Set Model
1 RRM1 Protein 1
2 SART3 Protein 1
3 SUGT1 Protein 2
4 VPS36 Protein 1
5 ATP6V1D Protein 1
6 GALNT2 Protein 2
7 GNAI3 Protein 2
8 SEC22B Protein 2
9 WDR77 Protein 2
10 KYNU Protein 2
11 PPIL3 Protein 1
12 CPOX Protein 2
13 MANBA Protein 1
14 BNIP1 Protein 1
15 CCT5 Protein 2
16 CYFIP2 Protein 1
17 MYO6 Protein 2
18 CUL1 Protein 2
19 VPS41 Protein 1
20 STOM Protein 1Where are the differential PAS in these genes:
#APAandPnotE_sel_gOnlyP
Meta=read.table("../data/PAS_doubleFilter/PAS_5perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = T) %>% dplyr::rename("ChimpUsage"=Chimp, "HumanUsage"=Human)
NucAPAres=read.table("../data/DiffIso_Nuclear_DF/AllPAS_withGeneSig.txt", header = T,stringsAsFactors = F) %>% inner_join(Meta, by=c("chr", "start","end", "gene"))Warning: Column `chr` joining character vector and factor, coercing into
character vectorWarning: Column `gene` joining character vector and factor, coercing into
character vectorNucAPAres_DP= NucAPAres %>% filter(gene %in%APAandPnotE_sel_gOnlyP$gene ) %>% filter(SigPAU2=="Yes")
NucAPAresSig=NucAPAres %>% filter(SigPAU2=="Yes")THere are 154 PAS in this set:
ggplot(NucAPAres_DP,aes(x=loc,fill=loc))+ geom_bar(stat="count") + scale_fill_brewer(palette = "RdYlBu")+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16), legend.position = "false") + labs(x="", y="Number of PAS", title="Expression independent PAS locations")
Enrichment for this:
Compare to all of the significant in that location.
NucAPAres_sig= NucAPAres %>% filter(SigPAU2=="Yes") %>% mutate(dPnotE=ifelse(PAS %in% NucAPAres_DP$PAS,"Yes", "No"))
enrich=c()
pval=c()
for (i in c("cds", "end", "intron", "utr3")){
x=nrow(NucAPAres_sig %>% filter(dPnotE=="Yes", loc==i))
m=nrow(NucAPAres_sig %>% filter( loc==i))
n=nrow(NucAPAres_sig %>% filter(loc!=i))
k=nrow(NucAPAres_sig %>% filter(dPnotE=="Yes"))
N=nrow(NucAPAres_sig)
pval=c(pval, phyper(x-1,m,n,k,lower.tail=F))
enrichval=(x/k)/(m/N)
enrich=c(enrich, enrichval)
}
enrich[1] 1.3100158 0.3521451 0.7012860 1.2145339pval[1] 0.144079246 0.993503594 0.976950002 0.005705065NucAPAres_DPLocEnrich=NucAPAres_DP %>% group_by(loc) %>% summarise(n=n()) %>% bind_cols(enrichment=enrich, pvalue=pval)
ggplot(NucAPAres_DPLocEnrich, aes(x=loc, y=n, fill=loc)) + geom_bar(stat="identity") + scale_fill_brewer(palette = "RdYlBu")+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16), legend.position = "false") + labs(x="", y="Number of PAS", title="Expression independent PAS locations")+ geom_text(aes(label=paste("Enrichment=",round(enrichment,2), "X", sep=""), vjust=0)) +geom_text(aes(label=paste("Pval=",round(pvalue,3), sep=""), vjust=2))
Interactions:
Are there differences in protien interactions for these.
