Test by number of PAS
Briana Mittleman
1/31/2020
Last updated: 2020-01-31
Checks: 7 0
Knit directory: apaQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.5.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190411) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/logs/
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: .Rprofile
Untracked: ._.DS_Store
Untracked: .gitignore
Untracked: @
Untracked: GEO_brimittleman/
Untracked: _workflowr.yml
Untracked: analysis/._PASdescriptiveplots.Rmd
Untracked: analysis/._cuttoffPercUsage.Rmd
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Nuclear.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Total.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/QTLexampleplots.Rmd
Untracked: analysis/cuttoffPercUsage.Rmd
Untracked: analysis/eQTLoverlap.Rmd
Untracked: analysis/interpret verify bam.Rmd
Untracked: analysis/interpret_verifybam.Rmd
Untracked: analysis/mergeRNA.Rmd
Untracked: analysis/oldstuffNotNeeded.Rmd
Untracked: analysis/remove_badlines.Rmd
Untracked: analysis/totSpecInNuclear.Rmd
Untracked: analysis/totSpecIncludenotTested.Rmd
Untracked: analysis/totalspec.Rmd
Untracked: apaQTL.Rproj
Untracked: checksumsfastq.txt.gz
Untracked: code/.NascentRNAdtPlotFirstintronicPAS.sh.swp
Untracked: code/._ApaQTL_nominalNonnorm.sh
Untracked: code/._BothFracDTPlotGeneRegions.sh
Untracked: code/._BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/._DistPAS2Sig_RandomIntron.py
Untracked: code/._EandPqtl_perm.sh
Untracked: code/._EandPqtls.sh
Untracked: code/._FC_NucintornUpandDown.sh
Untracked: code/._FC_UTR.sh
Untracked: code/._FC_intornUpandDownsteamPAS.sh
Untracked: code/._FC_nascentseq.sh
Untracked: code/._FC_newPeaks_olddata.sh
Untracked: code/._HMMpermuteTotal.py
Untracked: code/._HmmPermute.py
Untracked: code/._IntronicPASDT.sh
Untracked: code/._LC_samplegroups.py
Untracked: code/._LD_qtl.sh
Untracked: code/._LD_snpsproxy.sh
Untracked: code/._NascentRNAdtPlot.sh
Untracked: code/._NascentRNAdtPlot3UTRPAS.sh
Untracked: code/._NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/._NascentRNAdtPlotNucPAS.sh
Untracked: code/._NascentRNAdtPlotTotPAS.sh
Untracked: code/._NascentRNAdtPlotintronicPAS.sh
Untracked: code/._NascnetRNAdtPlotPAS.sh
Untracked: code/._NetSeq_fourthintronDT.sh
Untracked: code/._NomResfromPASSNP.py
Untracked: code/._NuclearPAS_5per.bed.py
Untracked: code/._NuclearandRNA5samp_dtplots.sh
Untracked: code/._PTTfacetboxplots.R
Untracked: code/._PrematureQTLNominal.sh
Untracked: code/._PrematureQTLPermuted.sh
Untracked: code/._QTL2bed.py
Untracked: code/._QTL2bed_withstrand.py
Untracked: code/._RNAbam2bw.sh
Untracked: code/._RNAseqDTplot.sh
Untracked: code/._RunRes2PAS.sh
Untracked: code/._SAF215upbed.py
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilefiltPAS
Untracked: code/._TESplots100bp.sh
Untracked: code/._TESplots150bp.sh
Untracked: code/._TESplots200bp.sh
Untracked: code/._TotalPAS_5perc.bed.py
Untracked: code/._Untitled
Untracked: code/._ZipandTabPheno.sh
Untracked: code/._aAPAqtl_nominal39ind.sh
Untracked: code/._allNucSpecQTLine.py
Untracked: code/._allNucSpecfromNonNorm.py
Untracked: code/._annotatePacBioPASregion.sh
Untracked: code/._annotatedPAS2bed.py
Untracked: code/._apaInPandE.py
Untracked: code/._apaQTLCorrectPvalMakeQQ.R
Untracked: code/._apaQTLCorrectpval_6or7a.R
Untracked: code/._apaQTL_Nominal.sh
Untracked: code/._apaQTL_nominalInclusive.sh
Untracked: code/._apaQTL_nominalv67.sh
Untracked: code/._apaQTL_permuted.sh
Untracked: code/._apaQTL_permuted_test6A7A.sh
Untracked: code/._apainRibo.py
Untracked: code/._assignNucIntonpeak2intronlocs.sh
Untracked: code/._assignTotIntronpeak2intronlocs.sh
Untracked: code/._bam2BW_5primemost.sh
Untracked: code/._bed2saf.py
Untracked: code/._bothFracDTplot1stintron.sh
Untracked: code/._bothFracDTplot4thintron.sh
Untracked: code/._bothFrac_FC.sh
Untracked: code/._callPeaksYL.py
Untracked: code/._changeRibonomQTLres2genename.py
Untracked: code/._changenomQTLres2geneName.py
Untracked: code/._chooseAnno2PAS_pacbio.py
Untracked: code/._chooseAnno2SAF.py
Untracked: code/._chooseSignalSite
Untracked: code/._chooseSignalSite.py
Untracked: code/._closestannotated.sh
Untracked: code/._closestannotated_byfrac.sh
Untracked: code/._cluster.json
Untracked: code/._clusterPAS.json
Untracked: code/._clusterfiltPAS.json
Untracked: code/._codingdms2bed.py
Untracked: code/._config.yaml
Untracked: code/._config2.yaml
Untracked: code/._configOLD.yaml
Untracked: code/._convertNominal2SNPLOC.py
Untracked: code/._convertNominal2SNPloc2Versions.py
Untracked: code/._convertNumeric.py
Untracked: code/._correctNomeqtl.R
Untracked: code/._createPlinkSampfile.py
Untracked: code/._dag.pdf
Untracked: code/._eQTL_switch2snploc.py
Untracked: code/._eQTLgenestestedapa.py
Untracked: code/._encodeRNADTplots.sh
Untracked: code/._extactPAS100meanphyloP.py
Untracked: code/._extractGenotypes.py
Untracked: code/._extractPACmeanPhyloP.py
Untracked: code/._extractseqfromqtlfastq.py
Untracked: code/._fc2leafphen.py
Untracked: code/._fc_filteredPAS6and7As.sh
Untracked: code/._fifteenBPupstreamPAS.py
Untracked: code/._fiftyBPupstreamPAS.py
Untracked: code/._filter5perc.R
Untracked: code/._filter5percPheno.py
Untracked: code/._filterLDsnps.py
Untracked: code/._filterMPPAS.py
Untracked: code/._filterMPPAS_15.py
Untracked: code/._filterMPPAS_15_7As.py
Untracked: code/._filterMPPAS_50.py
Untracked: code/._filterSAFforMP.py
Untracked: code/._filterpeaks.py
Untracked: code/._finalPASbed2SAF.py
Untracked: code/._fix4su304corr.py
Untracked: code/._fix4su604corr.py
Untracked: code/._fix4sukalisto.py
Untracked: code/._fixExandUnexeQTL
Untracked: code/._fixExandUnexeQTL.py
Untracked: code/._fixFChead.py
Untracked: code/._fixFChead_bothfrac.py
Untracked: code/._fixFChead_short.py
Untracked: code/._fixH3k12ac.py
Untracked: code/._fixPASregionSNPs.py
Untracked: code/._fixRNAhead4corr.py
Untracked: code/._fixRNAkalisto.py
Untracked: code/._fix_randomIntron.py
Untracked: code/._fixgroupedtranscript.py
Untracked: code/._fixhead_netseqfc.py
Untracked: code/._getAPAfromanyeQTL.py
Untracked: code/._getApapval4eqtl.py
Untracked: code/._getApapval4eqtl_unexp.py
Untracked: code/._getApapval4eqtl_version67.py
Untracked: code/._getDownstreamIntronNuclear.py
Untracked: code/._getIntronDownstreamPAS.py
Untracked: code/._getIntronUpstreamPAS.py
Untracked: code/._getQTLalleles.py
Untracked: code/._getQTLfastq.sh
Untracked: code/._getUpstreamIntronNuclear.py
Untracked: code/._grouptranscripts.py
Untracked: code/._intersectVCFandupPAS.sh
Untracked: code/._keep5perMAF.py
Untracked: code/._keepSNP_vcf.sh
Untracked: code/._make5percPeakbed.py
Untracked: code/._makeFileID.py
Untracked: code/._makePheno.py
Untracked: code/._makeSAFbothfrac5perc.py
Untracked: code/._makeSNP2rsidfile.py
Untracked: code/._makeeQTLempirical_unexp.py
Untracked: code/._makeeQTLempiricaldist.py
Untracked: code/._makegencondeTSSfile.py
Untracked: code/._mapSSsnps2PAS.sh
Untracked: code/._mergRNABam.sh
Untracked: code/._mergeAllBam.sh
Untracked: code/._mergeAnnotations.sh
Untracked: code/._mergeBW_norm.sh
Untracked: code/._mergeBamNascent.sh
Untracked: code/._mergeByFracBam.sh
Untracked: code/._mergePeaks.sh
Untracked: code/._mnase1stintron.sh
Untracked: code/._mnaseDT_fourthintron.sh
Untracked: code/._namePeaks.py
Untracked: code/._netseqDTplot1stIntron.sh
Untracked: code/._netseqFC.sh
Untracked: code/._nucQTLGWAS.py
Untracked: code/._nucSpecQTLineData.py
Untracked: code/._nucSpeceffectsize.py
Untracked: code/._nucspecnucPASine.py
Untracked: code/._pQTLsotherdata.py
Untracked: code/._pacbioDT.sh
Untracked: code/._pacbioIntronicDT.sh
Untracked: code/._parseBestbamid.py
Untracked: code/._peak2PAS.py
Untracked: code/._peakFC.sh
Untracked: code/._pheno2countonly.R
Untracked: code/._phenoQTLfromlist.py
Untracked: code/._processYRIgen.py
Untracked: code/._pttQTLsinapaQTL.py
Untracked: code/._qtlRegionseq.sh
Untracked: code/._qtlsPvalOppFrac.py
Untracked: code/._quantassign2parsedpeak.py
Untracked: code/._removeXfromHmm.py
Untracked: code/._removeloc_pheno.py
Untracked: code/._riboQTL.sh
Untracked: code/._runCorrectNomEqtl.sh
Untracked: code/._runHMMpermuteAPAqtls.sh
Untracked: code/._runHMMpermuteeQTLS.sh
Untracked: code/._runMakeEmpiricaleQTL_unexp.sh
Untracked: code/._runMakeeQTLempirical.sh
Untracked: code/._run_bam2bw_all3prime.sh
Untracked: code/._run_bam2bw_extra3.sh
Untracked: code/._run_bestbamid.sj
Untracked: code/._run_dist2sig_randomintron.sh
Untracked: code/._run_filtersnpLD.sh
Untracked: code/._run_getAPAfromeQTL_version6.7.sh
Untracked: code/._run_getApaPval4eqtl.sh
Untracked: code/._run_getapafromeQTL.py
Untracked: code/._run_getapafromeQTL.sh
Untracked: code/._run_getapapval4eqtl_unexp.sh
Untracked: code/._run_leafcutterDiffIso.sh
Untracked: code/._run_prxySNP.sh
Untracked: code/._run_pttfacetboxplot.sh
Untracked: code/._run_sepUsagephen.sh
Untracked: code/._run_sepgenobychrom.sh
Untracked: code/._run_verifybam.sh
Untracked: code/._selectNominalPvalues.py
Untracked: code/._sepUsagePhen.py
Untracked: code/._sepgenobychrom.py
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._sortindexRNAbam.sh
Untracked: code/._specAPAinE.py
Untracked: code/._submit-snakemakePAS.sh
Untracked: code/._submit-snakemakefiltPAS.sh
Untracked: code/._subsetAPAnotEorPgene.py
Untracked: code/._subsetAPAnotEorPgene_2versions.py
Untracked: code/._subsetApanoteGene.py
Untracked: code/._subsetApanoteGene_2versions.py
