5’ Splice site strength
Briana Mittleman
2/17/2020
Last updated: 2020-02-20
Checks: 6 1
Knit directory: apaQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190411) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /project2/gilad/briana/apaQTL/data/intron_analysis/transcriptsMinusExons.sort.bed | ../data/intron_analysis/transcriptsMinusExons.sort.bed |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: data/ProSeq/
Ignored: output/.DS_Store
Untracked files:
Untracked: .Rprofile
Untracked: ._.DS_Store
Untracked: .gitignore
Untracked: @
Untracked: GEO_brimittleman/
Untracked: _workflowr.yml
Untracked: analysis/._PASdescriptiveplots.Rmd
Untracked: analysis/._cuttoffPercUsage.Rmd
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Nuclear.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Total.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/QTLexampleplots.Rmd
Untracked: analysis/cuttoffPercUsage.Rmd
Untracked: analysis/eQTLoverlap.Rmd
Untracked: analysis/interpret verify bam.Rmd
Untracked: analysis/interpret_verifybam.Rmd
Untracked: analysis/mergeRNA.Rmd
Untracked: analysis/oldstuffNotNeeded.Rmd
Untracked: analysis/remove_badlines.Rmd
Untracked: analysis/totSpecInNuclear.Rmd
Untracked: analysis/totSpecIncludenotTested.Rmd
Untracked: analysis/totalspec.Rmd
Untracked: apaQTL.Rproj
Untracked: checksumsfastq.txt.gz
Untracked: code/.NascentRNAdtPlotFirstintronicPAS.sh.swp
Untracked: code/._Allsplicesite2fasta.py
Untracked: code/._ApaQTL_nominalNonnorm.sh
Untracked: code/._BothFracDTPlotGeneRegions.sh
Untracked: code/._BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/._ClosestTissuePAS.sh
Untracked: code/._CreateRNALZforeQTLs.sh
Untracked: code/._CreateRNALZnucAPAqtls.sh
Untracked: code/._DistPAS2Sig_RandomIntron.py
Untracked: code/._EandPqtl_perm.sh
Untracked: code/._EandPqtls.sh
Untracked: code/._ExtractGene4eQTLLZ.py
Untracked: code/._ExtractGene4eQTLLZpy
Untracked: code/._ExtractGeneRNAAssoc.py
Untracked: code/._ExtractPAS4LZeQTLs.py
Untracked: code/._ExtractPAS4eQTLsLZ.sh
Untracked: code/._ExtractPASforLZ.py
Untracked: code/._ExtractPASforLZ_run.sh
Untracked: code/._FC_NucintornUpandDown.sh
Untracked: code/._FC_UTR.sh
Untracked: code/._FC_intornUpandDownsteamPAS.sh
Untracked: code/._FC_nascentseq.sh
Untracked: code/._FC_newPeaks_olddata.sh
Untracked: code/._HMMpermuteTotal.py
Untracked: code/._HmmPermute.py
Untracked: code/._IntronicPASDT.sh
Untracked: code/._LC_samplegroups.py
Untracked: code/._LD_qtl.sh
Untracked: code/._LD_snpsproxy.sh
Untracked: code/._MapAllRBP.sh
Untracked: code/._NascentRNAdtPlot.sh
Untracked: code/._NascentRNAdtPlot3UTRPAS.sh
Untracked: code/._NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/._NascentRNAdtPlotNucPAS.sh
Untracked: code/._NascentRNAdtPlotTotPAS.sh
Untracked: code/._NascentRNAdtPlotintronicPAS.sh
Untracked: code/._NascnetRNAdtPlotPAS.sh
Untracked: code/._NetSeq_fourthintronDT.sh
Untracked: code/._NomResfromPASSNP.py
Untracked: code/._NuclearPAS_5per.bed.py
Untracked: code/._NuclearandRNA5samp_dtplots.sh
Untracked: code/._PTTfacetboxplots.R
Untracked: code/._PrematureQTLNominal.sh
Untracked: code/._PrematureQTLPermuted.sh
Untracked: code/._QTL2bed.py
Untracked: code/._QTL2bed_withstrand.py
Untracked: code/._RBPdisrupt.sh
Untracked: code/._RNAbam2bw.sh
Untracked: code/._RNAseqDTplot.sh
Untracked: code/._Randomsplicesite2fasta.py
Untracked: code/._Rplots.pdf
Untracked: code/._RunRes2PAS.sh
Untracked: code/._SAF215upbed.py
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilefiltPAS
Untracked: code/._TESplots100bp.sh
Untracked: code/._TESplots150bp.sh
Untracked: code/._TESplots200bp.sh
Untracked: code/._TotalPAS_5perc.bed.py
Untracked: code/._Untitled
Untracked: code/._ZipandTabPheno.sh
Untracked: code/._aAPAqtl_nominal39ind.sh
Untracked: code/._allNucSpecQTLine.py
Untracked: code/._allNucSpecfromNonNorm.py
Untracked: code/._annotatePacBioPASregion.sh
Untracked: code/._annotatedPAS2bed.py
Untracked: code/._apaInPandE.py
Untracked: code/._apaQTLCorrectPvalMakeQQ.R
Untracked: code/._apaQTLCorrectpval_6or7a.R
Untracked: code/._apaQTL_Nominal.sh
Untracked: code/._apaQTL_nominalInclusive.sh
Untracked: code/._apaQTL_nominalv67.sh
Untracked: code/._apaQTL_permuted.sh
Untracked: code/._apaQTL_permuted_test6A7A.sh
Untracked: code/._apainRibo.py
Untracked: code/._assignNucIntonpeak2intronlocs.sh
Untracked: code/._assignTotIntronpeak2intronlocs.sh
Untracked: code/._bam2BW_5primemost.sh
Untracked: code/._bed2saf.py
Untracked: code/._bothFracDTplot1stintron.sh
Untracked: code/._bothFracDTplot4thintron.sh
Untracked: code/._bothFrac_FC.sh
Untracked: code/._callPeaksYL.py
Untracked: code/._changeRibonomQTLres2genename.py
Untracked: code/._changenomQTLres2geneName.py
Untracked: code/._chooseAnno2PAS_pacbio.py
Untracked: code/._chooseAnno2SAF.py
Untracked: code/._chooseSignalSite
Untracked: code/._chooseSignalSite.py
Untracked: code/._closestannotated.sh
Untracked: code/._closestannotated_byfrac.sh
Untracked: code/._cluster.json
Untracked: code/._clusterPAS.json
Untracked: code/._clusterfiltPAS.json
Untracked: code/._codingdms2bed.py
Untracked: code/._config.yaml
Untracked: code/._config2.yaml
Untracked: code/._configOLD.yaml
Untracked: code/._convertNominal2SNPLOC.py
Untracked: code/._convertNominal2SNPloc2Versions.py
Untracked: code/._convertNumeric.py
Untracked: code/._correctNomeqtl.R
Untracked: code/._createPlinkSampfile.py
Untracked: code/._dag.pdf
Untracked: code/._eQTL_switch2snploc.py
Untracked: code/._eQTLgenestestedapa.py
Untracked: code/._encodeRNADTplots.sh
Untracked: code/._extactPAS100meanphyloP.py
Untracked: code/._extractGeneLZfiles.sh
Untracked: code/._extractGeneLZfileseQTLs.sh
Untracked: code/._extractGenotypes.py
Untracked: code/._extractPACmeanPhyloP.py
Untracked: code/._extractPhylop50up.py
Untracked: code/._extractPhylopextra50.py
Untracked: code/._extractRNApval4lz.py
Untracked: code/._extractseqfromqtlfastq.py
Untracked: code/._fc2leafphen.py
Untracked: code/._fc_filteredPAS6and7As.sh
Untracked: code/._fifteenBPupstreamPAS.py
Untracked: code/._fiftyBPupstreamPAS.py
Untracked: code/._filter5perc.R
Untracked: code/._filter5percPheno.py
Untracked: code/._filterLDsnps.py
Untracked: code/._filterMPPAS.py
Untracked: code/._filterMPPAS_15.py
Untracked: code/._filterMPPAS_15_7As.py
Untracked: code/._filterMPPAS_50.py
Untracked: code/._filterSAFforMP.py
Untracked: code/._filterpeaks.py
Untracked: code/._finalPASbed2SAF.py
Untracked: code/._fix4su304corr.py
Untracked: code/._fix4su604corr.py
Untracked: code/._fix4sukalisto.py
Untracked: code/._fixExandUnexeQTL
Untracked: code/._fixExandUnexeQTL.py
Untracked: code/._fixFChead.py
Untracked: code/._fixFChead_bothfrac.py
Untracked: code/._fixFChead_short.py
Untracked: code/._fixGWAS4Munge.py
Untracked: code/._fixH3k12ac.py
Untracked: code/._fixPASregionSNPs.py
Untracked: code/._fixRNAhead4corr.py
Untracked: code/._fixRNAkalisto.py
Untracked: code/._fix_randomIntron.py
Untracked: code/._fixgroupedtranscript.py
Untracked: code/._fixhead_netseqfc.py
Untracked: code/._getAPAfromanyeQTL.py
Untracked: code/._getApapval4eqtl.py
Untracked: code/._getApapval4eqtl_unexp.py
Untracked: code/._getApapval4eqtl_version67.py
Untracked: code/._getDownstreamIntronNuclear.py
Untracked: code/._getIntronDownstreamPAS.py
