apaQTL and PTTqtls
Briana Mittleman
7/8/2019
Last updated: 2019-09-06
Checks: 6 1
Knit directory: apaQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.4.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
The global environment had objects present when the code in the R Markdown file was run. These objects can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment. Use wflow_publish or wflow_build to ensure that the code is always run in an empty environment.
The following objects were defined in the global environment when these results were created:
| Name | Class | Size |
|---|---|---|
| data | environment | 56 bytes |
| env | environment | 56 bytes |
The command set.seed(20190411) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: docs/.DS_Store
Ignored: docs/figure/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: .Rprofile
Untracked: ._.DS_Store
Untracked: .gitignore
Untracked: @
Untracked: _workflowr.yml
Untracked: analysis/._PASdescriptiveplots.Rmd
Untracked: analysis/._cuttoffPercUsage.Rmd
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Nuclear.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Total.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/QTLexampleplots.Rmd
Untracked: analysis/cuttoffPercUsage.Rmd
Untracked: analysis/eQTLoverlap.Rmd
Untracked: analysis/interpret verify bam.Rmd
Untracked: analysis/interpret_verifybam.Rmd
Untracked: analysis/mergeRNA.Rmd
Untracked: analysis/oldstuffNotNeeded.Rmd
Untracked: analysis/remove_badlines.Rmd
Untracked: analysis/totalspec.Rmd
Untracked: apaQTL.Rproj
Untracked: code/.NascentRNAdtPlotFirstintronicPAS.sh.swp
Untracked: code/._ApaQTL_nominalNonnorm.sh
Untracked: code/._BothFracDTPlotGeneRegions.sh
Untracked: code/._BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/._EandPqtl_perm.sh
Untracked: code/._EandPqtls.sh
Untracked: code/._FC_NucintornUpandDown.sh
Untracked: code/._FC_UTR.sh
Untracked: code/._FC_intornUpandDownsteamPAS.sh
Untracked: code/._FC_nascentseq.sh
Untracked: code/._FC_newPeaks_olddata.sh
Untracked: code/._HMMpermuteTotal.py
Untracked: code/._HmmPermute.py
Untracked: code/._IntronicPASDT.sh
Untracked: code/._LC_samplegroups.py
Untracked: code/._LD_qtl.sh
Untracked: code/._LD_snpsproxy.sh
Untracked: code/._NascentRNAdtPlot.sh
Untracked: code/._NascentRNAdtPlot3UTRPAS.sh
Untracked: code/._NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/._NascentRNAdtPlotNucPAS.sh
Untracked: code/._NascentRNAdtPlotTotPAS.sh
Untracked: code/._NascentRNAdtPlotintronicPAS.sh
Untracked: code/._NascnetRNAdtPlotPAS.sh
Untracked: code/._NetSeq_fourthintronDT.sh
Untracked: code/._NomResfromPASSNP.py
Untracked: code/._NuclearPAS_5per.bed.py
Untracked: code/._PTTfacetboxplots.R
Untracked: code/._PrematureQTLNominal.sh
Untracked: code/._PrematureQTLPermuted.sh
Untracked: code/._QTL2bed.py
Untracked: code/._QTL2bed_withstrand.py
Untracked: code/._RNAbam2bw.sh
Untracked: code/._RNAseqDTplot.sh
Untracked: code/._RunRes2PAS.sh
Untracked: code/._SAF215upbed.py
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilefiltPAS
Untracked: code/._TESplots100bp.sh
Untracked: code/._TESplots150bp.sh
Untracked: code/._TESplots200bp.sh
Untracked: code/._TotalPAS_5perc.bed.py
Untracked: code/._Untitled
Untracked: code/._ZipandTabPheno.sh
Untracked: code/._aAPAqtl_nominal39ind.sh
Untracked: code/._annotatePacBioPASregion.sh
Untracked: code/._annotatedPAS2bed.py
Untracked: code/._apaInPandE.py
Untracked: code/._apaQTLCorrectPvalMakeQQ.R
Untracked: code/._apaQTLCorrectpval_6or7a.R
Untracked: code/._apaQTL_Nominal.sh
Untracked: code/._apaQTL_nominalv67.sh
Untracked: code/._apaQTL_permuted.sh
Untracked: code/._apaQTL_permuted_test6A7A.sh
Untracked: code/._apainRibo.py
Untracked: code/._assignNucIntonpeak2intronlocs.sh
Untracked: code/._assignTotIntronpeak2intronlocs.sh
Untracked: code/._bam2BW_5primemost.sh
Untracked: code/._bed2saf.py
Untracked: code/._bothFracDTplot1stintron.sh
Untracked: code/._bothFracDTplot4thintron.sh
Untracked: code/._bothFrac_FC.sh
Untracked: code/._callPeaksYL.py
Untracked: code/._changeRibonomQTLres2genename.py
Untracked: code/._changenomQTLres2geneName.py
Untracked: code/._chooseAnno2PAS_pacbio.py
Untracked: code/._chooseAnno2SAF.py
Untracked: code/._chooseSignalSite
Untracked: code/._chooseSignalSite.py
Untracked: code/._closestannotated.sh
Untracked: code/._closestannotated_byfrac.sh
Untracked: code/._cluster.json
Untracked: code/._clusterPAS.json
Untracked: code/._clusterfiltPAS.json
Untracked: code/._codingdms2bed.py
Untracked: code/._config.yaml
Untracked: code/._config2.yaml
Untracked: code/._configOLD.yaml
Untracked: code/._convertNominal2SNPLOC.py
Untracked: code/._convertNominal2SNPloc2Versions.py