Untracked: code/._subsetUnexplainedeQTLs.py
Untracked: code/._subsetVCF_SS.sh
Untracked: code/._subsetVCF_noSSregions.sh
Untracked: code/._subsetVCF_upstreamPAS.sh
Untracked: code/._subset_diffisopheno.py
Untracked: code/._subsetpermAPAwithGenelist.py
Untracked: code/._subsetpermAPAwithGenelist_2versions.py
Untracked: code/._subsetvcf_otherreg.sh
Untracked: code/._subsetvcf_permSS.sh
Untracked: code/._subtrachfiveprimeUTR.sh
Untracked: code/._subtractExons.sh
Untracked: code/._subtractfiveprimeUTR.sh
Untracked: code/._tabixSNPS.sh
Untracked: code/._tenBPupstreamPAS.py
Untracked: code/._testVerifyBam.sh
Untracked: code/._totSeceffectsize.py
Untracked: code/._twentyBPupstreamPAS.py
Untracked: code/._utrdms2saf.py
Untracked: code/._vcf2bed.py
Untracked: code/._verifyBam18517N.sh
Untracked: code/._verifyBam18517T.sh
Untracked: code/._verifyBam19128N.sh
Untracked: code/._verifyBam19128T.sh
Untracked: code/._wrap_verifybam.sh
Untracked: code/._writePTTexamplecode.py
Untracked: code/._writePTTexamplecode.sh
Untracked: code/.pversion
Untracked: code/.snakemake/
Untracked: code/1
Untracked: code/APAqtl_nominal.err
Untracked: code/APAqtl_nominal.out
Untracked: code/APAqtl_nominal_39.err
Untracked: code/APAqtl_nominal_39.out
Untracked: code/APAqtl_nominal_inclusive.err
Untracked: code/APAqtl_nominal_inclusive.out
Untracked: code/APAqtl_nominal_nonNorm.err
Untracked: code/APAqtl_nominal_nonNorm.out
Untracked: code/APAqtl_nominal_versions67.err
Untracked: code/APAqtl_nominal_versions67.out
Untracked: code/APAqtl_permuted.err
Untracked: code/APAqtl_permuted.out
Untracked: code/APAqtl_permuted_versions67.err
Untracked: code/APAqtl_permuted_versions67.out
Untracked: code/BothFracDTPlot1stintron.err
Untracked: code/BothFracDTPlot1stintron.out
Untracked: code/BothFracDTPlot4stintron.err
Untracked: code/BothFracDTPlot4stintron.out
Untracked: code/BothFracDTPlotGeneRegions.err
Untracked: code/BothFracDTPlotGeneRegions.out
Untracked: code/BothFracDTPlotGeneRegions_norm.err
Untracked: code/BothFracDTPlotGeneRegions_norm.out
Untracked: code/DistPAS2Sig_RandomIntron.py
Untracked: code/EandPqtl.err
Untracked: code/EandPqtl.out
Untracked: code/EncodeRNADTPlotGeneRegions.err
Untracked: code/EncodeRNADTPlotGeneRegions.out
Untracked: code/FC_NucintronPASupandDown.err
Untracked: code/FC_NucintronPASupandDown.out
Untracked: code/FC_UTR.err
Untracked: code/FC_UTR.out
Untracked: code/FC_intronPASupandDown.err
Untracked: code/FC_intronPASupandDown.out
Untracked: code/FC_nascent.err
Untracked: code/FC_nascentout
Untracked: code/FC_newPAS_olddata.err
Untracked: code/FC_newPAS_olddata.out
Untracked: code/HmmPermute.p
Untracked: code/IntronicPASDT.err
Untracked: code/IntronicPASDT.out
Untracked: code/LD_vcftools.hap.out
Untracked: code/NascentDTPlotGeneRegions.err
Untracked: code/NascentDTPlotGeneRegions.out
Untracked: code/NascentDTPlotPAS.err
Untracked: code/NascentDTPlotPAS.out
Untracked: code/NascentDTPlotPAS_3utr.err
Untracked: code/NascentDTPlotPAS_3utr.out
Untracked: code/NascentDTPlotPAS_firstintron.err
Untracked: code/NascentDTPlotPAS_firstintron.out
Untracked: code/NascentDTPlotPAS_intron.err
Untracked: code/NascentDTPlotPAS_intron.out
Untracked: code/NascentDTPlotPAS_nuc.err
Untracked: code/NascentDTPlotPAS_nuc.out
Untracked: code/NascentDTPlotPAS_tot.err
Untracked: code/NascentDTPlotPAS_tot.out
Untracked: code/Nuclear_example.err
Untracked: code/Nuclear_example.out
Untracked: code/NuclearandRNA5samp_dtplots.sh
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.err
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.out
Untracked: code/PACbioDT.err
Untracked: code/PACbioDT.out
Untracked: code/PACbioDTitronic.err
Untracked: code/PACbioDTitronic.out
Untracked: code/Prematureqtl_nominal.err
Untracked: code/Prematureqtl_nominal.out
Untracked: code/Prematureqtl_permuted.err
Untracked: code/Prematureqtl_permuted.out
Untracked: code/README.md
Untracked: code/RNABam2BW.err
Untracked: code/RNABam2BW.out
Untracked: code/RNAseqDTPlotGeneRegions.err
Untracked: code/RNAseqDTPlotGeneRegions.out
Untracked: code/Rplots.pdf
Untracked: code/TESplots100bp.err
Untracked: code/TESplots100bp.out
Untracked: code/TESplots150bp.err
Untracked: code/TESplots150bp.out
Untracked: code/TESplots200bp.err
Untracked: code/TESplots200bp.out
Untracked: code/Total_example.err
Untracked: code/Total_example.out
Untracked: code/Untitled
Untracked: code/YRI_LCL.vcf.gz
Untracked: code/YRI_LCL_chr1.vcf.gz.log
Untracked: code/YRI_LCL_chr1.vcf.gz.recode.vcf
Untracked: code/annotatedPASregion.err
Untracked: code/annotatedPASregion.out
Untracked: code/apaQTL_nominalInclusive.sh
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assigntotPeak2Intronicregion.err
Untracked: code/assigntotPeak2Intronicregion.out
Untracked: code/bam2bw.err
Untracked: code/bam2bw.out
Untracked: code/bam2bw_5primemost.err
Untracked: code/bam2bw_5primemost.out
Untracked: code/binary_fileset.log
Untracked: code/bothFrac_FC.err
Untracked: code/bothFrac_FC.out
Untracked: code/callSHscripts.txt
Untracked: code/closestannotated.err
Untracked: code/closestannotated.out
Untracked: code/closestannotatedbyfrac.err
Untracked: code/closestannotatedbyfrac.out
Untracked: code/dag.pdf
Untracked: code/dagPAS.pdf
Untracked: code/dagfiltPAS.pdf
Untracked: code/extactPAS100meanphyloP.py
Untracked: code/extractPACmeanPhyloP.py
Untracked: code/fixExandUnexeQTL
Untracked: code/fix_randomIntron.py
Untracked: code/genotypesYRI.gen.proc.keep.vcf.log
Untracked: code/genotypesYRI.gen.proc.keep.vcf.recode.vcf
Untracked: code/getseq100up.err
Untracked: code/getseq100up.out
Untracked: code/grouptranscripts.err
Untracked: code/grouptranscripts.out
Untracked: code/intersectPAS_ssSNPS.err
Untracked: code/intersectPAS_ssSNPS.out
Untracked: code/intersectVCFPAS.err
Untracked: code/intersectVCFPAS.out
Untracked: code/log/
Untracked: code/merge53PRIMEbam.err
Untracked: code/merge53PRIMEbam.out
Untracked: code/merge53RNAbam.err
Untracked: code/merge53prime.sh
Untracked: code/merge5RNABam.err
Untracked: code/merge5RNABam.out
Untracked: code/merge5RNAbam.out
Untracked: code/merge5RNAbam.sh
Untracked: code/mergeAnno.err
Untracked: code/mergeAnno.out
Untracked: code/mergeBWnorm.err
Untracked: code/mergeBWnorm.out
Untracked: code/mergeBamNacent.err
Untracked: code/mergeBamNacent.out
Untracked: code/mergeRNAbam.err
Untracked: code/mergeRNAbam.out
Untracked: code/mnaseDTPlot1stintron.err
Untracked: code/mnaseDTPlot1stintron.out
Untracked: code/mnaseDTPlot4thintron.err
Untracked: code/mnaseDTPlot4thintron.out
Untracked: code/netDTPlot4thintron.out
Untracked: code/netseqFC.err
Untracked: code/netseqFC.out
Untracked: code/neyDTPlot4thintron.err
Untracked: code/nucspecinE.py
Untracked: code/plink.log
Untracked: code/prxySNP.err
Untracked: code/prxySNP.out
Untracked: code/pttFacetBoxplots.err
Untracked: code/pttFacetBoxplots.out
Untracked: code/qtlFacetBoxplots.err
Untracked: code/qtlFacetBoxplots.out
Untracked: code/rLD_vcftools.hap.err
Untracked: code/riboqtl.err
Untracked: code/riboqtl.out
Untracked: code/runBestBamID.err
Untracked: code/runCorrectNomeqtl.err
Untracked: code/runCorrectNomeqtl.out
Untracked: code/runFilterLD.err
Untracked: code/runFilterLD.out
Untracked: code/runHMMpermute.err
Untracked: code/runHMMpermute.out
Untracked: code/runHMMpermuteeQTLs.err
Untracked: code/runHMMpermuteeQTLs.out
Untracked: code/runMakeEmpiricaleQTLs.err
Untracked: code/runMakeEmpiricaleQTLs.out
Untracked: code/runMakeEmpiricaleQTLsunex.err
Untracked: code/runMakeEmpiricaleQTLsunex.out
Untracked: code/run_DistPAS2Sig.err
Untracked: code/run_DistPAS2Sig.out
Untracked: code/run_DistPAS2Sig_intron.err
Untracked: code/run_DistPAS2Sig_intron.out
Untracked: code/run_bam2bw.err
Untracked: code/run_bam2bw.out
Untracked: code/run_bam2bwexta.err
Untracked: code/run_bam2bwexta.out
Untracked: code/run_dist2sig_randomintron.sh
Untracked: code/run_getAPAfromanyeQTL.err
Untracked: code/run_getAPAfromanyeQTL.out
Untracked: code/run_getApaPval4eQTLs.err
Untracked: code/run_getApaPval4eQTLs.out
Untracked: code/run_getApaPval4eQTLsunexplained.err
Untracked: code/run_getApaPval4eQTLsunexplained.out
Untracked: code/run_leafcutter_ds.err
Untracked: code/run_leafcutter_ds.out
Untracked: code/run_sepgenobychrom.err
Untracked: code/run_sepgenobychrom.out
Untracked: code/run_sepusage.err
Untracked: code/run_sepusage.out
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/run_verifybam128N.err
Untracked: code/run_verifybam128N.out
Untracked: code/run_verifybam128T.err
Untracked: code/run_verifybam128T.out
Untracked: code/run_verifybam517N.err
Untracked: code/run_verifybam517N.out
Untracked: code/run_verifybam517T.err
Untracked: code/run_verifybam517T.out
Untracked: code/runprxySNP.err
Untracked: code/runprxySNP.out
Untracked: code/runres2pas.err
Untracked: code/runres2pas.out
Untracked: code/scripts/
Untracked: code/scripts_PAS_500_Lymph/
Untracked: code/seqQTLfastq.err
Untracked: code/seqQTLfastq.out
Untracked: code/seqQTLregion.err
Untracked: code/seqQTLregion.out
Untracked: code/snakePASlog.out
Untracked: code/snakefiltPASlog.out
Untracked: code/sortindexRNABam.err
Untracked: code/sortindexRNABam.out
Untracked: code/specAPAinE.py
Untracked: code/subsetvcf_SS.err
Untracked: code/subsetvcf_SS.out
Untracked: code/subsetvcf_noSS.err
Untracked: code/subsetvcf_noSS.out
Untracked: code/subsetvcf_pas.err
Untracked: code/subsetvcf_pas.out
Untracked: code/subsetvcf_perm.err
Untracked: code/subsetvcf_perm.out
Untracked: code/subsetvcf_rand.err