Interactions=read.table("../data/bioGRID/GeneswInteractions.txt",stringsAsFactors = F, header = T)
OrthoUTR=read.table("../data/orthoUTR/HumanDistal3UTR.sort.bed", col.names = c("chr",'start','end','gene','score','strand'),stringsAsFactors = F) %>% mutate(length=end-start) %>% select(gene, length)
InteractionsAPA=Interactions %>%filter(gene %in% NucAPAresSig$gene) %>% mutate(dPnotE=ifelse(gene %in% NucAPAres_DP$gene, "Yes", "No"))%>% inner_join(OrthoUTR, by="gene") %>% mutate(density=nInt/length)
ggplot(InteractionsAPA,aes(x=dPnotE, y=log10(nInt+1),fill=dPnotE)) + geom_boxplot(notch = T) + stat_compare_means() + scale_fill_brewer(palette = "Set1")+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16), legend.position = "false") + labs(x="Gene in Expression independent set", y="log10(Number of Protein Interactions)", title="Protein Interactions for Expression \nindependent dAPA genes")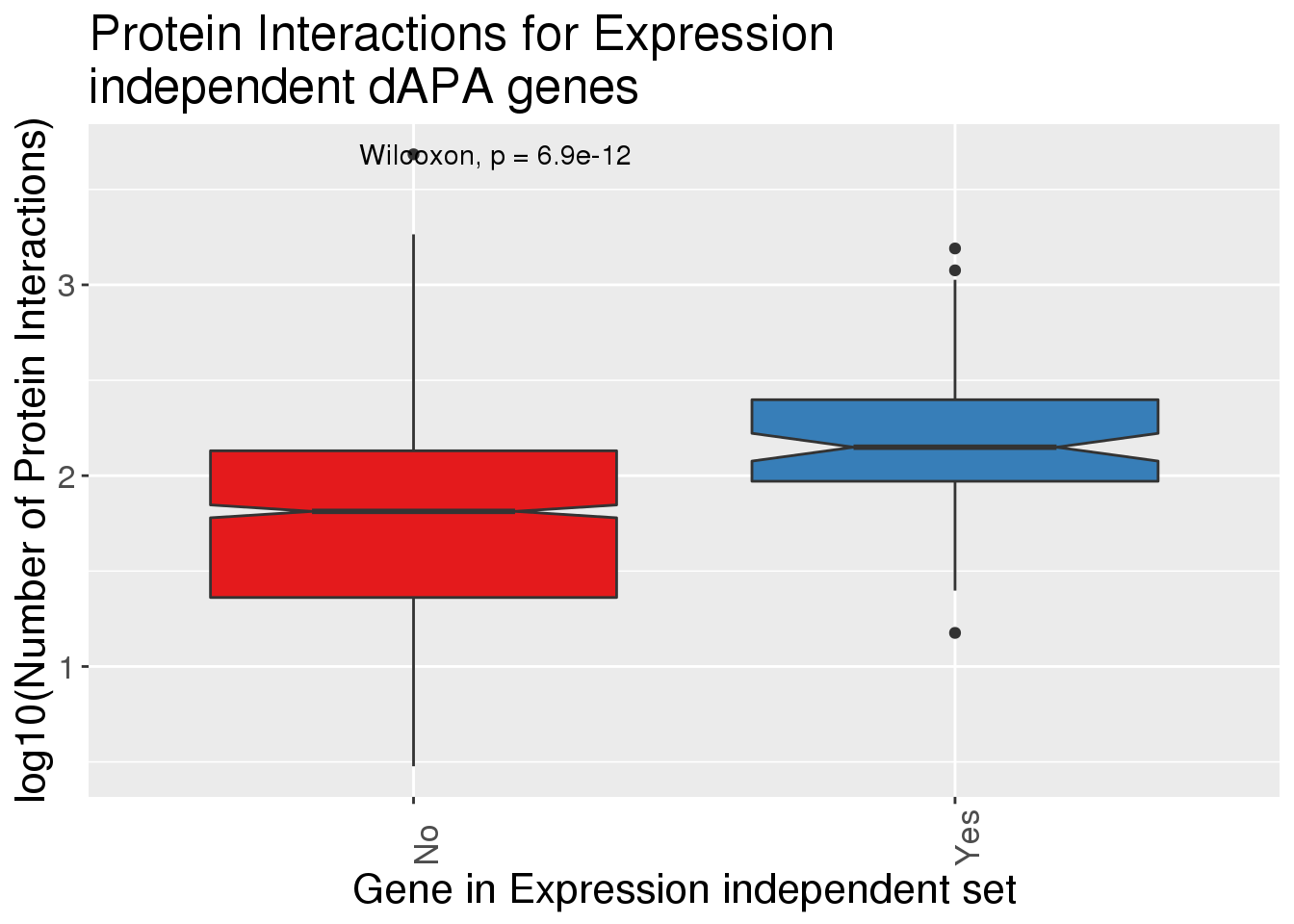
Plot density?