Untracked: code/._getIntronUpstreamPAS.py
Untracked: code/._getQTLalleles.py
Untracked: code/._getQTLfastq.sh
Untracked: code/._getUpstreamIntronNuclear.py
Untracked: code/._grouptranscripts.py
Untracked: code/._intersectVCFandupPAS.sh
Untracked: code/._keep5perMAF.py
Untracked: code/._keepSNP_vcf.sh
Untracked: code/._make5percPeakbed.py
Untracked: code/._makeFileID.py
Untracked: code/._makePheno.py
Untracked: code/._makeSAFbothfrac5perc.py
Untracked: code/._makeSNP2rsidfile.py
Untracked: code/._makeeQTLempirical_unexp.py
Untracked: code/._makeeQTLempiricaldist.py
Untracked: code/._makegencondeTSSfile.py
Untracked: code/._mapSSsnps2PAS.sh
Untracked: code/._mergRNABam.sh
Untracked: code/._mergeAllBam.sh
Untracked: code/._mergeAnnotations.sh
Untracked: code/._mergeBW_norm.sh
Untracked: code/._mergeBamNascent.sh
Untracked: code/._mergeByFracBam.sh
Untracked: code/._mergePeaks.sh
Untracked: code/._miRNAdisrupt.sh
Untracked: code/._mnase1stintron.sh
Untracked: code/._mnaseDT_fourthintron.sh
Untracked: code/._namePeaks.py
Untracked: code/._netseqDTplot1stIntron.sh
Untracked: code/._netseqFC.sh
Untracked: code/._nucQTLGWAS.py
Untracked: code/._nucSpecQTLineData.py
Untracked: code/._nucSpeceffectsize.py
Untracked: code/._nucspecnucPASine.py
Untracked: code/._pQTLsotherdata.py
Untracked: code/._pacbioDT.sh
Untracked: code/._pacbioIntronicDT.sh
Untracked: code/._parseALLSSres.py
Untracked: code/._parseBestbamid.py
Untracked: code/._parseLDRes.py
Untracked: code/._parseLDresBothPAS.sh
Untracked: code/._parseRanodmSSres.py
Untracked: code/._parseSSres.py
Untracked: code/._peak2PAS.py
Untracked: code/._peakFC.sh
Untracked: code/._pheno2countonly.R
Untracked: code/._phenoQTLfromlist.py
Untracked: code/._processYRIgen.py
Untracked: code/._pttQTLsinapaQTL.py
Untracked: code/._qtlRegionseq.sh
Untracked: code/._qtlsPvalOppFrac.py
Untracked: code/._quantassign2parsedpeak.py
Untracked: code/._removeXfromHmm.py
Untracked: code/._removeloc_pheno.py
Untracked: code/._riboQTL.sh
Untracked: code/._runCorrectNomEqtl.sh
Untracked: code/._runFixGWAS4Munge.sh
Untracked: code/._runHMMpermuteAPAqtls.sh
Untracked: code/._runHMMpermuteeQTLS.sh
Untracked: code/._runMakeEmpiricaleQTL_unexp.sh
Untracked: code/._runMakeeQTLempirical.sh
Untracked: code/._run_bam2bw_all3prime.sh
Untracked: code/._run_bam2bw_extra3.sh
Untracked: code/._run_bestbamid.sj
Untracked: code/._run_dist2sig_randomintron.sh
Untracked: code/._run_filtersnpLD.sh
Untracked: code/._run_getAPAfromeQTL_version6.7.sh
Untracked: code/._run_getApaPval4eqtl.sh
Untracked: code/._run_getapafromeQTL.py
Untracked: code/._run_getapafromeQTL.sh
Untracked: code/._run_getapapval4eqtl_unexp.sh
Untracked: code/._run_leafcutterDiffIso.sh
Untracked: code/._run_prxySNP.sh
Untracked: code/._run_pttfacetboxplot.sh
Untracked: code/._run_sepUsagephen.sh
Untracked: code/._run_sepgenobychrom.sh
Untracked: code/._run_verifybam.sh
Untracked: code/._selectNominalPvalues.py
Untracked: code/._sepUsagePhen.py
Untracked: code/._sepgenobychrom.py
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._sortindexRNAbam.sh
Untracked: code/._specAPAinE.py
Untracked: code/._splicesite2fasta.py
Untracked: code/._submit-snakemakePAS.sh
Untracked: code/._submit-snakemakefiltPAS.sh
Untracked: code/._subsetAPAnotEorPgene.py
Untracked: code/._subsetAPAnotEorPgene_2versions.py
Untracked: code/._subsetApanoteGene.py
Untracked: code/._subsetApanoteGene_2versions.py
Untracked: code/._subsetUnexplainedeQTLs.py
Untracked: code/._subsetVCF_SS.sh
Untracked: code/._subsetVCF_noSSregions.sh
Untracked: code/._subsetVCF_upstreamPAS.sh
Untracked: code/._subset_diffisopheno.py
Untracked: code/._subsetpermAPAwithGenelist.py
Untracked: code/._subsetpermAPAwithGenelist_2versions.py
Untracked: code/._subsetvcf_otherreg.sh
Untracked: code/._subsetvcf_permSS.sh
Untracked: code/._subtrachfiveprimeUTR.sh
Untracked: code/._subtractExons.sh
Untracked: code/._subtractfiveprimeUTR.sh
Untracked: code/._tabixSNPS.sh
Untracked: code/._tenBPupstreamPAS.py
Untracked: code/._test.pdf
Untracked: code/._testVerifyBam.sh
Untracked: code/._tissuePAS2hg19.sh
Untracked: code/._totSeceffectsize.py
Untracked: code/._twentyBPupstreamPAS.py
Untracked: code/._utrdms2saf.py
Untracked: code/._vcf2bed.py
Untracked: code/._verifyBam18517N.sh
Untracked: code/._verifyBam18517T.sh
Untracked: code/._verifyBam19128N.sh
Untracked: code/._verifyBam19128T.sh
Untracked: code/._wrap_verifybam.sh
Untracked: code/._writePTTexamplecode.py
Untracked: code/._writePTTexamplecode.sh
Untracked: code/.pversion
Untracked: code/.snakemake/
Untracked: code/1
Untracked: code/APAqtl_nominal.err
Untracked: code/APAqtl_nominal.out
Untracked: code/APAqtl_nominal_39.err
Untracked: code/APAqtl_nominal_39.out
Untracked: code/APAqtl_nominal_inclusive.err
Untracked: code/APAqtl_nominal_inclusive.out
Untracked: code/APAqtl_nominal_nonNorm.err
Untracked: code/APAqtl_nominal_nonNorm.out
Untracked: code/APAqtl_nominal_versions67.err
Untracked: code/APAqtl_nominal_versions67.out
Untracked: code/APAqtl_permuted.err
Untracked: code/APAqtl_permuted.out
Untracked: code/APAqtl_permuted_versions67.err
Untracked: code/APAqtl_permuted_versions67.out
Untracked: code/Allsplicesite2fasta.py
Untracked: code/BothFracDTPlot1stintron.err
Untracked: code/BothFracDTPlot1stintron.out
Untracked: code/BothFracDTPlot4stintron.err
Untracked: code/BothFracDTPlot4stintron.out
Untracked: code/BothFracDTPlotGeneRegions.err
Untracked: code/BothFracDTPlotGeneRegions.out
Untracked: code/BothFracDTPlotGeneRegions_norm.err
Untracked: code/BothFracDTPlotGeneRegions_norm.out
Untracked: code/ClosestTissuePAS.sh
Untracked: code/CreateRNALZforeQTLs.sh
Untracked: code/CreateRNALZnucAPAqtls.sh
Untracked: code/DistPAS2Sig_RandomIntron.py
Untracked: code/EandPqtl.err
Untracked: code/EandPqtl.out
Untracked: code/EncodeRNADTPlotGeneRegions.err
Untracked: code/EncodeRNADTPlotGeneRegions.out
Untracked: code/ExtractGene4eQTLLZ.py
Untracked: code/ExtractGene4eQTLLZpy
Untracked: code/ExtractGeneRNAAssoc.py
Untracked: code/ExtractPAS4LZeQTLs.py
Untracked: code/ExtractPAS4eQTLsLZ.sh
Untracked: code/ExtractPASforLZ.py
Untracked: code/ExtractPASforLZ_run.sh
Untracked: code/FC_NucintronPASupandDown.err
Untracked: code/FC_NucintronPASupandDown.out
Untracked: code/FC_UTR.err
Untracked: code/FC_UTR.out
Untracked: code/FC_intronPASupandDown.err
Untracked: code/FC_intronPASupandDown.out
Untracked: code/FC_nascent.err
Untracked: code/FC_nascentout
Untracked: code/FC_newPAS_olddata.err
Untracked: code/FC_newPAS_olddata.out
Untracked: code/HmmPermute.p
Untracked: code/IntronicPASDT.err
Untracked: code/IntronicPASDT.out
Untracked: code/LD_vcftools.hap.out
Untracked: code/MapAllRBP.sh
Untracked: code/MapRBP.err
Untracked: code/MapRBP.out
Untracked: code/NascentDTPlotGeneRegions.err
Untracked: code/NascentDTPlotGeneRegions.out
Untracked: code/NascentDTPlotPAS.err
Untracked: code/NascentDTPlotPAS.out
Untracked: code/NascentDTPlotPAS_3utr.err
Untracked: code/NascentDTPlotPAS_3utr.out
Untracked: code/NascentDTPlotPAS_firstintron.err
Untracked: code/NascentDTPlotPAS_firstintron.out
Untracked: code/NascentDTPlotPAS_intron.err
Untracked: code/NascentDTPlotPAS_intron.out
Untracked: code/NascentDTPlotPAS_nuc.err
Untracked: code/NascentDTPlotPAS_nuc.out