Untracked: code/subsetvcf_rand.out
Untracked: code/subtract5UTR.err
Untracked: code/subtract5UTR.out
Untracked: code/subtractExons.err
Untracked: code/subtractExons.out
Untracked: code/tabixSNPs.err
Untracked: code/tabixSNPs.out
Untracked: code/test_verifybam.err
Untracked: code/test_verifybam.out
Untracked: code/vcf_keepsnps.err
Untracked: code/vcf_keepsnps.out
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/zipandtabPhen.err
Untracked: code/zipandtabPhen.out
Untracked: data/._.DS_Store
Untracked: data/._MetaDataSequencing.txt
Untracked: data/AnnotatedPAS/
Untracked: data/ApaByEgene/
Untracked: data/ApaByPgene/
Untracked: data/BadLines/
Untracked: data/Battle_pQTL/
Untracked: data/CheckSums/
Untracked: data/CompareOldandNew/
Untracked: data/DTmatrix/
Untracked: data/DiffIso/
Untracked: data/EncodeRNA/
Untracked: data/ExampleQTLPlots/
Untracked: data/ExpressionIndependentapaQTLs.txt
Untracked: data/FiveMergedBW/
Untracked: data/FiveMergedBam/
Untracked: data/FlaggedPAS/
Untracked: data/GWAS_overlap/
Untracked: data/GeuvadisRNA/
Untracked: data/HMMqtls/
Untracked: data/LDSR_annotations/
Untracked: data/Li_eQTLs/
Untracked: data/NMD/
Untracked: data/NascentRNA/
Untracked: data/NucSpeceQTLeffect/
Untracked: data/PAS/
Untracked: data/PAS_Nuclear1000.1.log
Untracked: data/PAS_Nuclear1000.10.log
Untracked: data/PAS_Nuclear1000.11.log
Untracked: data/PAS_Nuclear1000.12.log
Untracked: data/PAS_Nuclear1000.13.log
Untracked: data/PAS_Nuclear1000.14.log
Untracked: data/PAS_Nuclear1000.15.log
Untracked: data/PAS_Nuclear1000.16.log
Untracked: data/PAS_Nuclear1000.17.log
Untracked: data/PAS_Nuclear1000.18.log
Untracked: data/PAS_Nuclear1000.19.log
Untracked: data/PAS_Nuclear1000.2.log
Untracked: data/PAS_Nuclear1000.20.log
Untracked: data/PAS_Nuclear1000.21.log
Untracked: data/PAS_Nuclear1000.22.log
Untracked: data/PAS_Nuclear1000.3.log
Untracked: data/PAS_Nuclear1000.4.log
Untracked: data/PAS_Nuclear1000.5.log
Untracked: data/PAS_Nuclear1000.6.log
Untracked: data/PAS_Nuclear1000.7.log
Untracked: data/PAS_Nuclear1000.8.log
Untracked: data/PAS_Nuclear1000.9.log
Untracked: data/PAS_Nuclear500.1.log
Untracked: data/PAS_Nuclear500.10.log
Untracked: data/PAS_Nuclear500.11.log
Untracked: data/PAS_Nuclear500.12.log
Untracked: data/PAS_Nuclear500.13.log
Untracked: data/PAS_Nuclear500.14.log
Untracked: data/PAS_Nuclear500.15.log
Untracked: data/PAS_Nuclear500.16.log
Untracked: data/PAS_Nuclear500.17.log
Untracked: data/PAS_Nuclear500.18.log
Untracked: data/PAS_Nuclear500.19.log
Untracked: data/PAS_Nuclear500.2.log
Untracked: data/PAS_Nuclear500.20.log
Untracked: data/PAS_Nuclear500.21.log
Untracked: data/PAS_Nuclear500.22.log
Untracked: data/PAS_Nuclear500.3.log
Untracked: data/PAS_Nuclear500.4.log
Untracked: data/PAS_Nuclear500.5.log
Untracked: data/PAS_Nuclear500.6.log
Untracked: data/PAS_Nuclear500.7.log
Untracked: data/PAS_Nuclear500.8.log
Untracked: data/PAS_Nuclear500.9.log
Untracked: data/PAS_postFlag/
Untracked: data/PolyA_DB/
Untracked: data/PreTerm_pheno/
Untracked: data/PrematureQTLNominal/
Untracked: data/PrematureQTLPermuted/
Untracked: data/QTLGenotypes/
Untracked: data/QTL_Nuclear1000.10.log
Untracked: data/QTL_Nuclear1000.11.log
Untracked: data/QTL_Nuclear1000.12.log
Untracked: data/QTL_Nuclear1000.13.log
Untracked: data/QTL_Nuclear1000.14.log
Untracked: data/QTL_Nuclear1000.15.log
Untracked: data/QTL_Nuclear1000.16.log
Untracked: data/QTL_Nuclear1000.17.log
Untracked: data/QTL_Nuclear1000.18.log
Untracked: data/QTL_Nuclear1000.19.log
Untracked: data/QTL_Nuclear1000.20.log
Untracked: data/QTL_Nuclear1000.21.log
Untracked: data/QTL_Nuclear1000.22.log
Untracked: data/QTL_Nuclear1000.4.log
Untracked: data/QTL_Nuclear1000.5.log
Untracked: data/QTL_Nuclear1000.7.log
Untracked: data/QTL_Nuclear1000.8.log
Untracked: data/QTL_Nuclear1000.9.log
Untracked: data/QTL_Nuclear500.10.log
Untracked: data/QTL_Nuclear500.11.log
Untracked: data/QTL_Nuclear500.12.log
Untracked: data/QTL_Nuclear500.13.log
Untracked: data/QTL_Nuclear500.14.log
Untracked: data/QTL_Nuclear500.15.log
Untracked: data/QTL_Nuclear500.16.log
Untracked: data/QTL_Nuclear500.17.log
Untracked: data/QTL_Nuclear500.18.log
Untracked: data/QTL_Nuclear500.19.log
Untracked: data/QTL_Nuclear500.20.log
Untracked: data/QTL_Nuclear500.21.log
Untracked: data/QTL_Nuclear500.22.log
Untracked: data/QTL_Nuclear500.5.log
Untracked: data/QTL_Nuclear500.7.log
Untracked: data/QTL_Nuclear500.8.log
Untracked: data/QTL_Nuclear500.9.log
Untracked: data/QTLoverlap/
Untracked: data/QTLoverlap_inclusive/
Untracked: data/QTLoverlap_nonNorm/
Untracked: data/README.md
Untracked: data/RNAseq/
Untracked: data/Reads2UTR/
Untracked: data/SNPinSS/
Untracked: data/SignalSiteFiles/
Untracked: data/TF_motifdisruption/
Untracked: data/ThirtyNineIndQtl_nominal/
Untracked: data/Version15bp6As/
Untracked: data/Version15bp7As/
Untracked: data/apaQTLNominal/
Untracked: data/apaQTLNominal_4pc/
Untracked: data/apaQTLNominal_inclusive/
Untracked: data/apaQTLPermuted/
Untracked: data/apaQTLPermuted_4pc/
Untracked: data/apaQTLs/
Untracked: data/assignedPeaks/
Untracked: data/assignedPeaks_15Up/
Untracked: data/bam/
Untracked: data/bam_clean/
Untracked: data/bam_waspfilt/
Untracked: data/bed_10up/
Untracked: data/bed_clean/
Untracked: data/bed_clean_sort/
Untracked: data/bed_waspfilter/
Untracked: data/bedsort_waspfilter/
Untracked: data/bothFrac_FC/
Untracked: data/bw/
Untracked: data/bw_norm/
Untracked: data/eQTLs/
Untracked: data/exampleQTLs/
Untracked: data/fastq/
Untracked: data/filterPeaks/
Untracked: data/fourSU/
Untracked: data/h3k27ac/
Untracked: data/highdiffsiggenes.txt
Untracked: data/inclusivePeaks/
Untracked: data/inclusivePeaks_FC/
Untracked: data/intronRNAratio/
Untracked: data/intron_analysis/
Untracked: data/locusZoom/
Untracked: data/mergedBG/
Untracked: data/mergedBW_byfrac/
Untracked: data/mergedBW_norm/
Untracked: data/mergedBam/
Untracked: data/mergedbyFracBam/
Untracked: data/molPhenos/
Untracked: data/molQTLs/
Untracked: data/motifdistrupt/
Untracked: data/nPAS/
Untracked: data/netseq/
Untracked: data/nonNorm_pheno/
Untracked: data/nuc_10up/
Untracked: data/nuc_10upclean/
Untracked: data/oldPASfiles/
Untracked: data/overlapeQTL_try2/
Untracked: data/overlapeQTLs/
Untracked: data/pQTLoverlap/
Untracked: data/pacbio/
Untracked: data/peakCoverage/
Untracked: data/peaks_5perc/
Untracked: data/phenotype/
Untracked: data/phenotype_5perc/
Untracked: data/phenotype_inclusivePAS/
Untracked: data/phylop/
Untracked: data/pttQTL/
Untracked: data/pttQTLplots/
Untracked: data/sigDiffGenes.txt
Untracked: data/sort/
Untracked: data/sort_clean/
Untracked: data/sort_waspfilter/
Untracked: data/twoMech/
Untracked: data/vareQTLvarAPAqtl/
Untracked: data/verifyBAM/
Untracked: data/verifyBAM_full/
Untracked: nohup.out
Untracked: output/._.DS_Store
Untracked: output/._AverageDiffHeatmap.Nuclear.png
Untracked: output/._AverageDiffHeatmap.Total.png
Untracked: output/._GeneswithAPApotential.png
Untracked: output/._GeneswithAPApotentialAllPAS.png
Untracked: output/._PASlocation.png
Untracked: output/._SignalSitePlot.png
Untracked: output/._meanCorrelationPhenotypes.svg
Untracked: output/._qqplot_Nuclear_APAperm.png
Untracked: output/._qqplot_Nuclear_APAperm_4pc.png
Untracked: output/._qqplot_Total_APAperm.png
Untracked: output/._qqplot_Total_APAperm_4pc.png
Untracked: output/AverageDiffHeatmap.Nuclear.png
Untracked: output/AverageDiffHeatmap.Total.png
Untracked: output/GeneswithAPApotential.png
Untracked: output/GeneswithAPApotentialAllPAS.png
Untracked: output/PASlocation.png
Untracked: output/SignalSitePlot.png
Untracked: output/SignalSitePlotbyLoc.png
Untracked: output/dtPlots/
Untracked: output/fastqc/
Untracked: output/meanCorrelationPhenotypes.svg
Untracked: output/newnuc.png
Untracked: output/newtot.png
Untracked: output/oldnuc.png
Untracked: output/oldtot.png
Untracked: output/qqplot_Nuclear_APAperm.png
Untracked: output/qqplot_Nuclear_APAperm_4pc.png
Untracked: output/qqplot_Total_APAperm.png
Untracked: output/qqplot_Total_APAperm_4pc.png
Untracked: run_verifybam517N.err
Untracked: run_verifybam517N.out
Unstaged changes:
Modified: analysis/LDregress.Rmd
Modified: analysis/NuclearSpecIncludeNotTested.Rmd
Modified: analysis/PASdescriptiveplots.Rmd
Modified: analysis/Readdistagainstfeatures.Rmd
Modified: analysis/nucSpecinEQTLs.Rmd
Modified: analysis/overlapapaqtlsandeqtls.Rmd
Modified: analysis/pQTLexampleplot.Rmd
Modified: analysis/propeQTLs_explained.Rmd
Modified: analysis/version15bpfilter.Rmd
Modified: code/DistPAS2Sig.py
Modified: code/apaQTLsnake.err
Deleted: code/test.txt
Deleted: reads_graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | a1607df | brimittleman | 2020-01-31 | look at tissue specifcity |
library(workflowr)This is workflowr version 1.5.0
Run ?workflowr for help getting startedlibrary(ggpubr)Loading required package: ggplot2Loading required package: magrittrlibrary(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ tibble 2.1.1 ✔ purrr 0.3.2
✔ tidyr 0.8.3 ✔ dplyr 0.8.0.1
✔ readr 1.3.1 ✔ stringr 1.3.1
✔ tibble 2.1.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ tidyr::extract() masks magrittr::extract()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
✖ purrr::set_names() masks magrittr::set_names()In this analysis I will answer the reviewer questions related to the number of PAS per gene.
First I want to get the number of PAS used at 5% per gene. I am doing this with the nuclear results.