ggplot(InteractionsAPA,aes(x=dPnotE, y=log10(density),fill=dPnotE)) + geom_boxplot(notch = T) + stat_compare_means() + scale_fill_brewer(palette = "Set1")+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16), legend.position = "false") + labs(x="Gene in Expression independent set", y="log10(UTR density of interactions)", title="Protein Interactions for Expression \nindependent dAPA genes")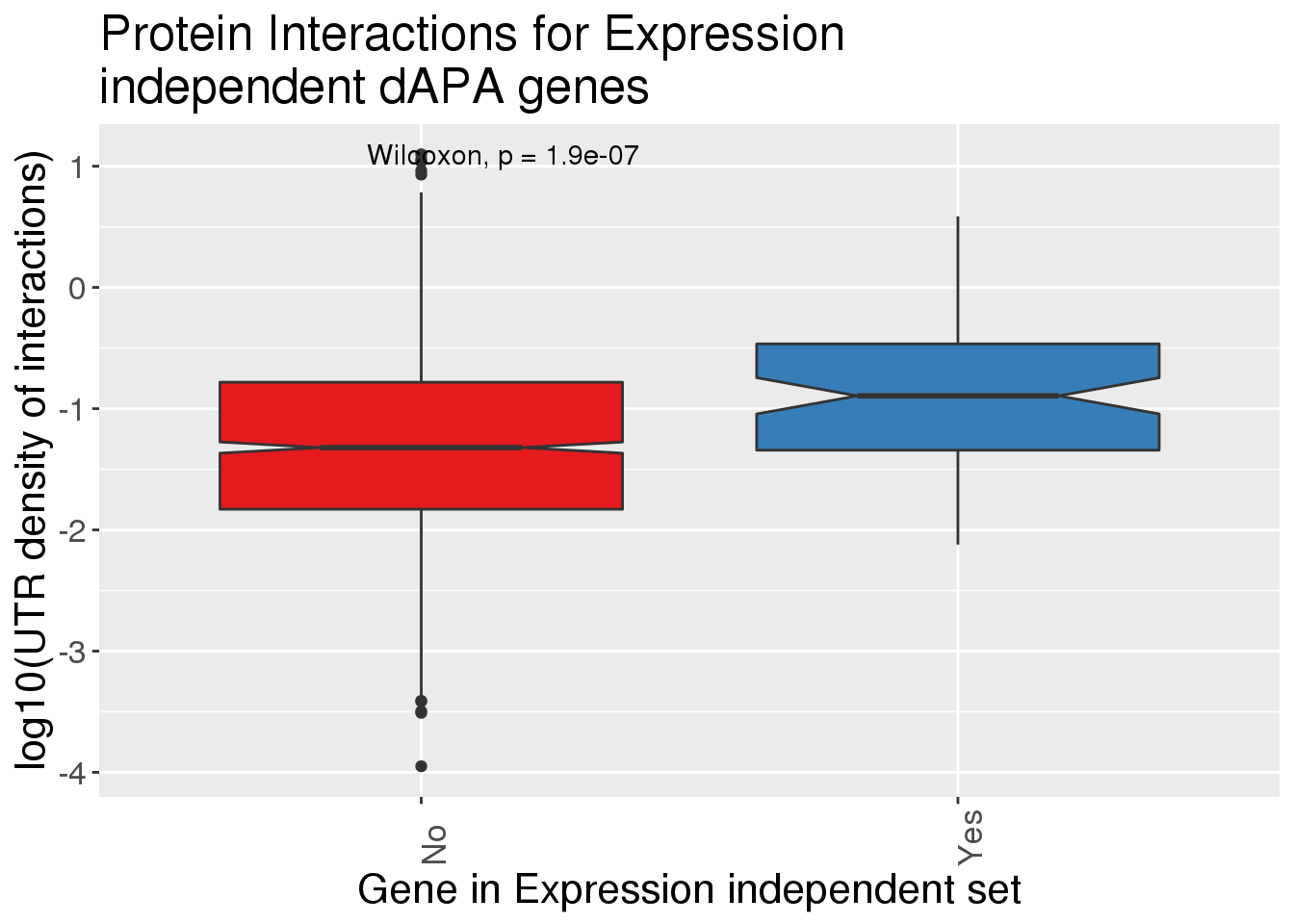
More likly to have one:
InteractionsAPA %>% mutate(HasInteraction=ifelse(nInt>0, "Yes", "No")) %>% group_by(dPnotE, HasInteraction) %>% summarise(nWithSet=n())# A tibble: 2 x 3
# Groups: dPnotE [2]
dPnotE HasInteraction nWithSet
<chr> <chr> <int>
1 No Yes 1292
2 Yes Yes 86Set should be the interaction set dAPA, de, and dP.
Alldiff=Protein %>% inner_join(DEgenes,by="gene") %>% inner_join(NucAPA, by="gene") %>% dplyr::select(gene)
#This is 101 genes.
geneAPAPnotEG=APAandPnotE %>% dplyr::select(gene)