Untracked: code/NascentDTPlotPAS_tot.err
Untracked: code/NascentDTPlotPAS_tot.out
Untracked: code/Nuclear_example.err
Untracked: code/Nuclear_example.out
Untracked: code/NuclearandRNA5samp_dtplots.sh
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.err
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.out
Untracked: code/PACbioDT.err
Untracked: code/PACbioDT.out
Untracked: code/PACbioDTitronic.err
Untracked: code/PACbioDTitronic.out
Untracked: code/Prematureqtl_nominal.err
Untracked: code/Prematureqtl_nominal.out
Untracked: code/Prematureqtl_permuted.err
Untracked: code/Prematureqtl_permuted.out
Untracked: code/RBPdisrupt.err
Untracked: code/RBPdisrupt.out
Untracked: code/RBPdisrupt.sh
Untracked: code/README.md
Untracked: code/RNABam2BW.err
Untracked: code/RNABam2BW.out
Untracked: code/RNAseqDTPlotGeneRegions.err
Untracked: code/RNAseqDTPlotGeneRegions.out
Untracked: code/Randomsplicesite2fasta.py
Untracked: code/Rplots.pdf
Untracked: code/TESplots100bp.err
Untracked: code/TESplots100bp.out
Untracked: code/TESplots150bp.err
Untracked: code/TESplots150bp.out
Untracked: code/TESplots200bp.err
Untracked: code/TESplots200bp.out
Untracked: code/Tissueclosestannotated.err
Untracked: code/Tissueclosestannotated.out
Untracked: code/Total_example.err
Untracked: code/Total_example.out
Untracked: code/Untitled
Untracked: code/YRI_LCL.vcf.gz
Untracked: code/YRI_LCL_chr1.vcf.gz.log
Untracked: code/YRI_LCL_chr1.vcf.gz.recode.vcf
Untracked: code/annotatedPASregion.err
Untracked: code/annotatedPASregion.out
Untracked: code/apaQTL_nominalInclusive.sh
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assigntotPeak2Intronicregion.err
Untracked: code/assigntotPeak2Intronicregion.out
Untracked: code/bam2bw.err
Untracked: code/bam2bw.out
Untracked: code/bam2bw_5primemost.err
Untracked: code/bam2bw_5primemost.out
Untracked: code/binary_fileset.log
Untracked: code/bothFrac_FC.err
Untracked: code/bothFrac_FC.out
Untracked: code/callSHscripts.txt
Untracked: code/closestannotated.err
Untracked: code/closestannotated.out
Untracked: code/closestannotatedbyfrac.err
Untracked: code/closestannotatedbyfrac.out
Untracked: code/dag.pdf
Untracked: code/dagPAS.pdf
Untracked: code/dagfiltPAS.pdf
Untracked: code/extactPAS100meanphyloP.py
Untracked: code/extractGeneLZfiles.err
Untracked: code/extractGeneLZfiles.out
Untracked: code/extractGeneLZfiles.sh
Untracked: code/extractGeneLZfileseQTLs.err
Untracked: code/extractGeneLZfileseQTLs.out
Untracked: code/extractGeneLZfileseQTLs.sh
Untracked: code/extractPACmeanPhyloP.py
Untracked: code/extractPASLZfiles.err
Untracked: code/extractPASLZfiles.out
Untracked: code/extractPASLZfileseQTLs.err
Untracked: code/extractPASLZfileseQTLs.out
Untracked: code/extractPhylop50up.py
Untracked: code/extractPhylopextra50.py
Untracked: code/extractRNApval4lz.py
Untracked: code/fixExandUnexeQTL
Untracked: code/fixGWAS4Munge.py
Untracked: code/fix_randomIntron.py
Untracked: code/fixmunge
Untracked: code/genotypesYRI.gen.proc.keep.vcf.log
Untracked: code/genotypesYRI.gen.proc.keep.vcf.recode.vcf
Untracked: code/getseq100up.err
Untracked: code/getseq100up.out
Untracked: code/grouptranscripts.err
Untracked: code/grouptranscripts.out
Untracked: code/intersectPAS_ssSNPS.err
Untracked: code/intersectPAS_ssSNPS.out
Untracked: code/intersectVCFPAS.err
Untracked: code/intersectVCFPAS.out
Untracked: code/liftoverPAShg38to19.err
Untracked: code/liftoverPAShg38to19.out
Untracked: code/log/
Untracked: code/logs/
Untracked: code/merge53PRIMEbam.err
Untracked: code/merge53PRIMEbam.out
Untracked: code/merge53RNAbam.err
Untracked: code/merge53prime.sh
Untracked: code/merge5RNABam.err
Untracked: code/merge5RNABam.out
Untracked: code/merge5RNAbam.out
Untracked: code/merge5RNAbam.sh
Untracked: code/mergeAnno.err
Untracked: code/mergeAnno.out
Untracked: code/mergeBWnorm.err
Untracked: code/mergeBWnorm.out
Untracked: code/mergeBamNacent.err
Untracked: code/mergeBamNacent.out
Untracked: code/mergeRNAbam.err
Untracked: code/mergeRNAbam.out
Untracked: code/miRNAdisrupt.err
Untracked: code/miRNAdisrupt.out
Untracked: code/miRNAdisrupt.sh
Untracked: code/mnaseDTPlot1stintron.err
Untracked: code/mnaseDTPlot1stintron.out
Untracked: code/mnaseDTPlot4thintron.err
Untracked: code/mnaseDTPlot4thintron.out
Untracked: code/netDTPlot4thintron.out
Untracked: code/netseqFC.err
Untracked: code/netseqFC.out
Untracked: code/neyDTPlot4thintron.err
Untracked: code/nucspecinE.py
Untracked: code/parseALLSSres.py
Untracked: code/parseLDRes.py
Untracked: code/parseLDres.err
Untracked: code/parseLDres.out
Untracked: code/parseLDresBothPAS.sh
Untracked: code/parseRanodmSSres.py
Untracked: code/parseSSres.py
Untracked: code/plink.log
Untracked: code/prxySNP.err
Untracked: code/prxySNP.out
Untracked: code/pttFacetBoxplots.err
Untracked: code/pttFacetBoxplots.out
Untracked: code/qtlFacetBoxplots.err
Untracked: code/qtlFacetBoxplots.out
Untracked: code/rLD_vcftools.hap.err
Untracked: code/riboqtl.err
Untracked: code/riboqtl.out
Untracked: code/runBestBamID.err
Untracked: code/runCorrectNomeqtl.err
Untracked: code/runCorrectNomeqtl.out
Untracked: code/runFilterLD.err
Untracked: code/runFilterLD.out
Untracked: code/runFixGWAS4Munge.sh
Untracked: code/runHMMpermute.err
Untracked: code/runHMMpermute.out
Untracked: code/runHMMpermuteeQTLs.err
Untracked: code/runHMMpermuteeQTLs.out
Untracked: code/runMakeEmpiricaleQTLs.err
Untracked: code/runMakeEmpiricaleQTLs.out
Untracked: code/runMakeEmpiricaleQTLsunex.err
Untracked: code/runMakeEmpiricaleQTLsunex.out
Untracked: code/run_DistPAS2Sig.err
Untracked: code/run_DistPAS2Sig.out
Untracked: code/run_DistPAS2Sig_intron.err
Untracked: code/run_DistPAS2Sig_intron.out
Untracked: code/run_bam2bw.err
Untracked: code/run_bam2bw.out
Untracked: code/run_bam2bwexta.err
Untracked: code/run_bam2bwexta.out
Untracked: code/run_dist2sig_randomintron.sh
Untracked: code/run_getAPAfromanyeQTL.err
Untracked: code/run_getAPAfromanyeQTL.out
Untracked: code/run_getApaPval4eQTLs.err
Untracked: code/run_getApaPval4eQTLs.out
Untracked: code/run_getApaPval4eQTLsunexplained.err
Untracked: code/run_getApaPval4eQTLsunexplained.out
Untracked: code/run_leafcutter_ds.err
Untracked: code/run_leafcutter_ds.out
Untracked: code/run_sepgenobychrom.err
Untracked: code/run_sepgenobychrom.out
Untracked: code/run_sepusage.err
Untracked: code/run_sepusage.out
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/run_verifybam128N.err
Untracked: code/run_verifybam128N.out
Untracked: code/run_verifybam128T.err
Untracked: code/run_verifybam128T.out
Untracked: code/run_verifybam517N.err
Untracked: code/run_verifybam517N.out
Untracked: code/run_verifybam517T.err
Untracked: code/run_verifybam517T.out
Untracked: code/runprxySNP.err
Untracked: code/runprxySNP.out
Untracked: code/runres2pas.err
Untracked: code/runres2pas.out
Untracked: code/scripts/
Untracked: code/scripts_PAS_500_Lymph/
Untracked: code/seqQTLfastq.err
Untracked: code/seqQTLfastq.out
Untracked: code/seqQTLregion.err
Untracked: code/seqQTLregion.out
Untracked: code/snakePASlog.out
Untracked: code/snakefiltPASlog.out
Untracked: code/sortindexRNABam.err
Untracked: code/sortindexRNABam.out
Untracked: code/specAPAinE.py
Untracked: code/splicesite2fasta.py
Untracked: code/subsetvcf_SS.err
Untracked: code/subsetvcf_SS.out