Untracked: code/._convertNumeric.py
Untracked: code/._correctNomeqtl.R
Untracked: code/._createPlinkSampfile.py
Untracked: code/._dag.pdf
Untracked: code/._eQTL_switch2snploc.py
Untracked: code/._eQTLgenestestedapa.py
Untracked: code/._encodeRNADTplots.sh
Untracked: code/._extractGenotypes.py
Untracked: code/._extractseqfromqtlfastq.py
Untracked: code/._fc2leafphen.py
Untracked: code/._fc_filteredPAS6and7As.sh
Untracked: code/._fifteenBPupstreamPAS.py
Untracked: code/._fiftyBPupstreamPAS.py
Untracked: code/._filter5perc.R
Untracked: code/._filter5percPheno.py
Untracked: code/._filterLDsnps.py
Untracked: code/._filterMPPAS.py
Untracked: code/._filterMPPAS_15.py
Untracked: code/._filterMPPAS_15_7As.py
Untracked: code/._filterMPPAS_50.py
Untracked: code/._filterSAFforMP.py
Untracked: code/._filterpeaks.py
Untracked: code/._finalPASbed2SAF.py
Untracked: code/._fix4su304corr.py
Untracked: code/._fix4su604corr.py
Untracked: code/._fix4sukalisto.py
Untracked: code/._fixExandUnexeQTL
Untracked: code/._fixExandUnexeQTL.py
Untracked: code/._fixFChead.py
Untracked: code/._fixFChead_bothfrac.py
Untracked: code/._fixFChead_short.py
Untracked: code/._fixH3k12ac.py
Untracked: code/._fixPASregionSNPs.py
Untracked: code/._fixRNAhead4corr.py
Untracked: code/._fixRNAkalisto.py
Untracked: code/._fixgroupedtranscript.py
Untracked: code/._fixhead_netseqfc.py
Untracked: code/._getAPAfromanyeQTL.py
Untracked: code/._getApapval4eqtl.py
Untracked: code/._getApapval4eqtl_unexp.py
Untracked: code/._getApapval4eqtl_version67.py
Untracked: code/._getDownstreamIntronNuclear.py
Untracked: code/._getIntronDownstreamPAS.py
Untracked: code/._getIntronUpstreamPAS.py
Untracked: code/._getQTLalleles.py
Untracked: code/._getQTLfastq.sh
Untracked: code/._getUpstreamIntronNuclear.py
Untracked: code/._grouptranscripts.py
Untracked: code/._intersectVCFandupPAS.sh
Untracked: code/._keep5perMAF.py
Untracked: code/._keepSNP_vcf.sh
Untracked: code/._make5percPeakbed.py
Untracked: code/._makeFileID.py
Untracked: code/._makePheno.py
Untracked: code/._makeSAFbothfrac5perc.py
Untracked: code/._makeSNP2rsidfile.py
Untracked: code/._makeeQTLempirical_unexp.py
Untracked: code/._makeeQTLempiricaldist.py
Untracked: code/._makegencondeTSSfile.py
Untracked: code/._mapSSsnps2PAS.sh
Untracked: code/._mergRNABam.sh
Untracked: code/._mergeAllBam.sh
Untracked: code/._mergeBW_norm.sh
Untracked: code/._mergeBamNascent.sh
Untracked: code/._mergeByFracBam.sh
Untracked: code/._mergePeaks.sh
Untracked: code/._mnase1stintron.sh
Untracked: code/._mnaseDT_fourthintron.sh
Untracked: code/._namePeaks.py
Untracked: code/._netseqDTplot1stIntron.sh
Untracked: code/._netseqFC.sh
Untracked: code/._nucQTLGWAS.py
Untracked: code/._nucSpecQTLineData.py
Untracked: code/._nucSpeceffectsize.py
Untracked: code/._pQTLsotherdata.py
Untracked: code/._pacbioDT.sh
Untracked: code/._pacbioIntronicDT.sh
Untracked: code/._parseBestbamid.py
Untracked: code/._peak2PAS.py
Untracked: code/._peakFC.sh
Untracked: code/._pheno2countonly.R
Untracked: code/._phenoQTLfromlist.py
Untracked: code/._processYRIgen.py
Untracked: code/._pttQTLsinapaQTL.py
Untracked: code/._qtlRegionseq.sh
Untracked: code/._qtlsPvalOppFrac.py
Untracked: code/._quantassign2parsedpeak.py
Untracked: code/._removeXfromHmm.py
Untracked: code/._removeloc_pheno.py
Untracked: code/._riboQTL.sh
Untracked: code/._runCorrectNomEqtl.sh
Untracked: code/._runHMMpermuteAPAqtls.sh
Untracked: code/._runHMMpermuteeQTLS.sh
Untracked: code/._runMakeEmpiricaleQTL_unexp.sh
Untracked: code/._runMakeeQTLempirical.sh
Untracked: code/._run_bam2bw_all3prime.sh
Untracked: code/._run_bam2bw_extra3.sh
Untracked: code/._run_bestbamid.sj
Untracked: code/._run_filtersnpLD.sh
Untracked: code/._run_getAPAfromeQTL_version6.7.sh
Untracked: code/._run_getApaPval4eqtl.sh
Untracked: code/._run_getapafromeQTL.py
Untracked: code/._run_getapafromeQTL.sh
Untracked: code/._run_getapapval4eqtl_unexp.sh
Untracked: code/._run_leafcutterDiffIso.sh
Untracked: code/._run_prxySNP.sh
Untracked: code/._run_pttfacetboxplot.sh
Untracked: code/._run_sepUsagephen.sh
Untracked: code/._run_sepgenobychrom.sh
Untracked: code/._run_verifybam.sh
Untracked: code/._selectNominalPvalues.py
Untracked: code/._sepUsagePhen.py
Untracked: code/._sepgenobychrom.py
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._sortindexRNAbam.sh
Untracked: code/._submit-snakemakePAS.sh
Untracked: code/._submit-snakemakefiltPAS.sh
Untracked: code/._subsetAPAnotEorPgene.py
Untracked: code/._subsetAPAnotEorPgene_2versions.py
Untracked: code/._subsetApanoteGene.py
Untracked: code/._subsetApanoteGene_2versions.py
Untracked: code/._subsetUnexplainedeQTLs.py
Untracked: code/._subsetVCF_SS.sh