GenesMatter= Alldiff %>% bind_rows(geneAPAPnotEG) %>% mutate(Ex=ifelse(gene %in% geneAPAPnotEG$gene, "No", "Yes")) %>% inner_join(Interactions, by="gene")ggplot(GenesMatter, aes(x=Ex, y=nInt, fill=Ex))+ geom_boxplot() + stat_compare_means() + scale_fill_brewer(palette = "RdYlBu")+theme(axis.text.x=element_text(angle=90, hjust=0), text= element_text(size=16), legend.position = "false") + labs(x="DE gene", y="Number of protein protein interactions", title="dAPA, DP, and DE")
Effect sizes :
Look at the PAS effect sizes here and in protien, translation, and expression.
NucAPAres_sig_dpnotE = NucAPAres_sig %>% filter(dPnotE =="Yes")
#nameID=read.table("../../genome_anotation_data/ensemble_to_genename.txt",sep="\t", header = T, stringsAsFactors = F) %>% dplyr::select(Gene_stable_ID, Gene.name)
#DE data
DE=read.table("../data/DiffExpression/DEtested_allres.txt",stringsAsFactors = F,header = F, col.names = c("Gene_stable_ID" ,"logFC" ,"AveExpr" , "t" , "P.Value" , "adj.P.Val", "B" )) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::rename('gene'=Gene.name) %>% dplyr::select(-Gene_stable_ID)
#translation