Untracked: code/subsetvcf_noSS.err
Untracked: code/subsetvcf_noSS.out
Untracked: code/subsetvcf_pas.err
Untracked: code/subsetvcf_pas.out
Untracked: code/subsetvcf_perm.err
Untracked: code/subsetvcf_perm.out
Untracked: code/subsetvcf_rand.err
Untracked: code/subsetvcf_rand.out
Untracked: code/subtract5UTR.err
Untracked: code/subtract5UTR.out
Untracked: code/subtractExons.err
Untracked: code/subtractExons.out
Untracked: code/tabixSNPs.err
Untracked: code/tabixSNPs.out
Untracked: code/test.pdf
Untracked: code/testFix.txt
Untracked: code/test_verifybam.err
Untracked: code/test_verifybam.out
Untracked: code/tissuePAS2hg19.sh
Untracked: code/vcf_keepsnps.err
Untracked: code/vcf_keepsnps.out
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/zipandtabPhen.err
Untracked: code/zipandtabPhen.out
Untracked: data/._.DS_Store
Untracked: data/._MetaDataSequencing.txt
Untracked: data/AnnotatedPAS/
Untracked: data/ApaByEgene/
Untracked: data/ApaByPgene/
Untracked: data/BadLines/
Untracked: data/BaseComp/
Untracked: data/Battle_pQTL/
Untracked: data/CheckSums/
Untracked: data/CompareOldandNew/
Untracked: data/DTmatrix/
Untracked: data/DiffIso/
Untracked: data/EncodeRNA/
Untracked: data/ExampleQTLPlots/
Untracked: data/ExampleQTLPlots_update/
Untracked: data/ExpressionIndependentapaQTLs.txt
Untracked: data/FiveMergedBW/
Untracked: data/FiveMergedBam/
Untracked: data/FlaggedPAS/
Untracked: data/GWAS_overlap/
Untracked: data/GeuvadisRNA/
Untracked: data/HMMqtls/
Untracked: data/LDSR_annotations/
Untracked: data/LZ_both/
Untracked: data/Li_eQTLs/
Untracked: data/NMD/
Untracked: data/NascentRNA/
Untracked: data/NucSpeceQTLeffect/
Untracked: data/PAS/
Untracked: data/PAS_postFlag/
Untracked: data/PolyA_DB/
Untracked: data/PreTerm_pheno/
Untracked: data/PrematureQTLNominal/
Untracked: data/PrematureQTLPermuted/
Untracked: data/QTLGenotypes/
Untracked: data/QTLoverlap/
Untracked: data/QTLoverlap_inclusive/
Untracked: data/QTLoverlap_nonNorm/
Untracked: data/README.md
Untracked: data/RNAseq/
Untracked: data/Reads2UTR/
Untracked: data/SNPinSS/
Untracked: data/SignalSiteFiles/
Untracked: data/TF_motifdisruption/
Untracked: data/TSS/
Untracked: data/ThirtyNineIndQtl_nominal/
Untracked: data/TissueData/
Untracked: data/Version15bp6As/
Untracked: data/Version15bp7As/
Untracked: data/apaQTLNominal/
Untracked: data/apaQTLNominal_4pc/
Untracked: data/apaQTLNominal_inclusive/
Untracked: data/apaQTLPermuted/
Untracked: data/apaQTLPermuted_4pc/
Untracked: data/apaQTLs/
Untracked: data/assignedPeaks/
Untracked: data/assignedPeaks_15Up/
Untracked: data/bam/
Untracked: data/bam_clean/
Untracked: data/bam_waspfilt/
Untracked: data/bed_10up/
Untracked: data/bed_clean/
Untracked: data/bed_clean_sort/
Untracked: data/bed_waspfilter/
Untracked: data/bedsort_waspfilter/
Untracked: data/bothFrac_FC/
Untracked: data/bw/
Untracked: data/bw_norm/
Untracked: data/coloc/
Untracked: data/eCLip/
Untracked: data/eQTL_LZ/
Untracked: data/eQTLs/
Untracked: data/exampleQTLs/
Untracked: data/exosome/
Untracked: data/fastq/
Untracked: data/filterPeaks/
Untracked: data/fourSU/
Untracked: data/h3k27ac/
Untracked: data/highdiffsiggenes.txt
Untracked: data/inclusivePeaks/
Untracked: data/inclusivePeaks_FC/
Untracked: data/intronRNAratio/
Untracked: data/intron_analysis/
Untracked: data/locusZoom/
Untracked: data/mergedBG/
Untracked: data/mergedBW_byfrac/
Untracked: data/mergedBW_norm/
Untracked: data/mergedBam/
Untracked: data/mergedbyFracBam/
Untracked: data/miRNAbinding/
Untracked: data/molPhenos/
Untracked: data/molQTLs/
Untracked: data/motifdistrupt/
Untracked: data/nPAS/
Untracked: data/netseq/
Untracked: data/nonNorm_pheno/
Untracked: data/nuc_10up/
Untracked: data/nuc_10upclean/
Untracked: data/oldPASfiles/
Untracked: data/overlapeQTL_try2/
Untracked: data/overlapeQTLs/
Untracked: data/pQTLoverlap/
Untracked: data/pacbio/
Untracked: data/peakCoverage/
Untracked: data/peaks_5perc/
Untracked: data/phenotype/
Untracked: data/phenotype_5perc/
Untracked: data/phenotype_inclusivePAS/
Untracked: data/phylop/
Untracked: data/pttQTL/
Untracked: data/pttQTLplots/
Untracked: data/sigDiffGenes.txt
Untracked: data/sort/
Untracked: data/sort_clean/
Untracked: data/sort_waspfilter/
Untracked: data/splicesite/
Untracked: data/twoMech/
Untracked: data/vareQTLvarAPAqtl/
Untracked: data/verifyBAM/
Untracked: data/verifyBAM_full/
Untracked: nohup.out
Untracked: output/._.DS_Store
Untracked: output/._AverageDiffHeatmap.Nuclear.png
Untracked: output/._AverageDiffHeatmap.Total.png
Untracked: output/._GeneswithAPApotential.png
Untracked: output/._GeneswithAPApotentialAllPAS.png
Untracked: output/._PASlocation.png
Untracked: output/._SignalSitePlot.png
Untracked: output/._meanCorrelationPhenotypes.svg
Untracked: output/._qqplot_Nuclear_APAperm.png
Untracked: output/._qqplot_Nuclear_APAperm_4pc.png
Untracked: output/._qqplot_Total_APAperm.png
Untracked: output/._qqplot_Total_APAperm_4pc.png
Untracked: output/AverageDiffHeatmap.Nuclear.png
Untracked: output/AverageDiffHeatmap.Total.png
Untracked: output/GeneswithAPApotential.png
Untracked: output/GeneswithAPApotentialAllPAS.png
Untracked: output/PASlocation.png
Untracked: output/SignalSitePlot.png
Untracked: output/SignalSitePlotbyLoc.png
Untracked: output/dtPlots/
Untracked: output/fastqc/
Untracked: output/meanCorrelationPhenotypes.svg
Untracked: output/newnuc.png
Untracked: output/newtot.png
Untracked: output/oldnuc.png
Untracked: output/oldtot.png
Untracked: output/qqplot_Nuclear_APAperm.png
Untracked: output/qqplot_Nuclear_APAperm_4pc.png
Untracked: output/qqplot_Total_APAperm.png
Untracked: output/qqplot_Total_APAperm_4pc.png
Untracked: run_verifybam517N.err
Untracked: run_verifybam517N.out
Unstaged changes:
Modified: analysis/ExploreNpas.Rmd
Modified: analysis/NuclearSpecIncludeNotTested.Rmd
Modified: analysis/PASdescriptiveplots.Rmd
Modified: analysis/Readdistagainstfeatures.Rmd
Modified: analysis/TSS.Rmd
Modified: analysis/decayAndStability.Rmd
Modified: analysis/miRNAdisrupt.Rmd
Modified: analysis/nascenttranscription.Rmd
Modified: analysis/nucSpecinEQTLs.Rmd
Modified: analysis/overlapapaqtlsandeqtls.Rmd
Modified: analysis/pQTLexampleplot.Rmd
Modified: analysis/version15bpfilter.Rmd
Modified: code/DistPAS2Sig.py
Modified: code/Script4NuclearQTLexamples.sh
Modified: code/Script4TotalQTLexamples.sh
Modified: code/apaQTLsnake.err
Modified: code/environment.yaml
Modified: code/run_qtlFacetBoxplots.sh
Deleted: code/test.txt
Deleted: reads_graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | d2056c1 | brimittleman | 2020-02-20 | add lcls, add coloc package, add 5’ss by decile |
| html | 38b532c | brimittleman | 2020-02-18 | Build site. |
| Rmd | fe3acb2 | brimittleman | 2020-02-18 | add res |
| html | 0fde09f | brimittleman | 2020-02-17 | Build site. |
| Rmd | f4a296f | brimittleman | 2020-02-17 | add initial res for splice site |
library(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(ggpubr)Loading required package: magrittr
Attaching package: 'magrittr'The following object is masked from 'package:purrr':
set_namesThe following object is masked from 'package:tidyr':
extractI will assess the 5’ splice site strength with maxentscore to see if this can tell us anything interesting about intronic polyadenylation.