Untracked: code/._subsetVCF_noSSregions.sh
Untracked: code/._subsetVCF_upstreamPAS.sh
Untracked: code/._subset_diffisopheno.py
Untracked: code/._subsetpermAPAwithGenelist.py
Untracked: code/._subsetpermAPAwithGenelist_2versions.py
Untracked: code/._subsetvcf_otherreg.sh
Untracked: code/._subsetvcf_permSS.sh
Untracked: code/._subtrachfiveprimeUTR.sh
Untracked: code/._subtractExons.sh
Untracked: code/._subtractfiveprimeUTR.sh
Untracked: code/._tabixSNPS.sh
Untracked: code/._tenBPupstreamPAS.py
Untracked: code/._testVerifyBam.sh
Untracked: code/._totSeceffectsize.py
Untracked: code/._twentyBPupstreamPAS.py
Untracked: code/._utrdms2saf.py
Untracked: code/._vcf2bed.py
Untracked: code/._verifyBam18517N.sh
Untracked: code/._verifyBam18517T.sh
Untracked: code/._verifyBam19128N.sh
Untracked: code/._verifyBam19128T.sh
Untracked: code/._wrap_verifybam.sh
Untracked: code/._writePTTexamplecode.py
Untracked: code/._writePTTexamplecode.sh
Untracked: code/.pversion
Untracked: code/.snakemake/
Untracked: code/1
Untracked: code/APAqtl_nominal.err
Untracked: code/APAqtl_nominal.out
Untracked: code/APAqtl_nominal_39.err
Untracked: code/APAqtl_nominal_39.out
Untracked: code/APAqtl_nominal_nonNorm.err
Untracked: code/APAqtl_nominal_nonNorm.out
Untracked: code/APAqtl_nominal_versions67.err
Untracked: code/APAqtl_nominal_versions67.out
Untracked: code/APAqtl_permuted.err
Untracked: code/APAqtl_permuted.out
Untracked: code/APAqtl_permuted_versions67.err
Untracked: code/APAqtl_permuted_versions67.out
Untracked: code/ApaQTL_nominalNonnorm.sh
Untracked: code/BothFracDTPlot1stintron.err
Untracked: code/BothFracDTPlot1stintron.out
Untracked: code/BothFracDTPlot4stintron.err
Untracked: code/BothFracDTPlot4stintron.out
Untracked: code/BothFracDTPlotGeneRegions.err
Untracked: code/BothFracDTPlotGeneRegions.out
Untracked: code/BothFracDTPlotGeneRegions_norm.err
Untracked: code/BothFracDTPlotGeneRegions_norm.out
Untracked: code/BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/DistPAS2Sig.py
Untracked: code/EandPqtl.err
Untracked: code/EandPqtl.out
Untracked: code/EandPqtl_perm.sh
Untracked: code/EandPqtls.sh
Untracked: code/EncodeRNADTPlotGeneRegions.err
Untracked: code/EncodeRNADTPlotGeneRegions.out
Untracked: code/FC_NucintornUpandDown.sh
Untracked: code/FC_NucintronPASupandDown.err
Untracked: code/FC_NucintronPASupandDown.out
Untracked: code/FC_UTR.err
Untracked: code/FC_UTR.out
Untracked: code/FC_UTR.sh
Untracked: code/FC_intornUpandDownsteamPAS.sh
Untracked: code/FC_intronPASupandDown.err
Untracked: code/FC_intronPASupandDown.out
Untracked: code/FC_nascent.err
Untracked: code/FC_nascentout
Untracked: code/FC_nascentseq.sh
Untracked: code/FC_newPAS_olddata.err
Untracked: code/FC_newPAS_olddata.out
Untracked: code/FC_newPeaks_olddata.sh
Untracked: code/HMMpermuteTotal.py
Untracked: code/HmmPermute.p
Untracked: code/HmmPermute.py
Untracked: code/IntronicPASDT.err
Untracked: code/IntronicPASDT.out
Untracked: code/IntronicPASDT.sh
Untracked: code/LC_samplegroups.py
Untracked: code/LD_qtl.sh
Untracked: code/LD_snpsproxy.sh
Untracked: code/LD_vcftools.hap.out
Untracked: code/NascentDTPlotGeneRegions.err
Untracked: code/NascentDTPlotGeneRegions.out
Untracked: code/NascentDTPlotPAS.err
Untracked: code/NascentDTPlotPAS.out
Untracked: code/NascentDTPlotPAS_3utr.err
Untracked: code/NascentDTPlotPAS_3utr.out
Untracked: code/NascentDTPlotPAS_firstintron.err
Untracked: code/NascentDTPlotPAS_firstintron.out
Untracked: code/NascentDTPlotPAS_intron.err
Untracked: code/NascentDTPlotPAS_intron.out
Untracked: code/NascentDTPlotPAS_nuc.err
Untracked: code/NascentDTPlotPAS_nuc.out
Untracked: code/NascentDTPlotPAS_tot.err
Untracked: code/NascentDTPlotPAS_tot.out
Untracked: code/NascentRNAdtPlot.sh
Untracked: code/NascentRNAdtPlot3UTRPAS.sh
Untracked: code/NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/NascentRNAdtPlotFirstintronicPAS.sh
Untracked: code/NascentRNAdtPlotNucPAS.sh
Untracked: code/NascentRNAdtPlotTotPAS.sh
Untracked: code/NascentRNAdtPlotintronicPAS.sh
Untracked: code/NascnetRNAdtPlotPAS.sh
Untracked: code/NetSeq_fourthintronDT.sh
Untracked: code/NomResfromPASSNP.py
Untracked: code/NuclearPAS_5per.bed.py
Untracked: code/Nuclear_example.err
Untracked: code/Nuclear_example.out
Untracked: code/PACbioDT.err
Untracked: code/PACbioDT.out
Untracked: code/PACbioDTitronic.err
Untracked: code/PACbioDTitronic.out
Untracked: code/PTTfacetboxplots.R
Untracked: code/PrematureQTLNominal.sh
Untracked: code/PrematureQTLPermuted.sh
Untracked: code/Prematureqtl_nominal.err
Untracked: code/Prematureqtl_nominal.out
Untracked: code/Prematureqtl_permuted.err
Untracked: code/Prematureqtl_permuted.out
Untracked: code/QTL2bed.py
Untracked: code/QTL2bed_withstrand.py