Ribo=read.table("../data/Wang_ribo/Additionaltable5_translationComparisons.txt",header = T, stringsAsFactors = F) %>% dplyr::rename("Gene_stable_ID"= ENSG) %>% inner_join(nameID,by="Gene_stable_ID") %>% dplyr::select(Gene.name, HvC.beta, HvC.pvalue, HvC.FDR) %>% dplyr::rename("gene"=Gene.name)
#prot
Prot=read.table("../data/Khan_prot/ProtData_effectSize.txt", header = T, stringsAsFactors = F)APAandE=NucAPAres_sig_dpnotE %>% inner_join(DE, by="gene")
ggplot(APAandE, aes(x=logFC, y=deltaPAU)) + geom_point(alpha=.3) + geom_smooth(method="lm") +stat_cor()
ggplot(APAandE, aes(x=logFC, y=deltaPAU, col=loc)) + geom_point(alpha=.3) + geom_smooth(method="lm") +stat_cor(label.x = 1)
APAandRibo=NucAPAres_sig_dpnotE %>% inner_join(Ribo, by="gene")
ggplot(APAandRibo, aes(x=HvC.beta, y=deltaPAU)) + geom_point(alpha=.3) + geom_smooth(method="lm") +stat_cor()
APAandprot=NucAPAres_sig_dpnotE %>% inner_join(Prot, by="gene")
ggplot(APAandprot, aes(x=logEf, y=deltaPAU))+ geom_point(alpha=.3) + geom_smooth(method="lm") +stat_cor( )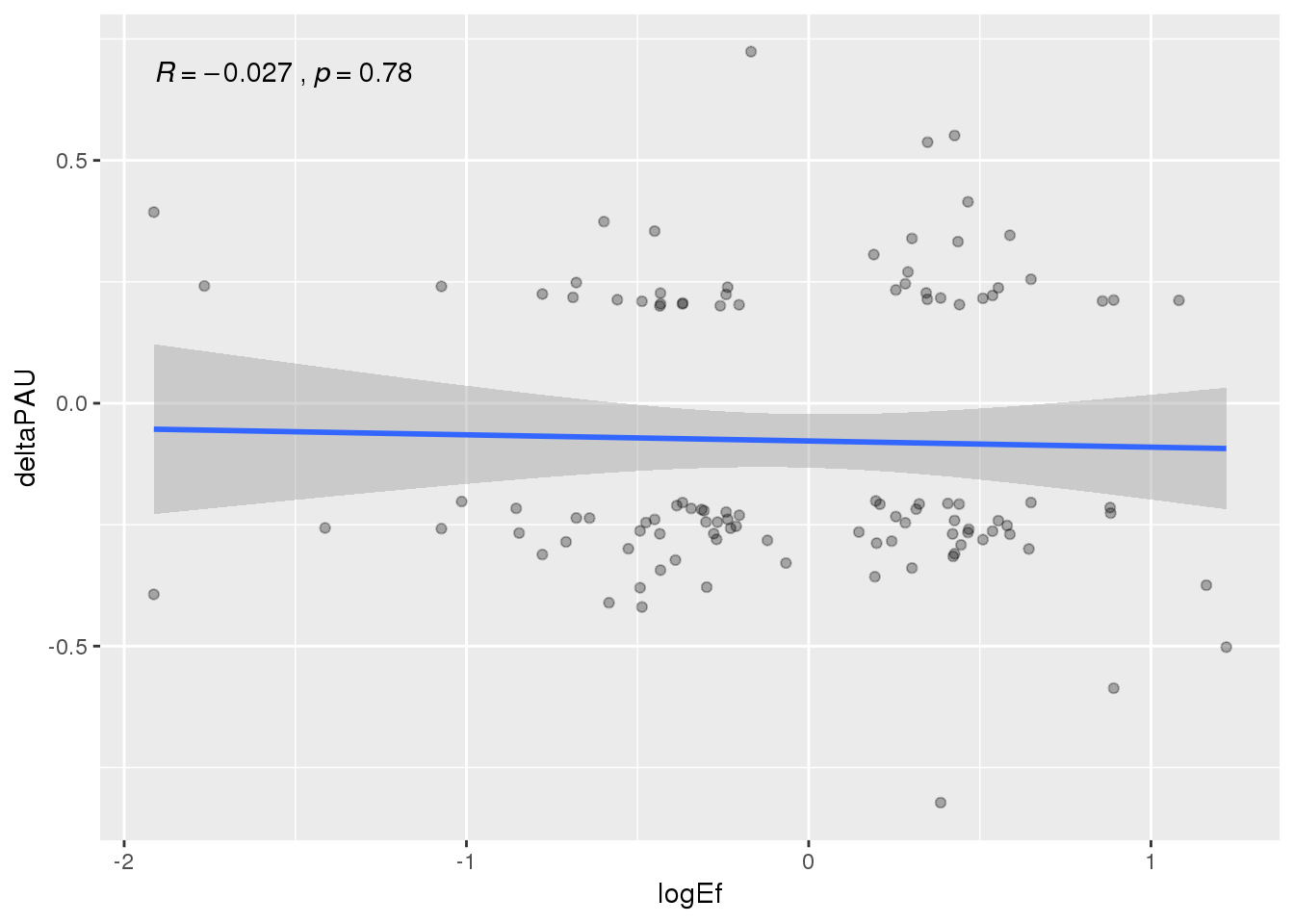
ggplot(APAandprot, aes(x=logEf, y=deltaPAU, col=loc))+ geom_point(alpha=.3) + geom_smooth(method="lm") +stat_cor( ) None of these are significant.