PAS=read.table("../data/PAS/APApeak_Peaks_GeneLocAnno.Nuclear.5perc.sort.bed",col.names = c("chr","start","end","name","score","strand")) %>% separate(name,into=c("pas", 'gene','loc'), sep=":") %>% group_by(gene) %>% summarise(nPAS=n())ggplot(PAS,aes(x=nPAS)) + geom_bar()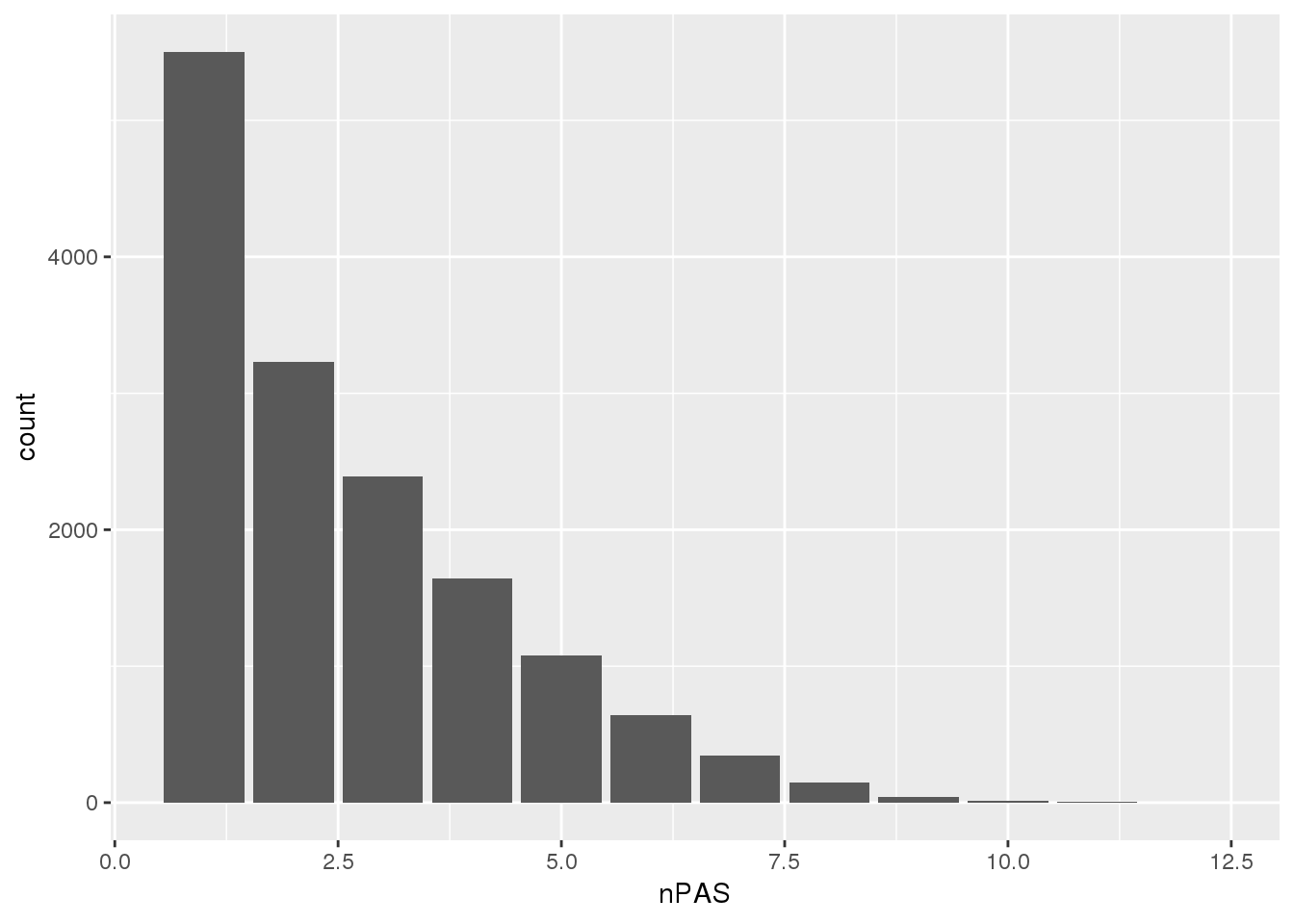 The data goes from 1-10 PAS per gene.
The data goes from 1-10 PAS per gene.
length of gene
I will start with AllTranscriptsbyName.Grouped.bed. For this I will used the longest annotated transcript by transcription start and end.
genes=read.table("../../genome_anotation_data/RefSeq_annotations/Hg19_refseq_genes.txt",header = T,stringsAsFactors = F) %>%
mutate(Genelength=txEnd-txStart) %>%
group_by(name2) %>%
arrange(desc(Genelength)) %>%
slice(1) %>%
select(name2, Genelength) %>%
rename("gene"=name2)PAS_wLength= PAS %>% inner_join(genes, by="gene")Check for correlation
cor.test(PAS_wLength$Genelength, PAS_wLength$nPAS)
Pearson's product-moment correlation
data: PAS_wLength$Genelength and PAS_wLength$nPAS
t = 22.118, df = 15041, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.1619636 0.1929179
sample estimates:
cor
0.1774846 PAS_wLength$nPAS=as.factor(PAS_wLength$nPAS)
ggplot(PAS_wLength, aes(x=nPAS,y=log10(Genelength), fill=nPAS)) + geom_boxplot() + labs(x="Number of PAS", y="log10(Length of Gene)", title="Relationship between gene length and Number of PAS") 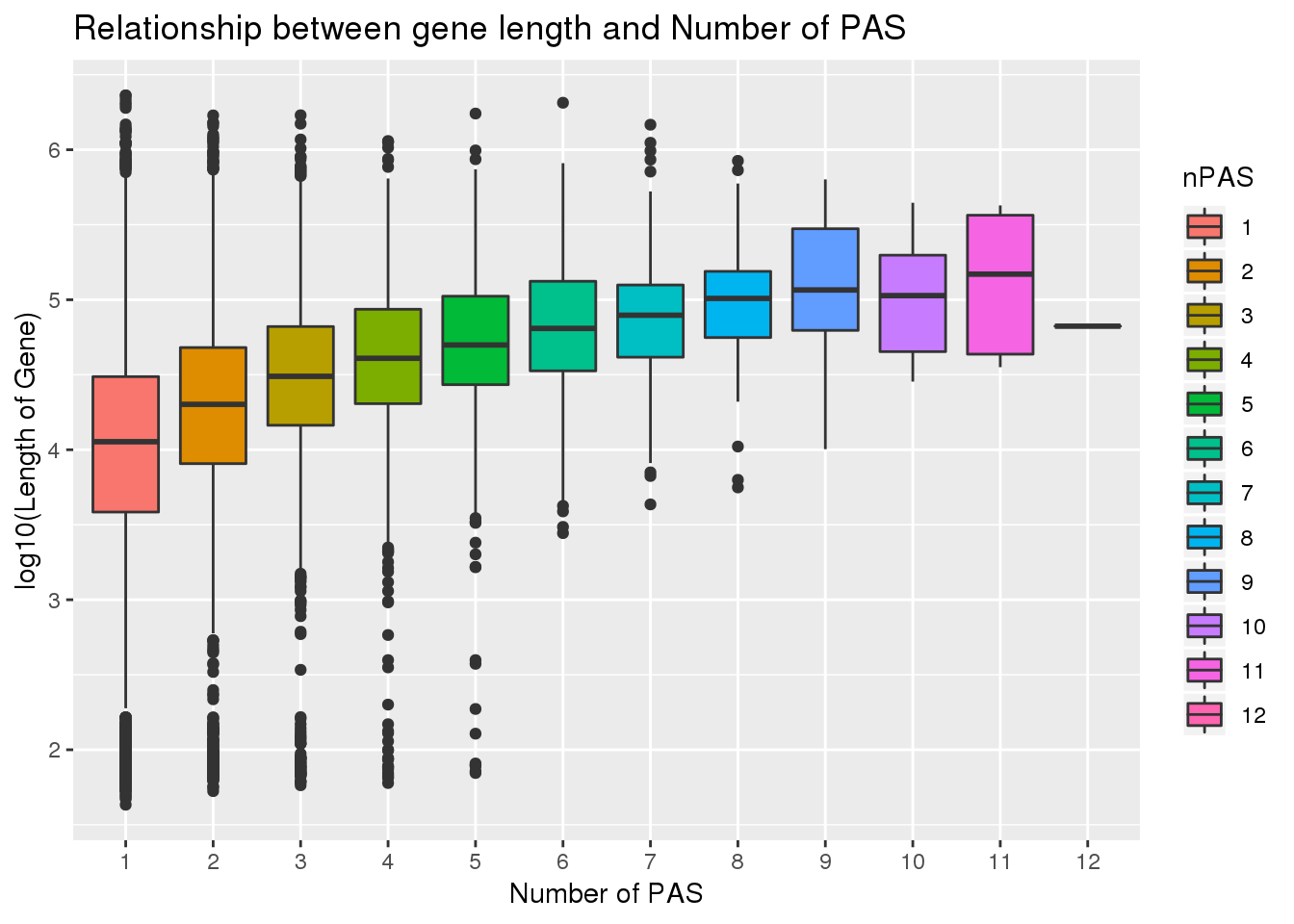
Seperate by only 1 pas vs multiple.