Untracked: code/README.md
Untracked: code/RNABam2BW.err
Untracked: code/RNABam2BW.out
Untracked: code/RNAbam2bw.sh
Untracked: code/RNAseqDTPlotGeneRegions.err
Untracked: code/RNAseqDTPlotGeneRegions.out
Untracked: code/RNAseqDTplot.sh
Untracked: code/Rplots.pdf
Untracked: code/RunRes2PAS.sh
Untracked: code/SAF215upbed.py
Untracked: code/SAF215upbed_gen.py
Untracked: code/Script4NuclearPTTqtlexamples.sh
Untracked: code/Script4NuclearQTLexamples.sh
Untracked: code/Script4TotalPTTqtlexamples.sh
Untracked: code/Script4TotalQTLexamples.sh
Untracked: code/TESplots100bp.err
Untracked: code/TESplots100bp.out
Untracked: code/TESplots100bp.sh
Untracked: code/TESplots150bp.err
Untracked: code/TESplots150bp.out
Untracked: code/TESplots150bp.sh
Untracked: code/TESplots200bp.err
Untracked: code/TESplots200bp.out
Untracked: code/TESplots200bp.sh
Untracked: code/TotalPAS_5perc.bed.py
Untracked: code/Total_example.err
Untracked: code/Total_example.out
Untracked: code/Untitled
Untracked: code/Upstream100Bases_general.py
Untracked: code/YRI_LCL.vcf.gz
Untracked: code/YRI_LCL_chr1.vcf.gz.log
Untracked: code/YRI_LCL_chr1.vcf.gz.recode.vcf
Untracked: code/ZipandTabPheno.sh
Untracked: code/aAPAqtl_nominal39ind.sh
Untracked: code/annotatePacBioPASregion.sh
Untracked: code/annotatedPAS2bed.py
Untracked: code/annotatedPASregion.err
Untracked: code/annotatedPASregion.out
Untracked: code/apaInPandE.py
Untracked: code/apaQTLCorrectPvalMakeQQ_4pc.R
Untracked: code/apaQTLCorrectpval_6or7a.R
Untracked: code/apaQTL_Nominal_4pc.sh
Untracked: code/apaQTL_nominalv67.sh
Untracked: code/apaQTL_permuted.4pc.sh
Untracked: code/apaQTL_permuted_test6A7A.sh
Untracked: code/apafacetboxplots.R
Untracked: code/apainRibo.py
Untracked: code/apaqtlfacetboxplots.R
Untracked: code/assignNucIntonpeak2intronlocs.sh
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignTotIntronpeak2intronlocs.sh
Untracked: code/assigntotPeak2Intronicregion.err
Untracked: code/assigntotPeak2Intronicregion.out
Untracked: code/bam2BW_5primemost.sh
Untracked: code/bam2bw.err
Untracked: code/bam2bw.out
Untracked: code/bam2bw_5primemost.err
Untracked: code/bam2bw_5primemost.out
Untracked: code/binary_fileset.log
Untracked: code/bothFracDTplot1stintron.sh
Untracked: code/bothFracDTplot4thintron.sh
Untracked: code/bothFrac_FC.err
Untracked: code/bothFrac_FC.out
Untracked: code/bothFrac_FC.sh
Untracked: code/callSHscripts.txt
Untracked: code/changePermQTLres2geneName.py
Untracked: code/changeRibonomQTLres2genename.py
Untracked: code/changenomQTLres2geneName.py
Untracked: code/chooseAnno2PAS_pacbio.py
Untracked: code/closestannotated.err
Untracked: code/closestannotated.out
Untracked: code/closestannotated.sh
Untracked: code/closestannotated_byfrac.sh
Untracked: code/closestannotatedbyfrac.err
Untracked: code/closestannotatedbyfrac.out
Untracked: code/codingdms2bed.py
Untracked: code/convertNominal2SNPLOC.py
Untracked: code/convertNominal2SNPloc2Versions.py
Untracked: code/correctNomeqtl.R
Untracked: code/createPlinkSampfile.py
Untracked: code/dag.pdf
Untracked: code/dagPAS.pdf
Untracked: code/dagfiltPAS.pdf
Untracked: code/eQTL_switch2snploc.py
Untracked: code/eQTLgenestestedapa.py
Untracked: code/encodeRNADTplots.sh
Untracked: code/environmentLeaf.yaml
Untracked: code/extractGenotypes.py
Untracked: code/extractseqfromqtlfastq.py
Untracked: code/fc2leafphen.py
Untracked: code/fc_filteredPAS6and7As.sh
Untracked: code/fifteenBPupstreamPAS.py
Untracked: code/fiftyBPupstreamPAS.py
Untracked: code/filterLDsnps.py
Untracked: code/filterMPPAS.py
Untracked: code/filterMPPAS_15.py
Untracked: code/filterMPPAS_15_7As.py
Untracked: code/filterMPPAS_50.py
Untracked: code/filterSAFforMP.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/finalPASbed2SAF.py
Untracked: code/findbuginpeaks.R
Untracked: code/fix4su304corr.py
Untracked: code/fix4su604corr.py
Untracked: code/fix4sukalisto.py
Untracked: code/fixExandUnexeQTL
Untracked: code/fixExandUnexeQTL.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFChead_short.py
Untracked: code/fixFChead_summary.py
Untracked: code/fixH3k12ac.py
Untracked: code/fixPASregionSNPs.py
Untracked: code/fixRNAhead4corr.py
Untracked: code/fixRNAkalisto.py
Untracked: code/fixgroupedtranscript.py
Untracked: code/fixhead_netseqfc.py
Untracked: code/genotypesYRI.gen.proc.keep.vcf.log
Untracked: code/genotypesYRI.gen.proc.keep.vcf.recode.vcf
Untracked: code/get100upPAS.py
Untracked: code/getAPAfromanyeQTL.py
Untracked: code/getApapval4eqtl.py
Untracked: code/getApapval4eqtl_unexp.py
Untracked: code/getApapval4eqtl_version67.py
Untracked: code/getDownstreamIntronNuclear.py
Untracked: code/getIntronDownstreamPAS.py
Untracked: code/getIntronUpstreamPAS.py