None of these are significant.
Check if any of these are genes with QTLs.
I will pull in the genes with nuclear apaQTLs first.
apaQTLs=read.table("../../apaQTL/data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.sort.bed",col.names = c('chr','start','end', 'PASid','score', 'strand')) %>% separate(PASid, into=c("gene", "PAS", "loc"),sep=":")
apaQTLGenes= apaQTLs %>% select(gene) %>% unique()APAandPnotE_apaQTL=APAandPnotE %>% mutate(apaQTL=ifelse(gene %in% apaQTLGenes$gene, "Yes", "No"))
APAandPnotE_apaQTL %>% group_by(apaQTL) %>% summarize(n=n())# A tibble: 2 x 2
apaQTL n
<chr> <int>
1 No 86
2 Yes 4APAandPnotE_apaQTL %>% filter(apaQTL=="Yes") gene HC.qvalues.protein ENSG apaQTL
1 STAT6 0.049128043 ENSG00000166888 Yes
2 RHOT1 0.009672323 ENSG00000126858 Yes
3 RNASEL 0.006755528 ENSG00000135828 Yes
4 BNIP1 0.021646516 ENSG00000113734 YesBackground for enrichment is all of the dAPA genes.
x= nrow(APAandPnotE_apaQTL %>% filter(apaQTL =="Yes"))
m= nrow(APAandPnotE_apaQTL)
n=nrow(NucAPA)- nrow(APAandPnotE_apaQTL)
k=nrow(apaQTLGenes)
x[1] 4#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 4phyper(x,m, n , k,lower.tail=F)[1] 1Not enriched for apaQTL.
pQTLs
Using protien specific QTLs from Battle et al.
pQTLs=read.table("../../apaQTL/data/Battle_pQTL/psQTLGeneNames.txt")
APAandPnotE_pQTL=APAandPnotE %>% mutate(pQTL=ifelse(gene %in% pQTLs$V1, "Yes", "No"))
APAandPnotE_pQTL %>% group_by(pQTL) %>% summarize(n=n())# A tibble: 2 x 2
pQTL n
<chr> <int>
1 No 86
2 Yes 4APAandPnotE_pQTL %>% filter(pQTL=="Yes") gene HC.qvalues.protein ENSG pQTL
1 TARS2 0.004796093 ENSG00000143374 Yes
2 ZBTB8OS 0.000021900 ENSG00000176261 Yes
3 NUP50 0.002061087 ENSG00000093000 Yes
4 UBA6 0.000154285 ENSG00000033178 Yesx= nrow(APAandPnotE_pQTL %>% filter(pQTL =="Yes"))
m= nrow(APAandPnotE_pQTL)
n=nrow(NucAPA)- nrow(APAandPnotE_pQTL)
k=nrow(pQTLs)
x[1] 4#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 4phyper(x,m, n , k,lower.tail=F)[1] 0.8959063Are any of the these the diff dom set? Test .4 first:
HumanRes=read.table("../data/DomDefGreaterX/Human_AllGenes_DiffTop.txt", col.names = c("Human_PAS", "gene","Human_DiffDom"),stringsAsFactors = F)
ChimpRes=read.table("../data/DomDefGreaterX/Chimp_AllGenes_DiffTop.txt", col.names = c("Chimp_PAS", "gene","Chimp_DiffDom"),stringsAsFactors = F)
BothRes=HumanRes %>% inner_join(ChimpRes,by="gene")