PAS_wLength$nPAS=as.numeric(as.character(PAS_wLength$nPAS))
PAS_wLength_apa= PAS_wLength %>% mutate(APA=ifelse(nPAS>1,"Yes","No"))
ggplot(PAS_wLength_apa,aes(x=APA, y=log10(Genelength))) + geom_boxplot() + stat_compare_means()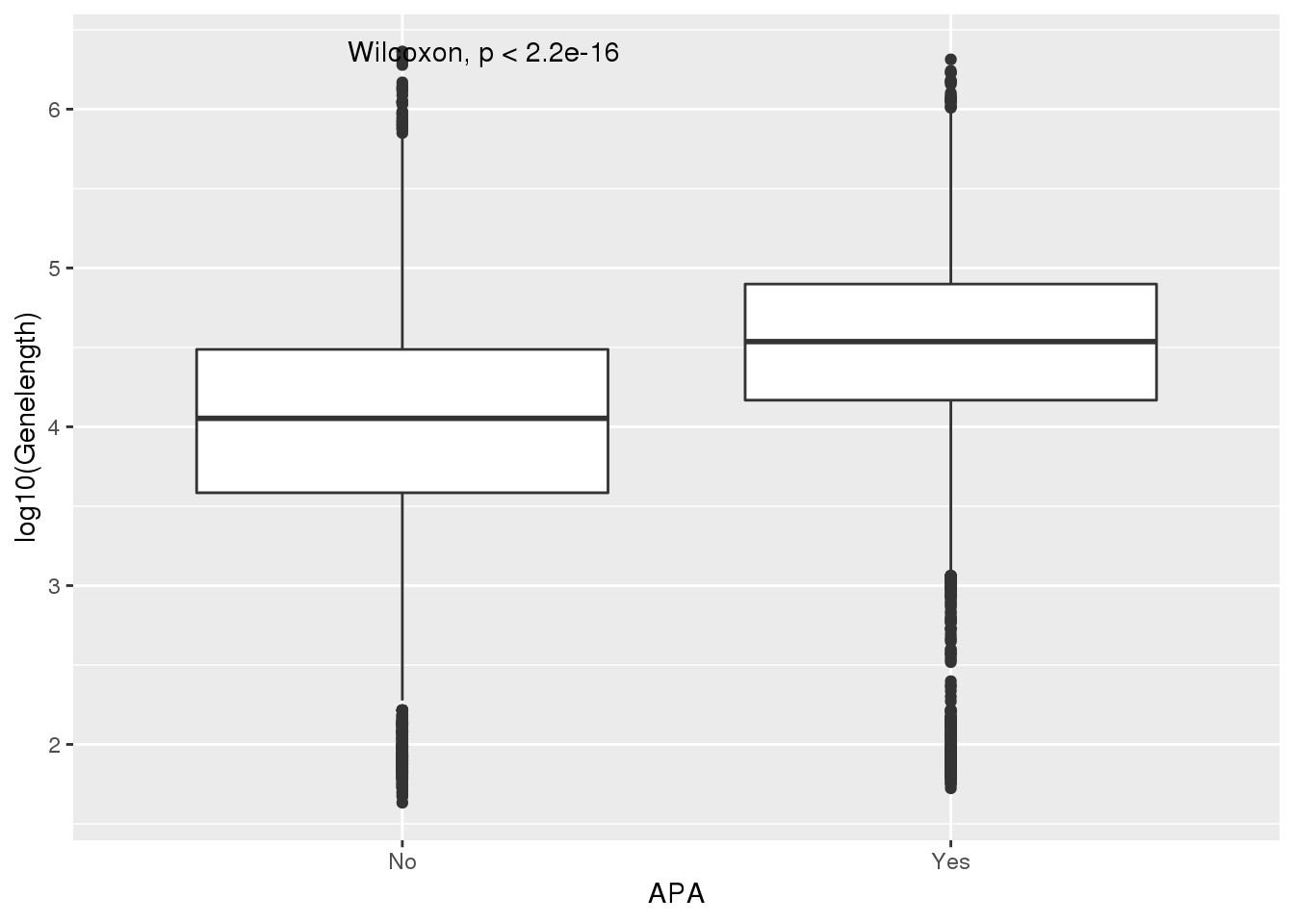 ###UTR length I also will look at length of the longest annotated 3’ UTR:
###UTR length I also will look at length of the longest annotated 3’ UTR:
UTR=read.table("../../genome_anotation_data/RefSeq_annotations/ncbiRefSeq_UTR3.sort.bed",col.names = c('chr','start','end','utr','gene', 'score','strand'),stringsAsFactors = F) %>%
mutate(UTRlength=end-start) %>%
group_by(gene) %>%
arrange(desc(UTRlength)) %>%
slice(1) %>%
select(gene, UTRlength) PAS_wUTRLength= PAS %>% inner_join(UTR, by="gene")Check for correlation
cor.test(PAS_wLength$Genelength, PAS_wLength$nPAS)
Pearson's product-moment correlation
data: PAS_wLength$Genelength and PAS_wLength$nPAS
t = 22.118, df = 15041, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.1619636 0.1929179
sample estimates:
cor
0.1774846 PAS_wUTRLength$nPAS=as.factor(PAS_wUTRLength$nPAS)
ggplot(PAS_wUTRLength, aes(x=nPAS,y=log10(UTRlength), fill=nPAS)) + geom_boxplot() + labs(x="Number of PAS", y="log10(Length of UTR)", title="Relationship between UTR length and Number of PAS") 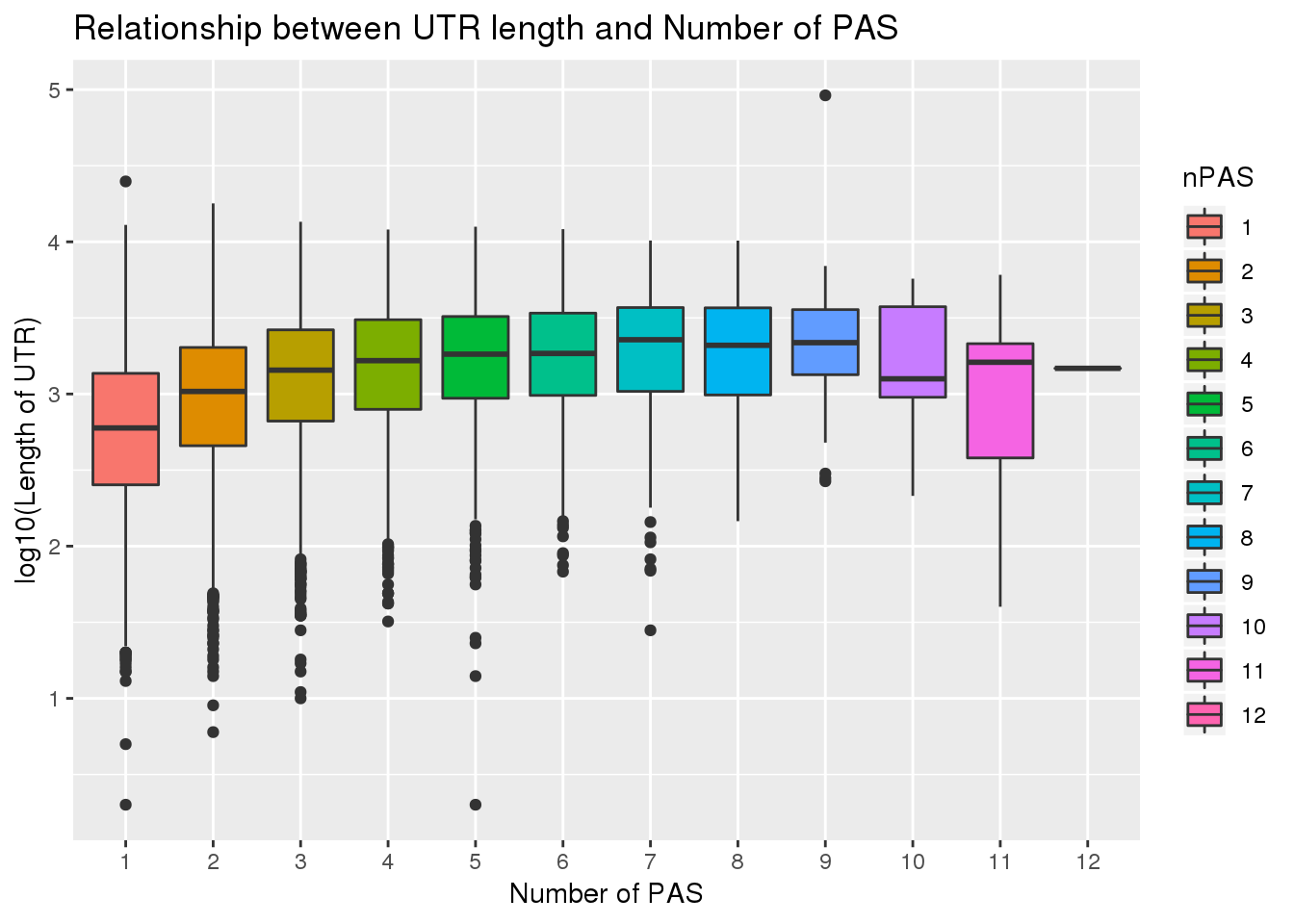
Seperate by only 1 pas vs multiple.
PAS_wUTRLength$nPAS=as.numeric(as.character(PAS_wUTRLength$nPAS))
PAS_wUTRLength_apa= PAS_wUTRLength %>% mutate(APA=ifelse(nPAS>1,"Yes","No"))
ggplot(PAS_wUTRLength_apa,aes(x=APA, y=log10(UTRlength))) + geom_boxplot() + stat_compare_means()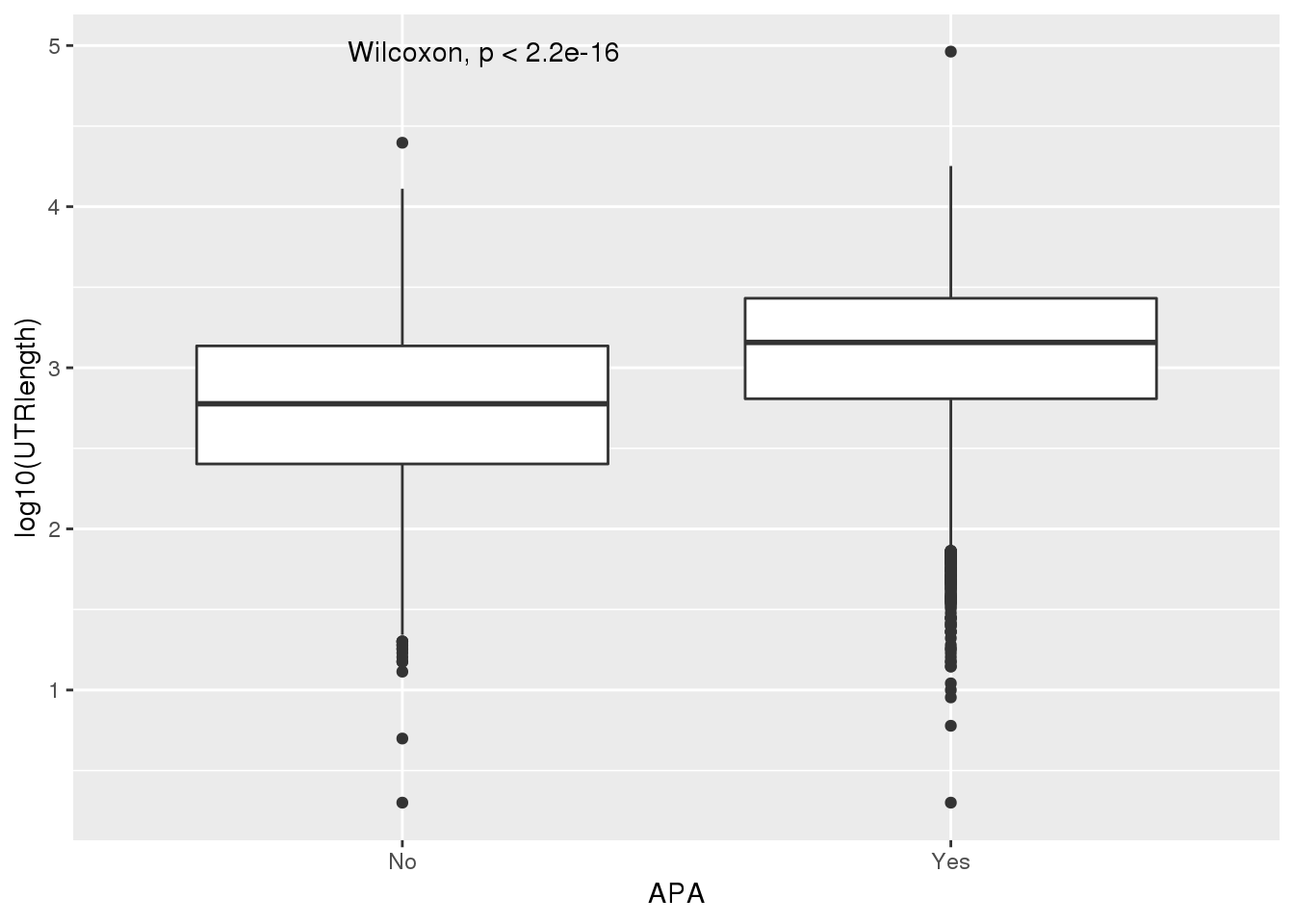
ggplot(PAS_wUTRLength_apa,aes(by=APA, fill=APA, x=log10(UTRlength))) + geom_density(alpha=.5)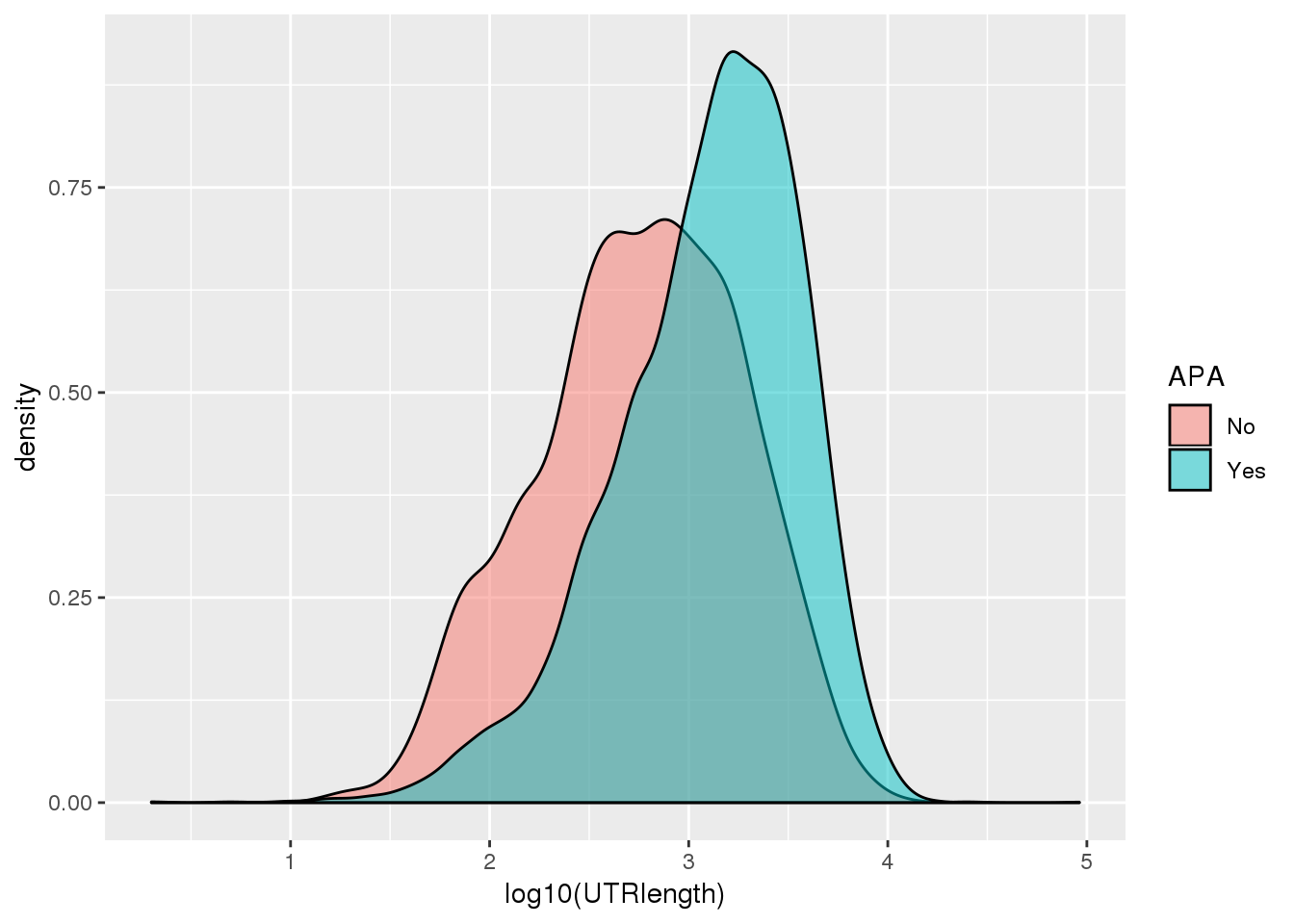
Only the UTR pas:
PAS_Utr= read.table("../data/PAS/APApeak_Peaks_GeneLocAnno.Nuclear.5perc.sort.bed",col.names = c("chr","start","end","name","score","strand")) %>%
separate(name,into=c("pas", 'gene','loc'), sep=":") %>%
filter(loc=="utr3") %>%
group_by(gene) %>%
summarise(nUTRPAS=n())UTRPAS_wUTRLength= PAS_Utr %>% inner_join(UTR, by="gene")Check for correlation
cor.test(UTRPAS_wUTRLength$UTRlength, UTRPAS_wUTRLength$nUTRPAS)
Pearson's product-moment correlation
data: UTRPAS_wUTRLength$UTRlength and UTRPAS_wUTRLength$nUTRPAS
t = 52.33, df = 12406, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.4107098 0.4395393
sample estimates:
cor
0.4252324 summary(lm(log10(UTRPAS_wUTRLength$UTRlength) ~ UTRPAS_wUTRLength$nUTRPAS))
Call:
lm(formula = log10(UTRPAS_wUTRLength$UTRlength) ~ UTRPAS_wUTRLength$nUTRPAS)