Untracked: code/getQTLalleles.py
Untracked: code/getQTLfastq.sh
Untracked: code/getSeq100up.sh
Untracked: code/getUpstreamIntronNuclear.py
Untracked: code/getseq100up.err
Untracked: code/getseq100up.out
Untracked: code/grouptranscripts.err
Untracked: code/grouptranscripts.out
Untracked: code/grouptranscripts.py
Untracked: code/intersectPAS_ssSNPS.err
Untracked: code/intersectPAS_ssSNPS.out
Untracked: code/intersectVCFPAS.err
Untracked: code/intersectVCFPAS.out
Untracked: code/intersectVCFandupPAS.sh
Untracked: code/keep5perMAF.py
Untracked: code/keepSNP_vcf.sh
Untracked: code/log/
Untracked: code/makeSAFbothfrac5perc.py
Untracked: code/makeSNP2rsidfile.py
Untracked: code/makeeQTLempirical_unexp.py
Untracked: code/makeeQTLempiricaldist.py
Untracked: code/makegencondeTSSfile.py
Untracked: code/mapSSsnps2PAS.sh
Untracked: code/mergRNABam.sh
Untracked: code/mergeBW_norm.sh
Untracked: code/mergeBWnorm.err
Untracked: code/mergeBWnorm.out
Untracked: code/mergeBamNacent.err
Untracked: code/mergeBamNacent.out
Untracked: code/mergeBamNascent.sh
Untracked: code/mergeRNAbam.err
Untracked: code/mergeRNAbam.out
Untracked: code/mnase1stintron.sh
Untracked: code/mnaseDTPlot1stintron.err
Untracked: code/mnaseDTPlot1stintron.out
Untracked: code/mnaseDTPlot4thintron.err
Untracked: code/mnaseDTPlot4thintron.out
Untracked: code/mnaseDT_fourthintron.sh
Untracked: code/netDTPlot4thintron.out
Untracked: code/netseqDTplot1stIntron.sh
Untracked: code/netseqFC.err
Untracked: code/netseqFC.out
Untracked: code/netseqFC.sh
Untracked: code/neyDTPlot4thintron.err
Untracked: code/nucQTLGWAS.py
Untracked: code/nucQTLGWAS_withLD.py
Untracked: code/nucSpecQTLineData.py
Untracked: code/nucSpeceffectsize.py
Untracked: code/pQTLsotherdata.py
Untracked: code/pacbioDT.sh
Untracked: code/pacbioIntronicDT.sh
Untracked: code/parseBestbamid.py
Untracked: code/phenoQTLfromlist.py
Untracked: code/plink.log
Untracked: code/processYRIgen.py
Untracked: code/prxySNP.err
Untracked: code/prxySNP.out
Untracked: code/pttFacetBoxplots.err
Untracked: code/pttFacetBoxplots.out
Untracked: code/pttQTLsinapaQTL.py
Untracked: code/pullTwoMechData.py
Untracked: code/qtlFacetBoxplots.err
Untracked: code/qtlFacetBoxplots.out
Untracked: code/qtlRegionseq.sh
Untracked: code/qtlsPvalOppFrac.py
Untracked: code/rLD_vcftools.hap.err
Untracked: code/removeXfromHmm.py
Untracked: code/removeloc_pheno.py
Untracked: code/riboQTL.sh
Untracked: code/riboqtl.err
Untracked: code/riboqtl.out
Untracked: code/runBestBamID.err
Untracked: code/runCorrectNomEqtl.sh
Untracked: code/runCorrectNomeqtl.err
Untracked: code/runCorrectNomeqtl.out
Untracked: code/runFilterLD.err
Untracked: code/runFilterLD.out
Untracked: code/runHMMpermute.err
Untracked: code/runHMMpermute.out
Untracked: code/runHMMpermuteAPAqtls.sh
Untracked: code/runHMMpermuteeQTLS.sh
Untracked: code/runHMMpermuteeQTLs.err
Untracked: code/runHMMpermuteeQTLs.out
Untracked: code/runMakeEmpiricaleQTL_unexp.sh
Untracked: code/runMakeEmpiricaleQTLs.err
Untracked: code/runMakeEmpiricaleQTLs.out
Untracked: code/runMakeEmpiricaleQTLsunex.err
Untracked: code/runMakeEmpiricaleQTLsunex.out
Untracked: code/runMakeeQTLempirical.sh
Untracked: code/run_DistPAS2Sig.err
Untracked: code/run_DistPAS2Sig.out
Untracked: code/run_bam2bw.err
Untracked: code/run_bam2bw.out
Untracked: code/run_bam2bw_all3prime.sh
Untracked: code/run_bam2bw_extra3.sh
Untracked: code/run_bam2bwexta.err
Untracked: code/run_bam2bwexta.out
Untracked: code/run_bestbamid.sh
Untracked: code/run_distPAS2Sig.sh
Untracked: code/run_filtersnpLD.sh
Untracked: code/run_getAPAfromanyeQTL.err
Untracked: code/run_getAPAfromanyeQTL.out
Untracked: code/run_getAPAfromeQTL_version6.7.sh
Untracked: code/run_getApaPval4eQTLs.err
Untracked: code/run_getApaPval4eQTLs.out
Untracked: code/run_getApaPval4eQTLsunexplained.err
Untracked: code/run_getApaPval4eQTLsunexplained.out
Untracked: code/run_getApaPval4eqtl.sh
Untracked: code/run_getapafromeQTL.sh
Untracked: code/run_getapapval4eqtl_unexp.sh
Untracked: code/run_leafcutterDiffIso.sh
Untracked: code/run_leafcutter_ds.err
Untracked: code/run_leafcutter_ds.out
Untracked: code/run_prxySNP.sh
Untracked: code/run_pttfacetboxplot.sh
Untracked: code/run_qtlFacetBoxplots.sh
Untracked: code/run_sepUsagephen.sh
Untracked: code/run_sepgenobychrom.err
Untracked: code/run_sepgenobychrom.out
Untracked: code/run_sepgenobychrom.sh
Untracked: code/run_sepusage.err
Untracked: code/run_sepusage.out
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/run_verifybam.sh
Untracked: code/run_verifybam128N.err
Untracked: code/run_verifybam128N.out
Untracked: code/run_verifybam128T.err
Untracked: code/run_verifybam128T.out
Untracked: code/run_verifybam517N.err