http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html
How to use MaxEntScan::score5ss
Each sequence must be 9 bases long. [3 bases in exon][6 bases in intron] Input sequences as a FastA file with one sequence per line (no linebreaks). Non-ACGT sequences will not be processed.
Example Fasta File
> dummy1
cagGTAAGT
> dummy2
gagGTAAGT
> dummy3
taaATAAGTI assigned PAS to introns. in https://brimittleman.github.io/apaQTL/nucintronicanalysis.html
pas2intron=read.table("../data/intron_analysis/IntronPeaksontoIntrons.bed",col.names = c("intronCHR", "intronStart", "intronEnd", "gene", "score", "strand", "peakCHR", "peakStart", "peakEnd", "PeakID", "meanUsage", "peakStrand"))
#%>% mutate(PASloc=ifelse(strand=="+", peakEnd, peakStart)) %>% dplyr::select(intronStart, intronEnd, gene, strand, PeakID, PASloc ,meanUsage) %>% mutate(intronLength=intronEnd-intronStart , distance2PAS= ifelse(strand=="+", PASloc-intronStart, intronEnd-PASloc), propIntron=distance2PAS/intronLength)I need a file with the PAS and the 5’ splice site. For negative strand the 5’ is the end and postitive strand PAS it is the start.
postive: start= start-3 end= start + 6
negative: start= end -6 end= end + 3
mkdir ../data/splicesitePAS_5SS_pos= pas2intron %>% filter(strand=="+") %>% mutate(start=intronStart-3, end= intronStart +6) %>% select(intronCHR, start,end, PeakID,meanUsage, strand)
PAS_5SS_neg=pas2intron %>% filter(strand=="-") %>% mutate(start=intronEnd-6, end= intronEnd +3) %>% select(intronCHR, start,end, PeakID,meanUsage, strand)
PAS_5SS_both= PAS_5SS_neg %>% bind_rows(PAS_5SS_pos)
write.table(PAS_5SS_pos, "../data/splicesite/TestPosSS.bed", col.names = F, row.names = F, quote=F, sep="\t")
write.table(PAS_5SS_neg, "../data/splicesite/TestNegSS.bed", col.names = F, row.names = F, quote=F, sep="\t")
write.table(PAS_5SS_both, "../data/splicesite/AllPASSS.bed", col.names = F, row.names = F, quote=F, sep="\t")Merge and sort these to get the nucleotides:
sort -k1,1 -k2,2n ../data/splicesite/AllPASSS.bed > ../data/splicesite/AllPASSS.sort.bed
#cut chr
sed 's/^chr//' ../data/splicesite/AllPASSS.sort.bed > ../data/splicesite/AllPASSS.sort.noChr.bed
#bedtools nuc
bedtools nuc -fi /project2/gilad/briana/genome_anotation_data/genome/Homo_sapiens.GRCh37.75.dna_sm.all.fa -bed ../data/splicesite/AllPASSS.sort.noChr.bed -seq -s > ../data/splicesite/AllPASSS.sort.Nuc.txtThis works and it flips the strand. the first 3 bases are the exon and the next 6 are the intron.
I need to turn this into a FA file. with the first 3 lower case and second 6 upper like the example. I can do this in python.