Residuals:
Min 1Q Median 3Q Max
-1.80819 -0.28373 0.03181 0.31534 1.40264
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 2.579931 0.008362 308.54 <2e-16 ***
UTRPAS_wUTRLength$nUTRPAS 0.269648 0.004808 56.09 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.4431 on 12406 degrees of freedom
Multiple R-squared: 0.2023, Adjusted R-squared: 0.2022
F-statistic: 3146 on 1 and 12406 DF, p-value: < 2.2e-16UTRPAS_wUTRLength$nUTRPAS=as.factor(UTRPAS_wUTRLength$nUTRPAS)
ggplot(UTRPAS_wUTRLength, aes(x=nUTRPAS,y=log10(UTRlength), fill=nUTRPAS)) + geom_boxplot() + labs(x="Number of 3' UTR PAS", y="log10(Length of UTR)", title="Relationship between UTR length and Number of PAS") 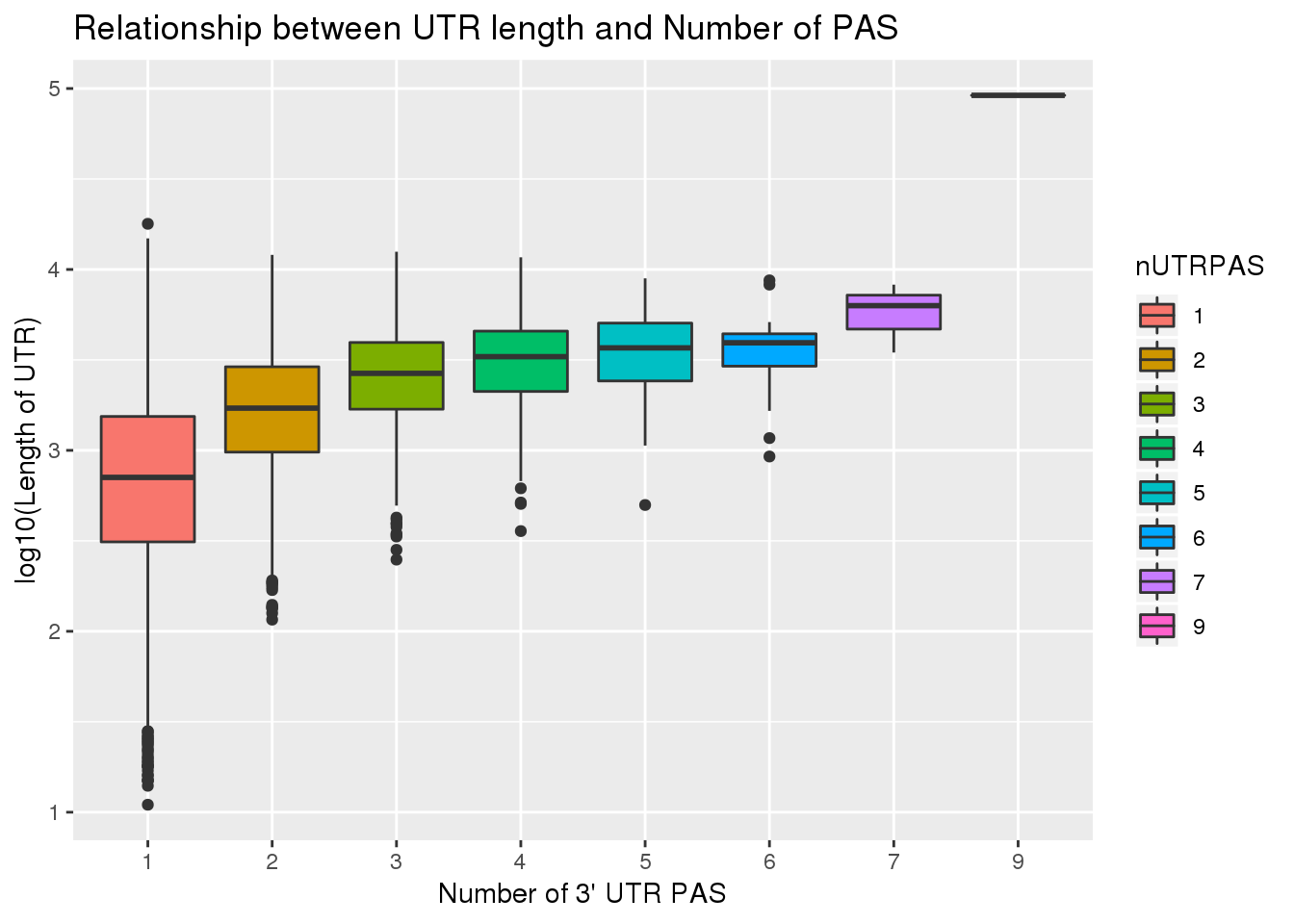
Expression level
Expression level by number of PAS
Calculate mean normalized gene expression values per gene.
geneNames=read.table("../../genome_anotation_data/ensemble_to_genename.txt", sep="\t", col.names = c('gene_id', 'gene', 'source' ),stringsAsFactors = F, header = T) %>% select(gene_id, gene)Rnames=colnames(read.table("../data/molPhenos/RNAhead.txt", header = T))
Expression=read.table("../data/molPhenos/fastqtl_qqnorm_RNAseq_phase2.fixed.noChr.txt.gz",col.names = Rnames) %>%
separate(ID,into=c("gene_id","extra"), sep="\\.") %>%
inner_join(geneNames,by = "gene_id") %>%
select(-Chr,-start,-end,-gene_id, -extra) %>%
gather("ind", "exp", -gene) %>%
group_by(gene) %>%
summarise(MeanExp=mean(exp))PAS_wExp= PAS %>% inner_join(Expression, by="gene")
cor.test(PAS_wExp$MeanExp, PAS_wExp$nPAS)
Pearson's product-moment correlation
data: PAS_wExp$MeanExp and PAS_wExp$nPAS
t = 5.7763, df = 10337, p-value = 7.859e-09
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.03748661 0.07591472
sample estimates:
cor
0.05672167 summary(lm(PAS_wExp$MeanExp ~ PAS_wExp$nPAS))
Call:
lm(formula = PAS_wExp$MeanExp ~ PAS_wExp$nPAS)
Residuals:
Min 1Q Median 3Q Max
-0.15218 -0.03221 0.00099 0.03222 0.15212
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.0032918 0.0008791 -3.744 0.000182 ***
PAS_wExp$nPAS 0.0015122 0.0002618 5.776 7.86e-09 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.04714 on 10337 degrees of freedom
Multiple R-squared: 0.003217, Adjusted R-squared: 0.003121
F-statistic: 33.37 on 1 and 10337 DF, p-value: 7.859e-09PAS_wExp$nPAS=as.factor(PAS_wExp$nPAS)
ggplot(PAS_wExp, aes(x=nPAS,y=MeanExp, fill=nPAS)) + geom_boxplot() + labs(x="Number of PAS", y="Mean normalized expression", title="Relationship between expression and Number of PAS") 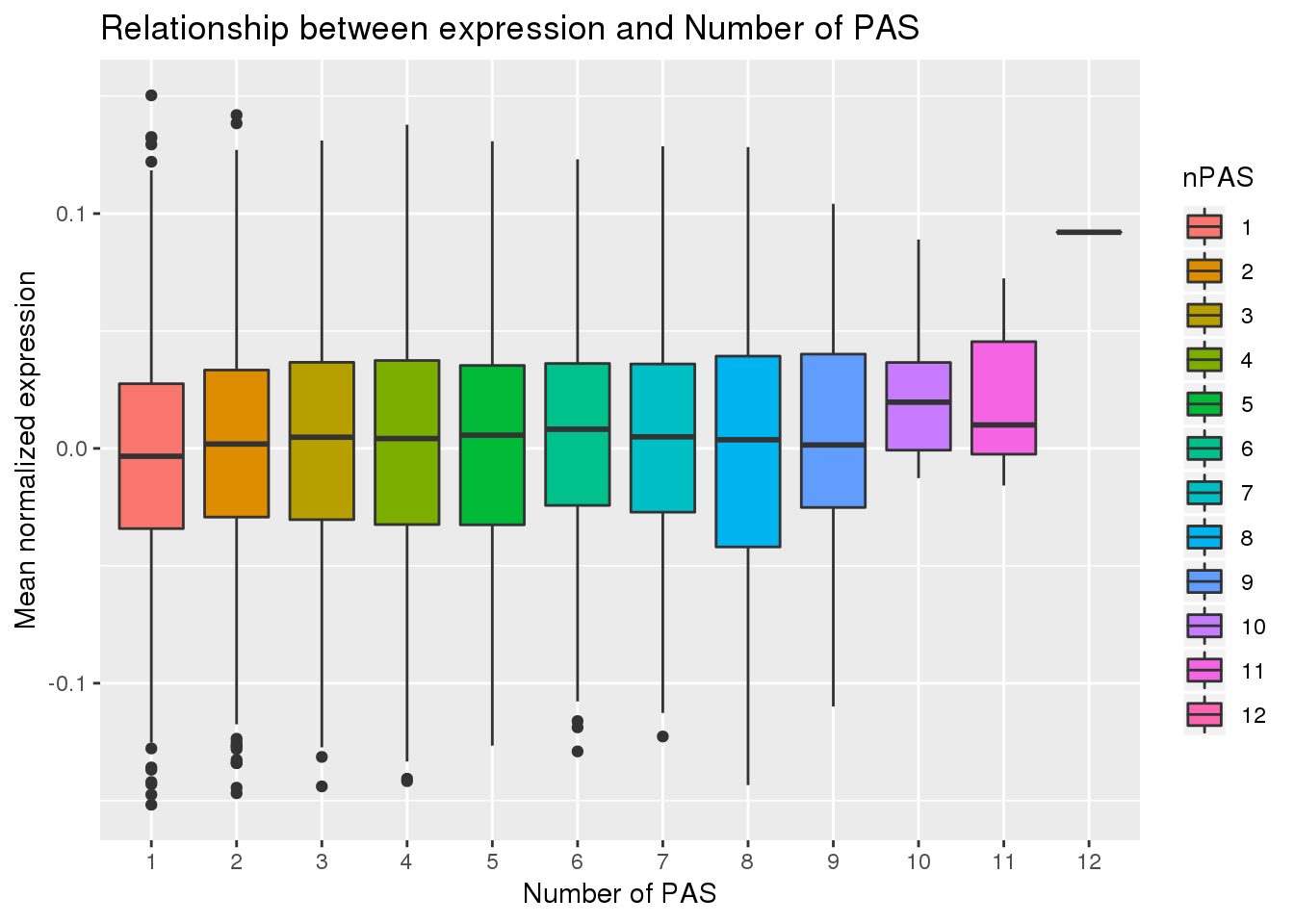
No aparent difference here. I will remove the 12 and test correlation again.