Untracked: code/run_verifybam517N.out
Untracked: code/run_verifybam517T.err
Untracked: code/run_verifybam517T.out
Untracked: code/run_verifybam_fullVCF.sh
Untracked: code/runprxySNP.err
Untracked: code/runprxySNP.out
Untracked: code/runres2pas.err
Untracked: code/runres2pas.out
Untracked: code/selectNominalPvalues.py
Untracked: code/sepUsagePhen.py
Untracked: code/sepgenobychrom.py
Untracked: code/seqQTLfastq.err
Untracked: code/seqQTLfastq.out
Untracked: code/seqQTLregion.err
Untracked: code/seqQTLregion.out
Untracked: code/snakePASlog.out
Untracked: code/snakefiltPASlog.out
Untracked: code/sortindexRNABam.err
Untracked: code/sortindexRNABam.out
Untracked: code/sortindexRNAbam.sh
Untracked: code/subsetAPAnotEorPgene.py
Untracked: code/subsetAPAnotEorPgene_2versions.py
Untracked: code/subsetApanoteGene.py
Untracked: code/subsetApanoteGene_2versions.py
Untracked: code/subsetUnexplainedeQTLs.py
Untracked: code/subsetVCF_SS.sh
Untracked: code/subsetVCF_noSSregions.sh
Untracked: code/subsetVCF_upstreamPAS.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subsetpermAPAwithGenelist.py
Untracked: code/subsetpermAPAwithGenelist_2versions.py
Untracked: code/subsetvcf_SS.err
Untracked: code/subsetvcf_SS.out
Untracked: code/subsetvcf_noSS.err
Untracked: code/subsetvcf_noSS.out
Untracked: code/subsetvcf_otherreg.sh
Untracked: code/subsetvcf_pas.err
Untracked: code/subsetvcf_pas.out
Untracked: code/subsetvcf_perm.err
Untracked: code/subsetvcf_perm.out
Untracked: code/subsetvcf_permSS.sh
Untracked: code/subsetvcf_rand.err
Untracked: code/subsetvcf_rand.out
Untracked: code/subtract5UTR.err
Untracked: code/subtract5UTR.out
Untracked: code/subtractExons.err
Untracked: code/subtractExons.out
Untracked: code/subtractExons.sh
Untracked: code/subtractfiveprimeUTR.sh
Untracked: code/tabixSNPS.sh
Untracked: code/tabixSNPs.err
Untracked: code/tabixSNPs.out
Untracked: code/tenBPupstreamPAS.py
Untracked: code/testVerifyBam.sh
Untracked: code/test_verifybam.err
Untracked: code/test_verifybam.out
Untracked: code/totSeceffectsize.py
Untracked: code/transcriptdm2bed.py
Untracked: code/twentyBPupstreamPAS.py
Untracked: code/utrdms2saf.py
Untracked: code/vcf2bed.py
Untracked: code/vcf_keepsnps.err
Untracked: code/vcf_keepsnps.out
Untracked: code/verifyBam18517N.sh
Untracked: code/verifyBam18517T.sh
Untracked: code/verifyBam19128N.sh
Untracked: code/verifyBam19128T.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/wrap_verifybam.sh
Untracked: code/wrap_verifybam_full.sh
Untracked: code/writeExampleQTLcode.py
Untracked: code/writePTTexamplecode.py
Untracked: code/zipandtabPhen.err
Untracked: code/zipandtabPhen.out
Untracked: data/._.DS_Store
Untracked: data/._MetaDataSequencing.txt
Untracked: data/AnnotatedPAS/
Untracked: data/ApaByEgene/
Untracked: data/ApaByPgene/
Untracked: data/BadLines/
Untracked: data/Battle_pQTL/
Untracked: data/CheckSums/
Untracked: data/CompareOldandNew/
Untracked: data/DTmatrix/
Untracked: data/DiffIso/
Untracked: data/EncodeRNA/
Untracked: data/ExampleQTLPlots/
Untracked: data/FlaggedPAS/
Untracked: data/GWAS_overlap/
Untracked: data/GeuvadisRNA/
Untracked: data/HMMqtls/
Untracked: data/Li_eQTLs/
Untracked: data/NascentRNA/
Untracked: data/NucSpeceQTLeffect/
Untracked: data/PAS/
Untracked: data/PAS_postFlag/
Untracked: data/PolyA_DB/
Untracked: data/PreTerm_pheno/
Untracked: data/PrematureQTLNominal/
Untracked: data/PrematureQTLPermuted/
Untracked: data/QTLGenotypes/
Untracked: data/QTLoverlap/
Untracked: data/QTLoverlap_nonNorm/
Untracked: data/README.md
Untracked: data/RNAseq/
Untracked: data/Reads2UTR/
Untracked: data/SNPinSS/
Untracked: data/SignalSiteFiles/
Untracked: data/TF_motifdisruption/
Untracked: data/ThirtyNineIndQtl_nominal/
Untracked: data/Version15bp6As/
Untracked: data/Version15bp7As/
Untracked: data/apaQTLNominal/
Untracked: data/apaQTLNominal_4pc/
Untracked: data/apaQTLPermuted/
Untracked: data/apaQTLPermuted_4pc/
Untracked: data/apaQTLs/
Untracked: data/assignedPeaks/
Untracked: data/assignedPeaks_15Up/
Untracked: data/bam/
Untracked: data/bam_clean/
Untracked: data/bam_waspfilt/
Untracked: data/bed_10up/
Untracked: data/bed_clean/
Untracked: data/bed_clean_sort/
Untracked: data/bed_waspfilter/
Untracked: data/bedsort_waspfilter/
Untracked: data/bothFrac_FC/
Untracked: data/bw/
Untracked: data/bw_norm/
Untracked: data/eQTLs/
Untracked: data/exampleQTLs/
Untracked: data/fastq/
Untracked: data/filterPeaks/
Untracked: data/fourSU/
Untracked: data/h3k27ac/
Untracked: data/highdiffsiggenes.txt
Untracked: data/inclusivePeaks/
Untracked: data/inclusivePeaks_FC/
Untracked: data/intronRNAratio/
Untracked: data/intron_analysis/
Untracked: data/locusZoom/
Untracked: data/mergedBG/