BothRes_40=BothRes %>% filter(Chimp_DiffDom >=0.4 | Human_DiffDom>=0.4) %>% mutate(Set= ifelse(Human_PAS==Chimp_PAS,"Same", "Different"),cut=40)
NucAPAres_sig_sm= NucAPAres_sig %>% filter(dPnotE=="Yes")
BothRes_40_dp= BothRes_40 %>% filter(gene %in% NucAPAres_sig_sm$gene)
BothRes_40_dp %>% group_by(Set) %>% summarise(n())# A tibble: 2 x 2
Set `n()`
<chr> <int>
1 Different 8
2 Same 32metaSm= Meta %>% select(loc, PAS)
DiffHuman= BothRes_40_dp %>% filter(Set=="Different") %>% select(gene, Human_PAS) %>% rename(PAS= Human_PAS)%>% inner_join(metaSm, by="PAS")Warning: Column `PAS` joining character vector and factor, coercing into
character vectorDiffChimp= BothRes_40_dp %>% filter(Set=="Different") %>% select(gene, Chimp_PAS)%>% rename(PAS= Chimp_PAS)%>% inner_join(metaSm, by="PAS")Warning: Column `PAS` joining character vector and factor, coercing into
character vectorDiffHuman gene PAS loc
1 SEC22B human18938 end
2 EIF4G2 human54877 utr3
3 TUBGCP3 human100208 intron
4 IRF3 human170101 utr3
5 HK2 human183666 intron
6 PPIL3 chimp195902 intron
7 FLNB human233016 utr3
8 CPOX human235691 utr3DiffChimp gene PAS loc
1 SEC22B chimp17094 end
2 EIF4G2 human54890 cds
3 TUBGCP3 chimp91832 utr3
4 IRF3 human170093 utr3
5 HK2 human183677 utr3
6 PPIL3 human199028 utr3
7 FLNB human233019 utr3
8 CPOX human235678 utr3
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggpubr_0.2 magrittr_1.5 UpSetR_1.3.3 forcats_0.3.0
[5] stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1
[9] tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38
[4] colorspace_1.3-2 generics_0.0.2 htmltools_0.3.6
[7] yaml_2.2.0 utf8_1.1.4 rlang_0.4.0
[10] later_0.7.5 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 RColorBrewer_1.1-2 modelr_0.1.2
[16] readxl_1.1.0 plyr_1.8.4 munsell_0.5.0
[19] gtable_0.2.0 workflowr_1.6.0 cellranger_1.1.0
[22] rvest_0.3.2 evaluate_0.12 labeling_0.3
[25] knitr_1.20 httpuv_1.4.5 fansi_0.4.0
[28] broom_0.5.1 Rcpp_1.0.2 promises_1.0.1
[31] scales_1.0.0 backports_1.1.2 jsonlite_1.6
[34] fs_1.3.1 gridExtra_2.3 hms_0.4.2
[37] digest_0.6.18 stringi_1.2.4 grid_3.5.1
[40] rprojroot_1.3-2 cli_1.1.0 tools_3.5.1
[43] lazyeval_0.2.1 crayon_1.3.4 whisker_0.3-2
[46] pkgconfig_2.0.2 xml2_1.2.0 lubridate_1.7.4
[49] assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[52] rstudioapi_0.10 R6_2.3.0 nlme_3.1-137
[55] git2r_0.26.1 compiler_3.5.1