PAS_wExpFilt= PAS %>% inner_join(Expression, by="gene") %>% filter(nPAS <10)
cor.test(PAS_wExpFilt$MeanExp, PAS_wExpFilt$nPAS)
Pearson's product-moment correlation
data: PAS_wExpFilt$MeanExp and PAS_wExpFilt$nPAS
t = 5.4617, df = 10320, p-value = 4.825e-08
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.03442997 0.07290264
sample estimates:
cor
0.05368623 summary(lm(PAS_wExpFilt$MeanExp ~ PAS_wExpFilt$nPAS))
Call:
lm(formula = PAS_wExpFilt$MeanExp ~ PAS_wExpFilt$nPAS)
Residuals:
Min 1Q Median 3Q Max
-0.151852 -0.032263 0.000969 0.032277 0.152032
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.0031448 0.0008866 -3.547 0.000391 ***
PAS_wExpFilt$nPAS 0.0014524 0.0002659 5.462 4.82e-08 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.04715 on 10320 degrees of freedom
Multiple R-squared: 0.002882, Adjusted R-squared: 0.002786
F-statistic: 29.83 on 1 and 10320 DF, p-value: 4.825e-08PAS_wExpFilt$nPAS=as.numeric(as.character(PAS_wExpFilt$nPAS))
PAS_wExpFilt_apa= PAS_wExpFilt %>% mutate(APA=ifelse(nPAS>1,"Yes","No"))
ggplot(PAS_wExpFilt_apa,aes(x=APA, y=MeanExp)) + geom_boxplot() + stat_compare_means()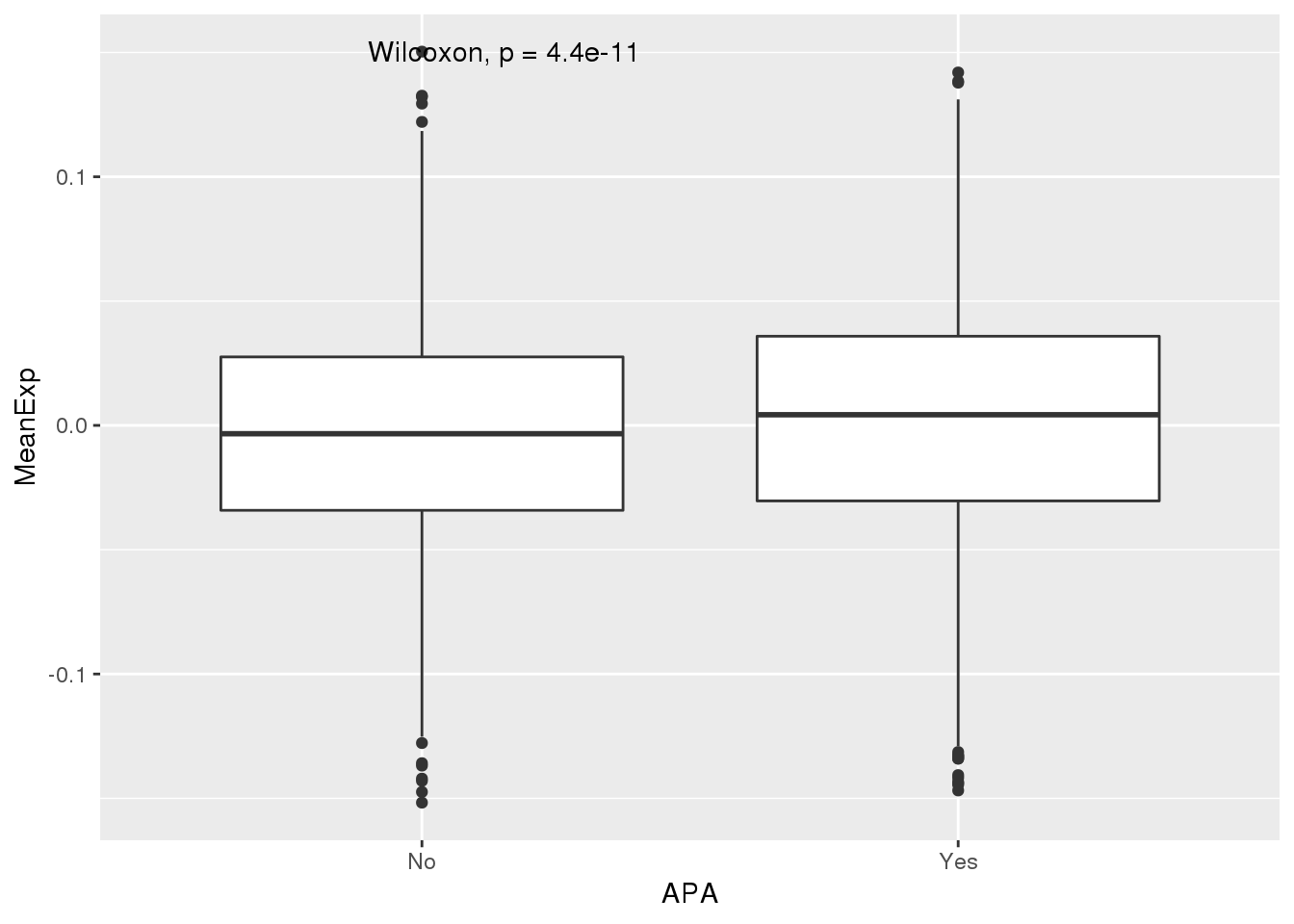
ggplot(PAS_wExpFilt_apa,aes(by=APA, fill=APA, x=MeanExp)) + geom_density(alpha=.5)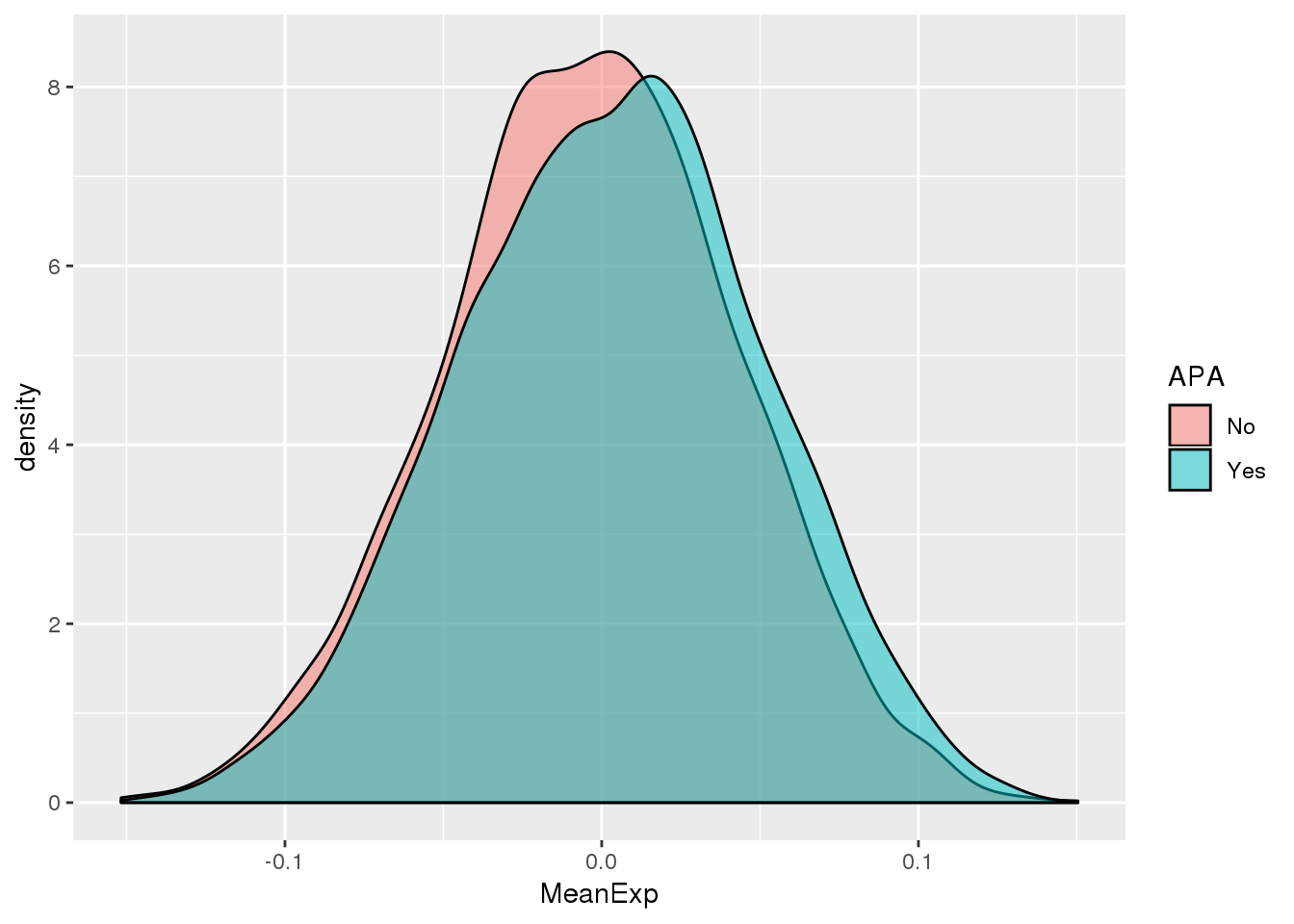
It looks like there is a significant difference here between genes with APA and those without, but visualy it doesnt look like number of PAS is driven by expression.
Go terms
I will write out seperate lists for genes with 1 PAS and those with more than one. I will use GOrilla to test for gene set inforamtion
PAS_noapa= PAS %>% filter(nPAS==1) %>% select(gene)
PAS_apa= PAS %>% filter(nPAS>1)%>%arrange(desc(nPAS)) %>% select(gene)I will use 1 PAS as backgroun and with APA as the set.
mkdir ../data/nPAS/write.table(PAS_noapa,"../data/nPAS/GenesNoAPA.txt", col.names = F, row.names = F, quote = F)
write.table(PAS_apa,"../data/nPAS/GenesAPA.txt", col.names = F, row.names = F, quote = F)Significant processes : FDR q <10^-9:
regulation of nucleobase-containing compound metabolic process
regulation of cellular macromolecule biosynthetic process
nucleic acid metabolic process
regulation of macromolecule biosynthetic process
regulation of cellular biosynthetic process regulation of nucleic acid-templated transcription
regulation of RNA biosynthetic process
regulation of transcription, DNA-templated
regulation of biosynthetic process
regulation of nitrogen compound metabolic process
regulation of primary metabolic process regulation of cellular metabolic process
RNA processing
Significant function : FDR q <10^-9:
heterocyclic compound binding
organic cyclic compound binding nucleic acid binding
DNA binding
Significant component : FDR q <10^-9:
intracellular part nucleoplasm nuclear part
intracellular organelle nucleus intracellular membrane-bounded organelle
intracellular organelle part
nucleoplasm part
organelle part
organelle
Not really sure what to do with this. I don’t have an expectation for this. These are key ceullualar processes, functions, and regions. Most genes in this analysis have APA.