Untracked: data/mergedBW_byfrac/
Untracked: data/mergedBW_norm/
Untracked: data/mergedBam/
Untracked: data/mergedbyFracBam/
Untracked: data/molPhenos/
Untracked: data/molQTLs/
Untracked: data/motifdistrupt/
Untracked: data/netseq/
Untracked: data/nonNorm_pheno/
Untracked: data/nuc_10up/
Untracked: data/nuc_10upclean/
Untracked: data/oldPASfiles/
Untracked: data/overlapeQTL_try2/
Untracked: data/overlapeQTLs/
Untracked: data/pQTLoverlap/
Untracked: data/pacbio/
Untracked: data/peakCoverage/
Untracked: data/peaks_5perc/
Untracked: data/phenotype/
Untracked: data/phenotype_5perc/
Untracked: data/pttQTL/
Untracked: data/pttQTLplots/
Untracked: data/sigDiffGenes.txt
Untracked: data/sort/
Untracked: data/sort_clean/
Untracked: data/sort_waspfilter/
Untracked: data/twoMech/
Untracked: data/verifyBAM/
Untracked: data/verifyBAM_full/
Untracked: docs/._.DS_Store
Untracked: docs/figure/._.DS_Store
Untracked: nohup.out
Untracked: output/._.DS_Store
Untracked: output/._meanCorrelationPhenotypes.svg
Untracked: output/dtPlots/
Untracked: output/fastqc/
Untracked: output/meanCorrelationPhenotypes.svg
Untracked: run_verifybam517N.err
Untracked: run_verifybam517N.out
Unstaged changes:
Modified: analysis/Readdistagainstfeatures.Rmd
Modified: analysis/overlapapaqtlsandeqtls.Rmd
Modified: analysis/version15bpfilter.Rmd
Modified: code/BothFracDTPlotGeneRegions.sh
Modified: code/Snakefile
Modified: code/SnakefilefiltPAS
Modified: code/apaQTLCorrectPvalMakeQQ.R
Modified: code/apaQTL_Nominal.sh
Modified: code/apaQTL_permuted.sh
Modified: code/apaQTLsnake.err
Modified: code/bam2bw.sh
Modified: code/bed2saf.py
Modified: code/cluster.json
Modified: code/clusterfiltPAS.json
Modified: code/config.yaml
Modified: code/environment.yaml
Modified: code/makePheno.py
Modified: code/mergeAllBam.sh
Modified: code/mergeByFracBam.sh
Modified: code/mergePeaks.sh
Modified: code/peakFC.sh
Modified: code/snakemake.batch
Modified: code/snakemakePAS.batch
Modified: code/snakemakefiltPAS.batch
Modified: code/submit-snakemake.sh
Modified: code/submit-snakemakePAS.sh
Modified: code/submit-snakemakefiltPAS.sh
Deleted: code/test.txt
Modified: data/MetaDataSequencing.txt
Deleted: reads_graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 00fe2b4 | brimittleman | 2019-07-26 | Build site. |
| Rmd | cee6ce0 | brimittleman | 2019-07-26 | get pvalues form <-16 tests |
| html | 33a700a | brimittleman | 2019-07-25 | Build site. |
| Rmd | f3e3e16 | brimittleman | 2019-07-25 | small edits from paper writing and add gwas analysis |
| html | 272b0b4 | brimittleman | 2019-07-08 | Build site. |
| Rmd | b1e6dd1 | brimittleman | 2019-07-08 | update ptt analysis |
I am interesting in understanding the PTTqtls a bit more. I want to ask if pttQTLs are apaQTLs. I will ask if the PTTqtl genes are also apaQTL genes.
library(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(workflowr)This is workflowr version 1.4.0
Run ?workflowr for help getting startedPTT
totptt=read.table("../data/PrematureQTLPermuted/Total_preterminationPheno.txt.gz.qqnorm_AllChrBH.txt", stringsAsFactors = F, header = T) %>% filter(-log10(bh)>1) %>% separate(pid,into = c("chr", "start", "end", "geneID"), sep=":") %>% separate(geneID, into=c("gene", "Frac"),sep="_") %>% select(gene) %>% unique()
totpttQTL=read.table("../data/PrematureQTLPermuted/Total_preterminationPheno.txt.gz.qqnorm_AllChrBH.txt", stringsAsFactors = F, header = T) %>% filter(-log10(bh)>1)
write.table(totpttQTL, file="../data/pttQTL/TotalPttQTL.txt", col.names = T, row.names = F, quote = F, sep="\t")
nucptt=read.table("../data/PrematureQTLPermuted/Nuclear_preterminationPheno.txt.gz.qqnorm_AllChrBH.txt", stringsAsFactors = F, header = T) %>% filter(-log10(bh)>1) %>% separate(pid,into = c("chr", "start", "end", "geneID"), sep=":") %>% separate(geneID, into=c("gene", "Frac"),sep="_") %>% select(gene) %>% unique()
nucpttQTL=read.table("../data/PrematureQTLPermuted/Nuclear_preterminationPheno.txt.gz.qqnorm_AllChrBH.txt", stringsAsFactors = F, header = T) %>% filter(-log10(bh)>1)
write.table(nucpttQTL, file="../data/pttQTL/NuclearPttQTL.txt", col.names = T, row.names = F, quote = F, sep="\t")APA
nucAPA=read.table("../data/apaQTLs/NuclearapaQTLGenes.txt", col.names = c("gene"),stringsAsFactors = F)
totAPA=read.table("../data/apaQTLs/TotalapaQTLGenes.txt", col.names = c("gene"),stringsAsFactors = F)totptt %>% inner_join(totAPA,by="gene") %>% nrow()[1] 20totptt %>% anti_join(totAPA,by="gene") %>% nrow()[1] 4nucptt %>% inner_join(nucAPA,by="gene") %>% nrow()[1] 54nucptt %>% anti_join(nucAPA,by="gene") %>% nrow()[1] 15There are 8 total ptt genes that are not apaQTL genes and there are 15 nuclear ptt genes that are not apaQTL genes.