For each PAS i will have the name then the bases in the next
python splicesite2fasta.pyScore online with site and use Maximum Entropy Model.
splice result to keep every other line. Then I can join the reults with the initial bed.
python parseSSres.pyres=read.table("../data/splicesite/MaxIntResParsed.txt", col.names=c("splicesite", "maxentscore"), header=F, stringsAsFactors = F)
bothSS=read.table("../data/splicesite/AllPASSS.sort.noChr.bed", header = F, col.names = c("chr", 'start','end','PAS', "NuclearUsage", 'strand'))
bothandres=bothSS %>% bind_cols(res)Plot usage and score:
cor.test(bothandres$NuclearUsage, bothandres$maxentscore)
Pearson's product-moment correlation
data: bothandres$NuclearUsage and bothandres$maxentscore
t = -3.547, df = 12534, p-value = 0.0003911
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
-0.04914437 -0.01416833
sample estimates:
cor
-0.03166605 ggplot(bothandres, aes(x=maxentscore, y=NuclearUsage)) + geom_point() + geom_density2d(col="red")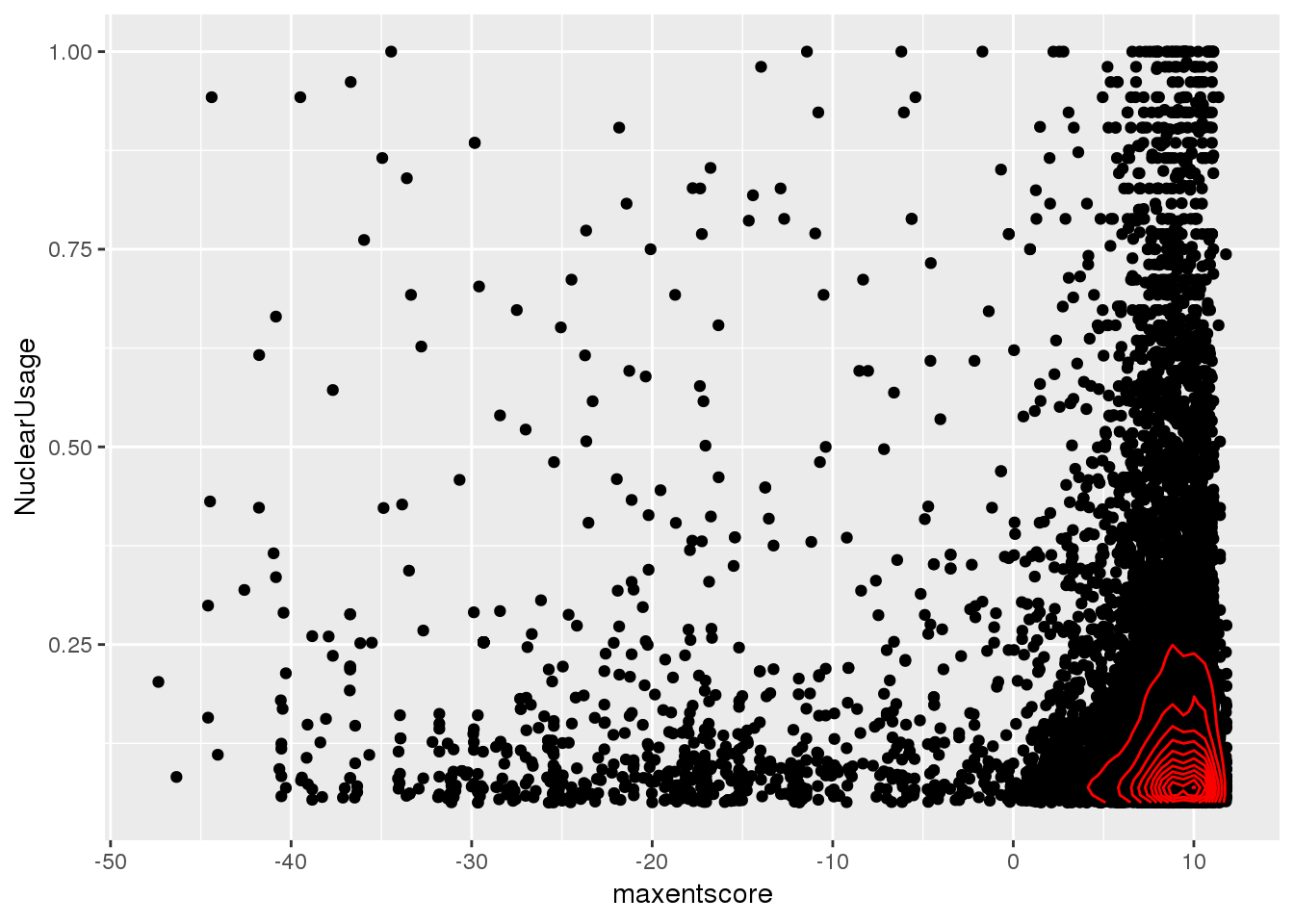
| Version | Author | Date |
|---|---|---|
| 0fde09f | brimittleman | 2020-02-17 |
Filter usage higher (25%) and score above 0
bothandres_filt= bothandres %>% filter(NuclearUsage>0.25, maxentscore>0)
ggplot(bothandres_filt, aes(x=maxentscore, y=NuclearUsage)) + geom_point() + geom_density2d(col="red") + geom_smooth(method="lm")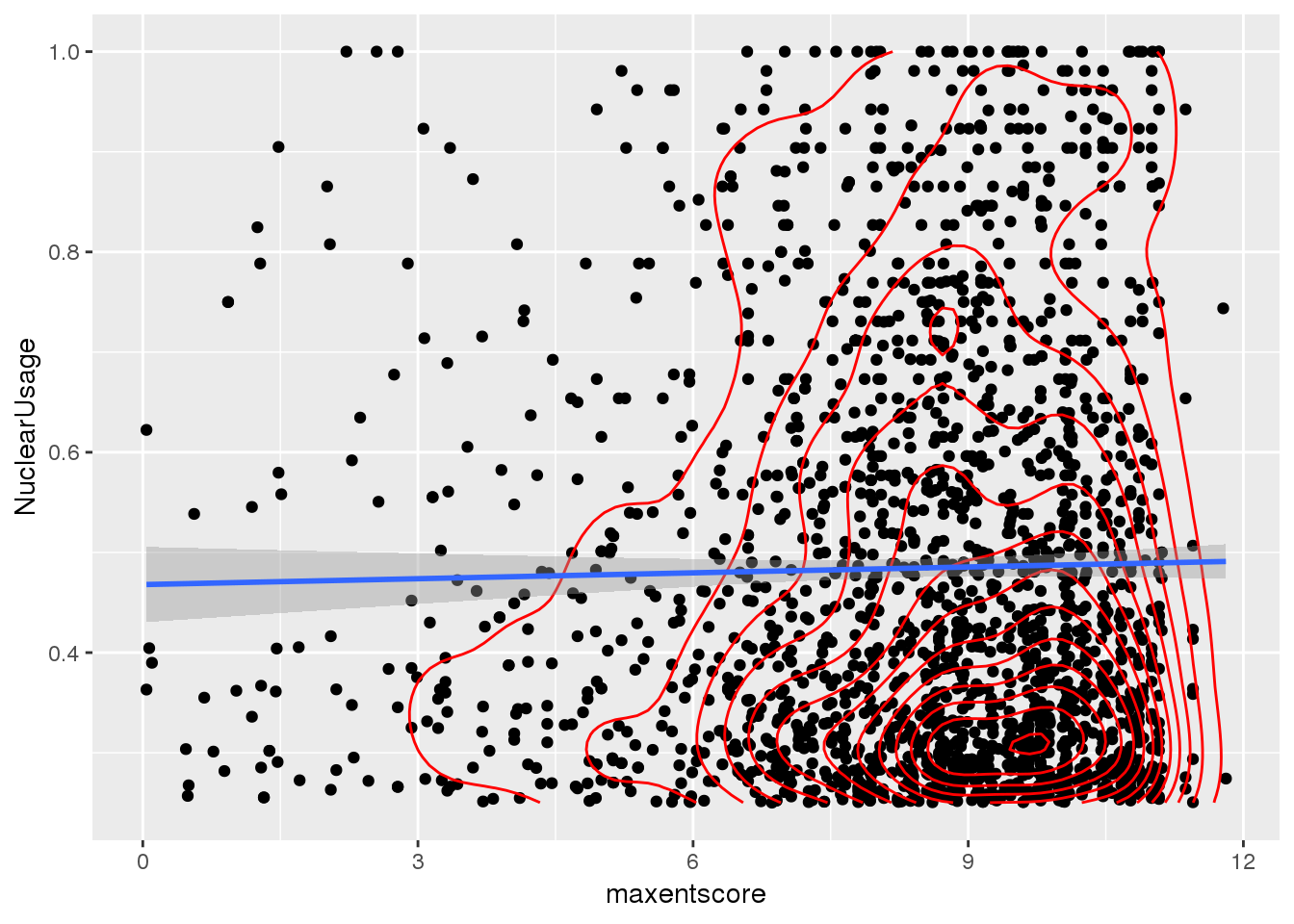
| Version | Author | Date |
|---|---|---|
| 0fde09f | brimittleman | 2020-02-17 |
Does not look like there is a relationship here.
Expectation is a stronger 5’ SS means lower intronic usage. I will compare top 10% usage and bottom 10% usage
quantile(bothandres$NuclearUsage,probs=c(.1,.9)) 10% 90%
0.05653846 0.37990385 bothandres_topbottom = bothandres %>% filter(NuclearUsage<= 0.056 | NuclearUsage >=0.38) %>% mutate(Usage=ifelse(NuclearUsage <=.15, "Low","High"))
ggplot(bothandres_topbottom,aes(x=Usage, y=maxentscore))+ geom_boxplot()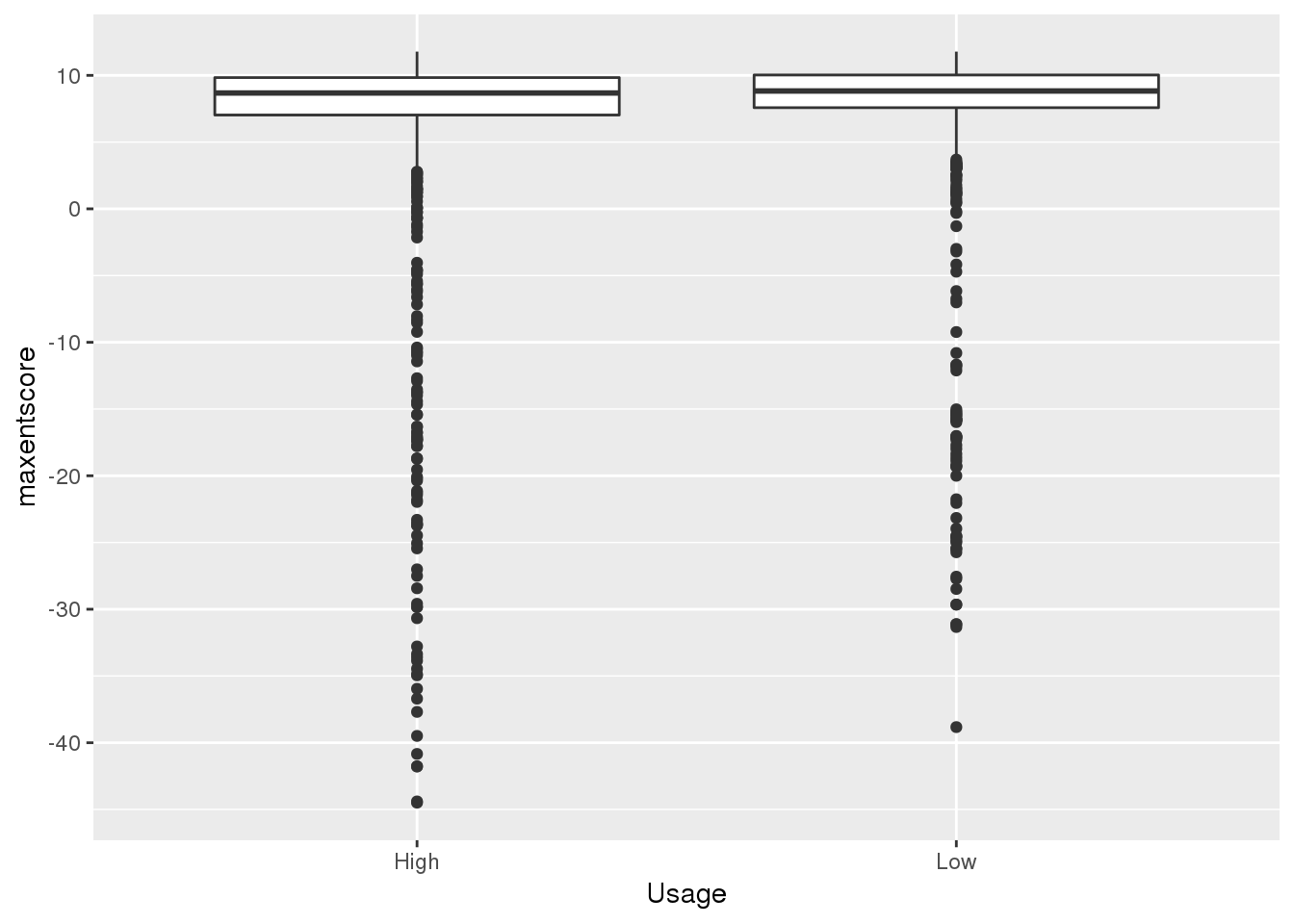
| Version | Author | Date |
|---|---|---|
| 38b532c | brimittleman | 2020-02-18 |
bothandres_top=bothandres_topbottom %>% filter(Usage=="High")
bothandres_bottom=bothandres_topbottom %>% filter(Usage=="Low")
#x to the left of y
wilcox.test(bothandres_top$maxentscore, bothandres_bottom$maxentscore, alternative="less")
Wilcoxon rank sum test with continuity correction
data: bothandres_top$maxentscore and bothandres_bottom$maxentscore
W = 681970, p-value = 0.00136
alternative hypothesis: true location shift is less than 0top used have lower scores. This is in line with expectation.