Tissue specificity
Median gene-level TPM by tissue. Median expression was calculated from the file GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_tpm.gct.gz.
I will download information from gtex. I can then set a TPM cutoff and look at for each gene how many tissues it is expressed.
GTEX_test<-read.table("../data/nPAS/GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_median_tpm.gct", header = T, skip=2, sep = '\t') %>%
separate(Name,into=c("gene_id","extra"), sep="\\.") %>%
inner_join(geneNames, by="gene_id") %>%
select(-gene_id,-Description,-extra) %>%
gather("tissue", "TPM",-gene)ggplot(GTEX_test,aes(y=log10(TPM), by=tissue, fill=tissue)) + geom_boxplot()+theme(legend.position = "none") Warning: Removed 1456429 rows containing non-finite values (stat_boxplot).
Try logTPM of 2 - 100
Filter genes that come up with more than 54 due to gene name issues.
GTEX=read.table("../data/nPAS/GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_median_tpm.gct", header = T, skip=2, sep = '\t') %>%
separate(Name,into=c("gene_id","extra"), sep="\\.") %>%
inner_join(geneNames, by="gene_id") %>%
select(-gene_id,-Description,-extra) %>%
gather("tissue", "TPM",-gene) %>%
filter(TPM >=100 )%>%
group_by(gene) %>%
summarise(nTissue=n()) %>%
filter(nTissue<=54)Join this with the PAS info:
PAS_tissue=PAS %>% inner_join(GTEX,by="gene")cor.test(PAS_tissue$nPAS, PAS_tissue$nTissue)
Pearson's product-moment correlation
data: PAS_tissue$nPAS and PAS_tissue$nTissue
t = -6.8873, df = 3589, p-value = 6.685e-12
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
-0.14637420 -0.08180869
sample estimates:
cor
-0.114212 PAS_tissue$nPAS= as.factor(PAS_tissue$nPAS)
ggplot(PAS_tissue, aes(x=nPAS,y=nTissue, fill=nPAS)) + geom_boxplot() + labs(x="Number of PAS", y="Number of tissues median TPM>2", title="Relationship tissue specificity and Number of PAS") 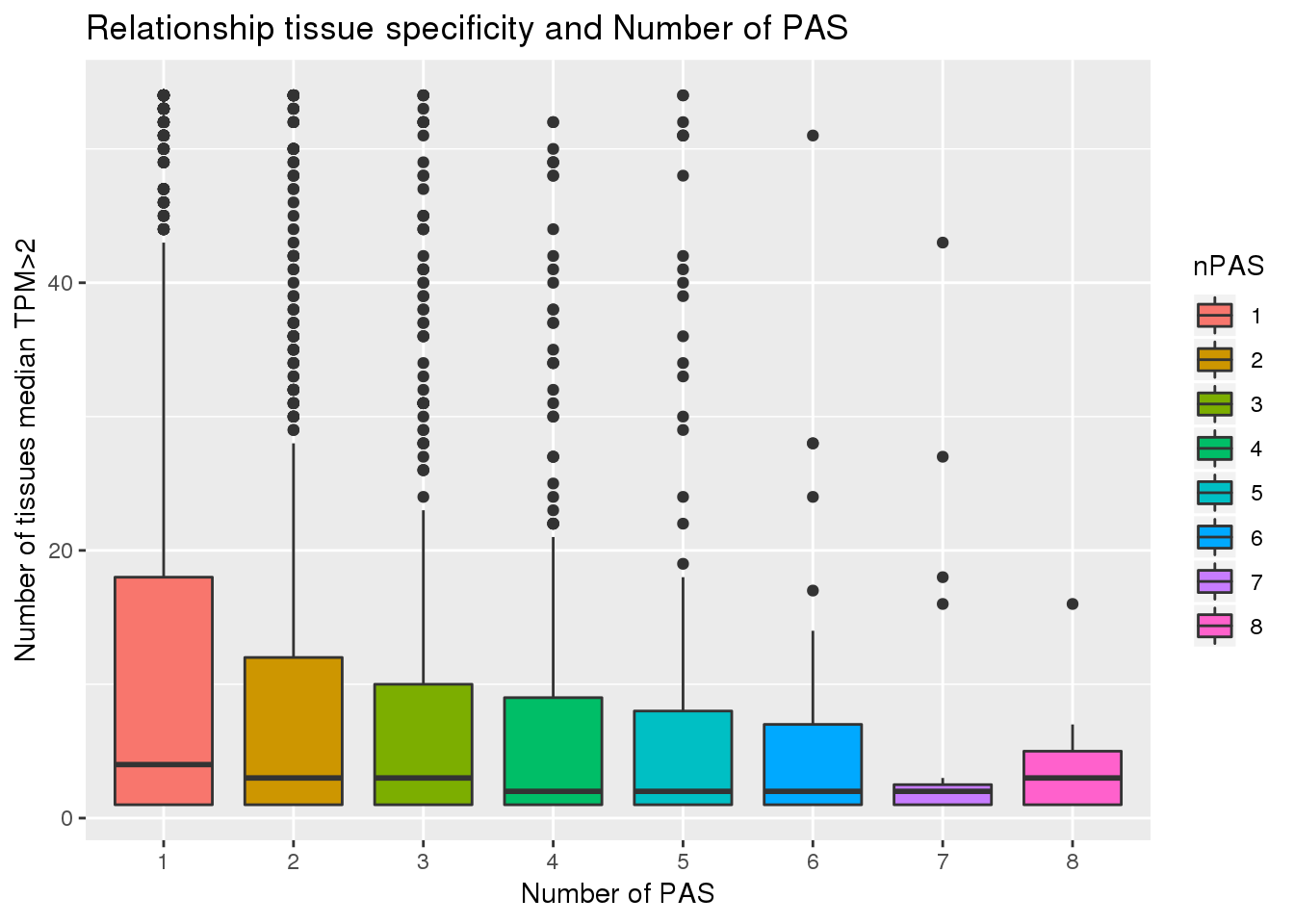 With and without APA
With and without APA
PAS_tissue$nPAS=as.numeric(as.character(PAS_tissue$nPAS))
PAS_tissue_apa= PAS_tissue %>% mutate(APA=ifelse(nPAS>1,"Yes","No"))
ggplot(PAS_tissue_apa,aes(by=APA, x=nTissue,fill=APA)) + geom_density(alpha=.4)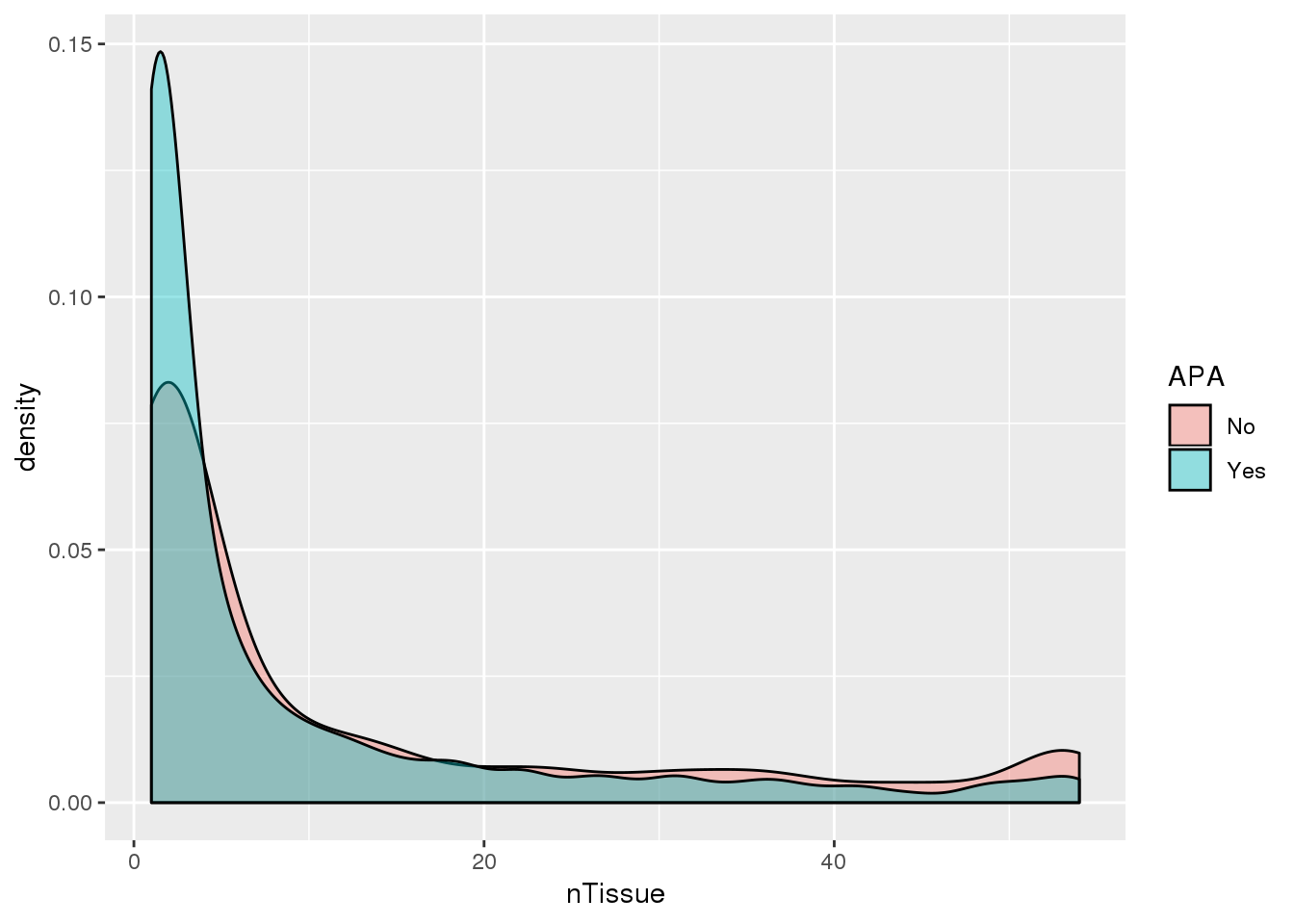
Looks like genes with apa are a bit more specific.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.3.0 stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2
[5] readr_1.3.1 tidyr_0.8.3 tibble_2.1.1 tidyverse_1.2.1
[9] ggpubr_0.2 magrittr_1.5 ggplot2_3.1.1 workflowr_1.5.0
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38 colorspace_1.3-2
[5] generics_0.0.2 htmltools_0.3.6 yaml_2.2.0 rlang_0.4.0
[9] later_0.7.5 pillar_1.3.1 glue_1.3.0 withr_2.1.2
[13] modelr_0.1.2 readxl_1.1.0 plyr_1.8.4 munsell_0.5.0
[17] gtable_0.2.0 cellranger_1.1.0 rvest_0.3.2 evaluate_0.12
[21] labeling_0.3 knitr_1.20 httpuv_1.4.5 broom_0.5.1
[25] Rcpp_1.0.2 promises_1.0.1 scales_1.0.0 backports_1.1.2
[29] jsonlite_1.6 fs_1.3.1 hms_0.4.2 digest_0.6.18
[33] stringi_1.2.4 grid_3.5.1 rprojroot_1.3-2 cli_1.1.0
[37] tools_3.5.1 lazyeval_0.2.1 crayon_1.3.4 whisker_0.3-2
[41] pkgconfig_2.0.2 xml2_1.2.0 lubridate_1.7.4 assertthat_0.2.0
[45] rmarkdown_1.10 httr_1.3.1 rstudioapi_0.10 R6_2.3.0
[49] nlme_3.1-137 git2r_0.26.1 compiler_3.5.1