Compare to a random set of splice sites. Select 12536
chroms=c("chr1", 'chr2', 'chr3', 'chr4', 'chr5', 'chr6', 'chr7', 'chr8', 'chr9', 'chr10', 'chr11', 'chr12', 'chr13', 'chr14', 'chr15', 'chr16', 'chr17', 'chr18', 'chr19', 'chr20', 'chr21', 'chr22')
allIntron=read.table("/project2/gilad/briana/apaQTL/data/intron_analysis/transcriptsMinusExons.sort.bed", col.names = c("chr","start","end", 'gene', 'score','strand'),header = T, stringsAsFactors = F) %>% filter(chr %in% chroms)
#sampleIntron= allIntron %>% sample_n(12536, replace = F)Get the 5’ splice site for these:
#randPAS_5SS_pos= sampleIntron %>% filter(strand=="+") %>% mutate(newStart=start-3, newEnd= start +6) %>% select(chr, newStart,newEnd, gene,score, strand)
#randPAS_5SS_neg=sampleIntron %>% filter(strand=="-") %>% mutate(newStart=end-6, newEnd= end +3) %>% select(chr, newStart,newEnd, gene,score, strand)
#randPAS_both= randPAS_5SS_pos %>% bind_rows(randPAS_5SS_neg)
#write.table(randPAS_both,"../data/splicesite/RandomIntronSS.bed",sep="\t", col.names = F, row.names = F, quote = F)sort -k1,1 -k2,2n ../data/splicesite/RandomIntronSS.bed | sed 's/^chr//' > ../data/splicesite/RandomIntronSS_noChr.bed
#bedtools nuc
bedtools nuc -fi /project2/gilad/briana/genome_anotation_data/genome/Homo_sapiens.GRCh37.75.dna_sm.all.fa -bed ../data/splicesite/RandomIntronSS_noChr.bed -seq -s > ../data/splicesite/RandomIntronSS_noChr.Nuc.bed
python Randomsplicesite2fasta.py
python parseRanodmSSres.pyEval:
RandomSites=read.table("../data/splicesite/RandomIntronSS.bed",col.names = c('chr','start','end','name','score','strand'))
RandomRes= read.table("../data/splicesite/RandomSSMaxentParsed.txt", col.names = c("splicesite", "maxentscore_cont"), stringsAsFactors = F, header = F)
RandomSitewRes=RandomSites %>% bind_cols(RandomRes)Compare these to the actual:
RealandCont=as.data.frame(cbind(Control=RandomSitewRes$maxentscore_cont, PAS=bothandres$maxentscore))
RealandContG=RealandCont %>% gather("set", "score")ggplot(RealandContG, aes(x=set, y = score,fill=set)) + geom_boxplot() + stat_compare_means()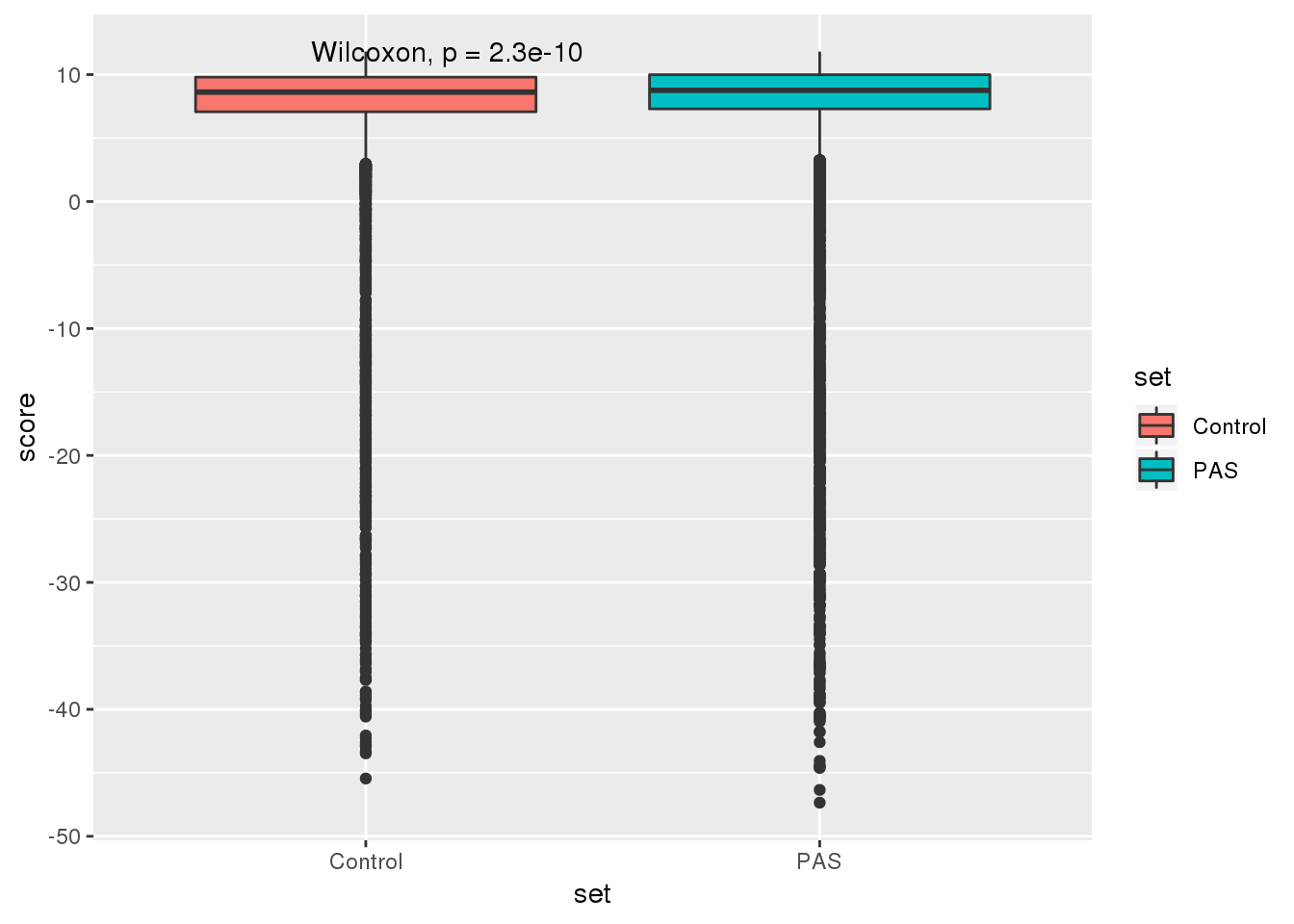
| Version | Author | Date |
|---|---|---|
| 38b532c | brimittleman | 2020-02-18 |
summary(RandomSitewRes$maxentscore_cont) Min. 1st Qu. Median Mean 3rd Qu. Max.
-45.450 7.070 8.620 7.477 9.800 11.810 summary(bothandres$maxentscore) Min. 1st Qu. Median Mean 3rd Qu. Max.
-47.350 7.298 8.760 6.872 9.990 11.810 wilcox.test(RandomSitewRes$maxentscore_cont,bothandres$maxentscore, alternative = "less")
Wilcoxon rank sum test with continuity correction
data: RandomSitewRes$maxentscore_cont and bothandres$maxentscore
W = 74942000, p-value = 1.133e-10
alternative hypothesis: true location shift is less than 0Test if any of the QTLs fall in 5’ splice sites. For this I will look at the 5’ site for every intron:
allIntron_sspos= allIntron %>% filter(strand=="+") %>% mutate(newStart=start-3, newEnd= start +6) %>% select(chr, newStart,newEnd, gene,score, strand)
allIntron_ssneg= allIntron %>% filter(strand=="-") %>% mutate(newStart=end-6, newEnd= end +3) %>% select(chr, newStart,newEnd, gene,score, strand)
AllIntron_both=allIntron_ssneg %>% bind_rows(allIntron_sspos)
write.table(AllIntron_both, "../data/splicesite/AllIntron5primeSS.bed", col.names = F, row.names = F, quote = F, sep="\t")sort and intersect with qtl snps.
sort -k1,1 -k2,2n ../data/splicesite/AllIntron5primeSS.bed| sed 's/^chr//' > ../data/splicesite/AllIntron5primeSS_sort.bed
sort -k1,1 -k2,2n ../data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.WITHSTRAND.bed | sed '1d' | head -n -1 > ../data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.WITHSTRAND.sort.bed
bedtools intersect -wo -a ../data/splicesite/AllIntron5primeSS_sort.bed -b ../data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.WITHSTRAND.sort.bed -s > ../data/splicesite/QTLin5SS.txt
1 example.
15 31229459 31229468 FAN1 . + 15 31229462 31229463 FAN1:peak42822:utr3 83 + 1
Intron by presence of intronic
Seperate the introns by splice site strength. First run the nuc on all of them
#../data/splicesite/AllIntron5primeSS_sort.bed
#bedtools nuc
bedtools nuc -fi /project2/gilad/briana/genome_anotation_data/genome/Homo_sapiens.GRCh37.75.dna_sm.all.fa -bed ../data/splicesite/AllIntron5primeSS_sort.bed -seq -s > ../data/splicesite/AllIntron5primeSS_sort_Nuc.bed
python Allsplicesite2fasta.py
python parseALLSSres.pyIntronSites=read.table("../data/splicesite/AllIntron5primeSS_sort.bed", col.names =c('chr','start','end','name','score','strand'))
IntronRes=read.table("../data/splicesite/AllIntron_Parsed.txt", col.names=c("splicesite", "maxentscore") )
bothandres_g= bothandres %>% mutate(intron=paste(chr,start,end,sep=":")) %>% group_by(intron) %>% summarise(nPAS=n())
IntronSiteswRes=IntronSites %>% bind_cols(IntronRes) %>% mutate(intron=paste(chr,start,end,sep=":")) %>% full_join(bothandres_g,by="intron") %>% replace_na(list(nPAS = 0))Now I need to decide the cutoffs:
ggplot(IntronSiteswRes, aes(x=maxentscore)) + geom_density()
summary(IntronSiteswRes$maxentscore) Min. 1st Qu. Median Mean 3rd Qu. Max.
-48.470 7.130 8.680 7.514 9.800 11.810 quantile(IntronSiteswRes$maxentscore, seq(0,1, by=.1)) 0% 10% 20% 30% 40% 50% 60% 70% 80% 90%
-48.47 4.83 6.64 7.54 8.23 8.68 9.10 9.60 10.07 10.51
100%
11.81 IntronSiteswRes_dec= IntronSiteswRes %>% mutate(decile_rank = ntile(IntronSiteswRes$maxentscore,10))
IntronSiteswRes_decG= IntronSiteswRes_dec%>% group_by(decile_rank) %>% summarise(PAS=sum(nPAS))ggplot(IntronSiteswRes_decG, aes(x=decile_rank, y=PAS)) +geom_bar(stat="identity") + labs(x="5' Splice Site strength Decile", y= "Number of Intronic PAS", title="Number of intronic PAS by 5' Splice site strength")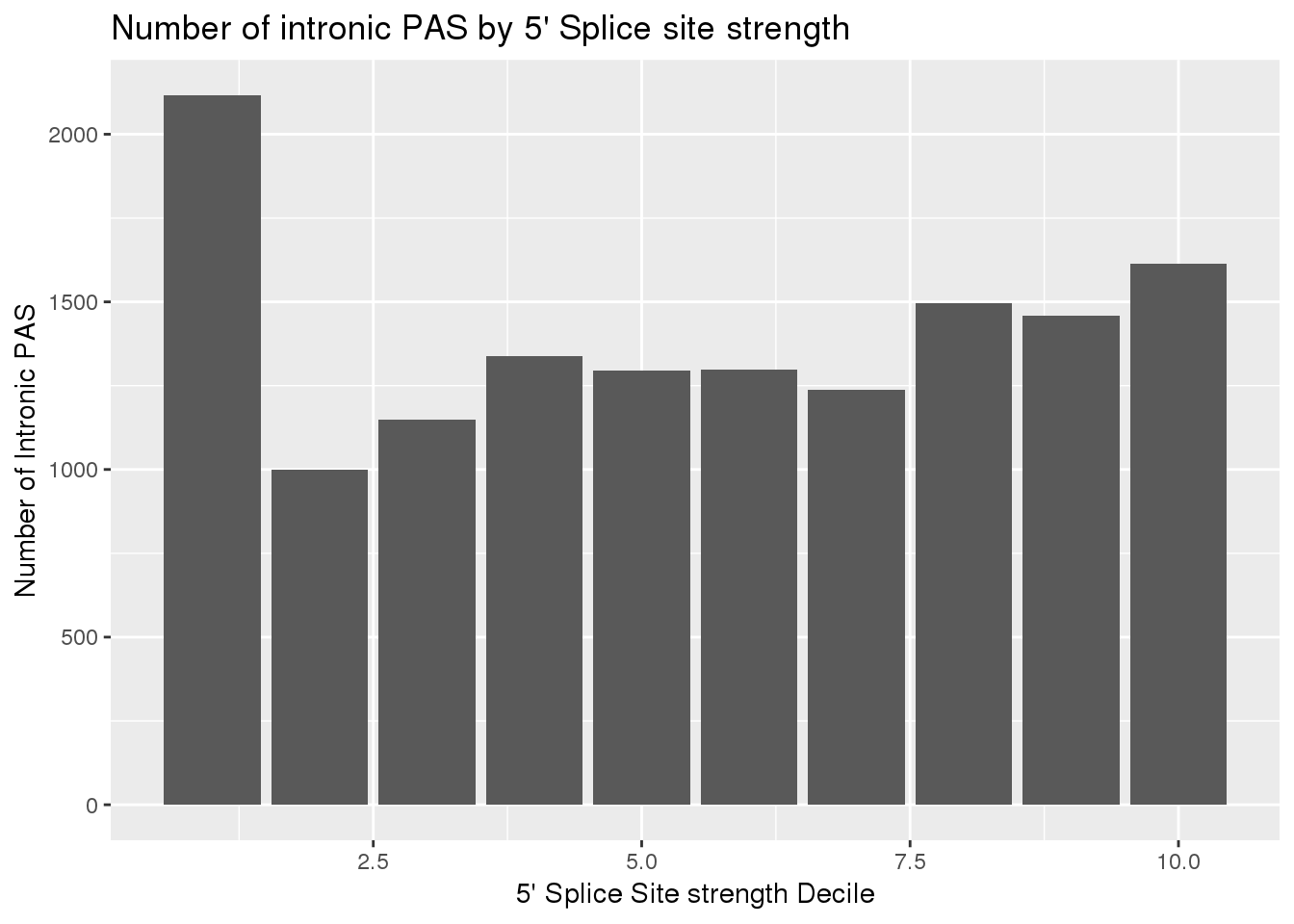
hypergeometric significance
x=2116
m=IntronSiteswRes_dec %>% filter(decile_rank == 1) %>% nrow()
n=IntronSiteswRes_dec %>% filter(decile_rank != 1) %>% nrow()
k=sum(IntronSiteswRes_dec$nPAS)
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 1400x[1] 2116phyper(x, m, n, k,lower.tail=F)[1] 8.065256e-87This means there is significant enrichment for PAS in introns with the weakest 5’ splice sites.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggpubr_0.2 magrittr_1.5 forcats_0.3.0 stringr_1.3.1
[5] dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1 tidyr_0.8.3
[9] tibble_2.1.1 ggplot2_3.1.1 tidyverse_1.2.1 workflowr_1.6.0
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38 colorspace_1.3-2
[5] generics_0.0.2 htmltools_0.3.6 yaml_2.2.0 rlang_0.4.0
[9] later_0.7.5 pillar_1.3.1 glue_1.3.0 withr_2.1.2
[13] modelr_0.1.2 readxl_1.1.0 plyr_1.8.4 munsell_0.5.0
[17] gtable_0.2.0 cellranger_1.1.0 rvest_0.3.2 evaluate_0.12
[21] labeling_0.3 knitr_1.20 httpuv_1.4.5 highr_0.7
[25] broom_0.5.1 Rcpp_1.0.2 promises_1.0.1 scales_1.0.0
[29] backports_1.1.2 jsonlite_1.6 fs_1.3.1 hms_0.4.2
[33] digest_0.6.18 stringi_1.2.4 grid_3.5.1 rprojroot_1.3-2
[37] cli_1.1.0 tools_3.5.1 lazyeval_0.2.1 crayon_1.3.4
[41] whisker_0.3-2 pkgconfig_2.0.2 MASS_7.3-51.1 xml2_1.2.0
[45] lubridate_1.7.4 assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[49] rstudioapi_0.10 R6_2.3.0 nlme_3.1-137 git2r_0.26.1
[53] compiler_3.5.1