Look at the distribution. To do this I will pull out the ptt associations from the apa data.
I will right a script that takes a the fraction, pulls the ptt associations into 2 files, one for the associations and one for the non associations.
python pttQTLsinapaQTL.py Total
python pttQTLsinapaQTL.py Nuclearnames=c('pid' ,'nvar' ,'shape1' ,'shape2', 'dummy' ,'sid', 'dist', 'npval', 'slope', 'ppval' ,'bpval', 'bh')
totapaPTT=read.table("../data/pttQTL/Totalapa_TotalPttQTL.txt", col.names = names, stringsAsFactors = F)
totapaNotPTT=read.table("../data/pttQTL/Totalapa_NOT_TotalPttQTL.txt", col.names = names, stringsAsFactors = F)qqplot(-log10(runif(nrow(totapaNotPTT))), -log10(totapaNotPTT$bpval),ylab="-log10 expression permuted pvalue", xlab="Uniform expectation", main="Total PTT genes in Total apaQTL")
points(sort(-log10(runif(nrow(totapaPTT)))), sort(-log10(totapaPTT$bpval)),col= alpha("Red"))
abline(0,1)
legend("topleft", legend=c("ptt gene", "All other genes"),col=c("red", "black"), pch=16,bty = 'n')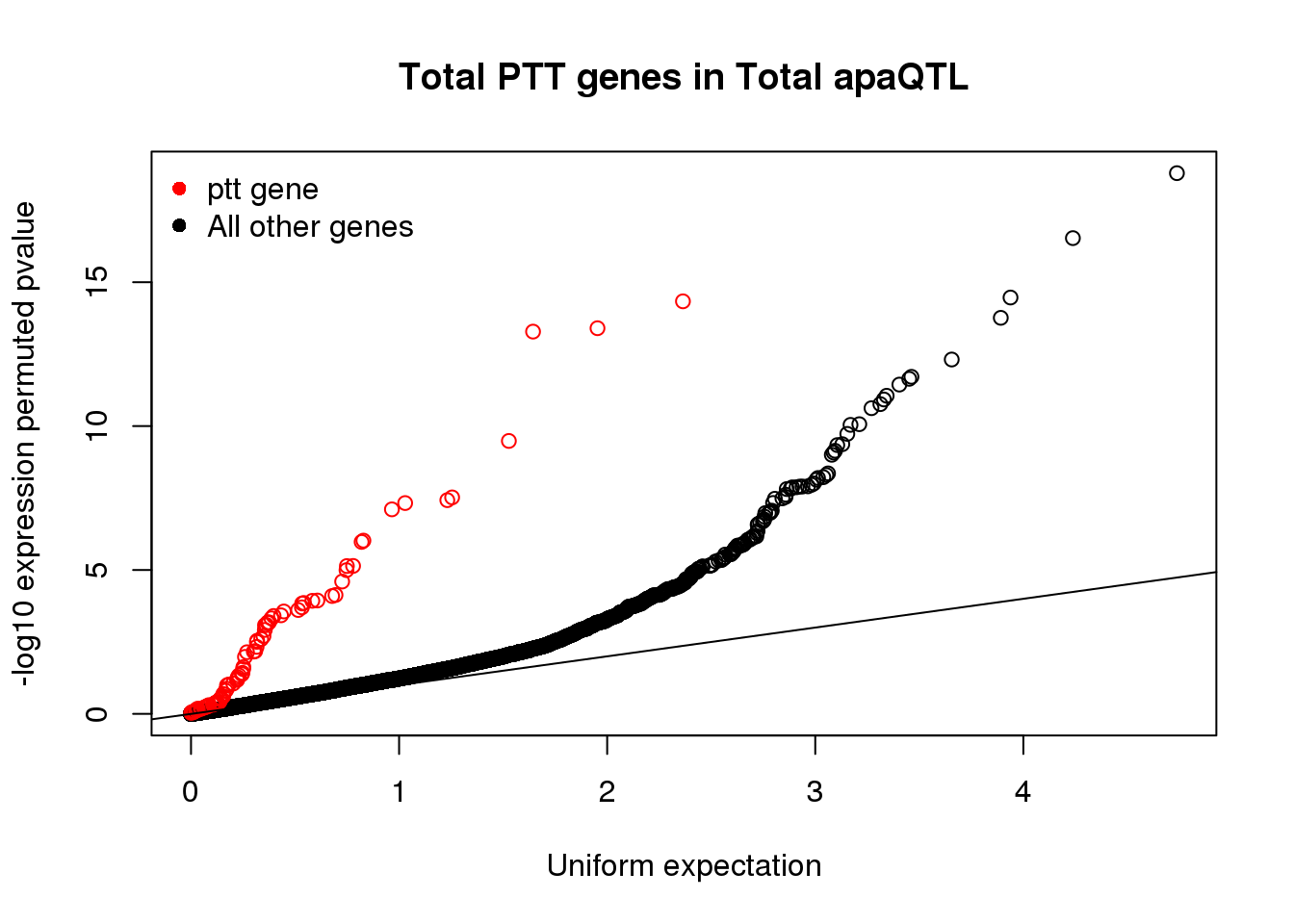
| Version | Author | Date |
|---|---|---|
| 272b0b4 | brimittleman | 2019-07-08 |
nucapaPTT=read.table("../data/pttQTL/Nuclearapa_NuclearPttQTL.txt", col.names = names, stringsAsFactors = F)
nucapaNotPTT=read.table("../data/pttQTL/Nuclearapa_NOT_NuclearPttQTL.txt", col.names = names, stringsAsFactors = F)qqplot(-log10(runif(nrow(nucapaNotPTT))), -log10(nucapaNotPTT$bpval),ylab="-log10 expression permuted pvalue", xlab="Uniform expectation", main="Nuclear PTT genes in Nuclear apaQTL")
points(sort(-log10(runif(nrow(nucapaPTT)))), sort(-log10(nucapaPTT$bpval)),col= alpha("Red"))
abline(0,1)
legend("topleft", legend=c("ptt gene", "All other genes"),col=c("red", "black"), pch=16,bty = 'n')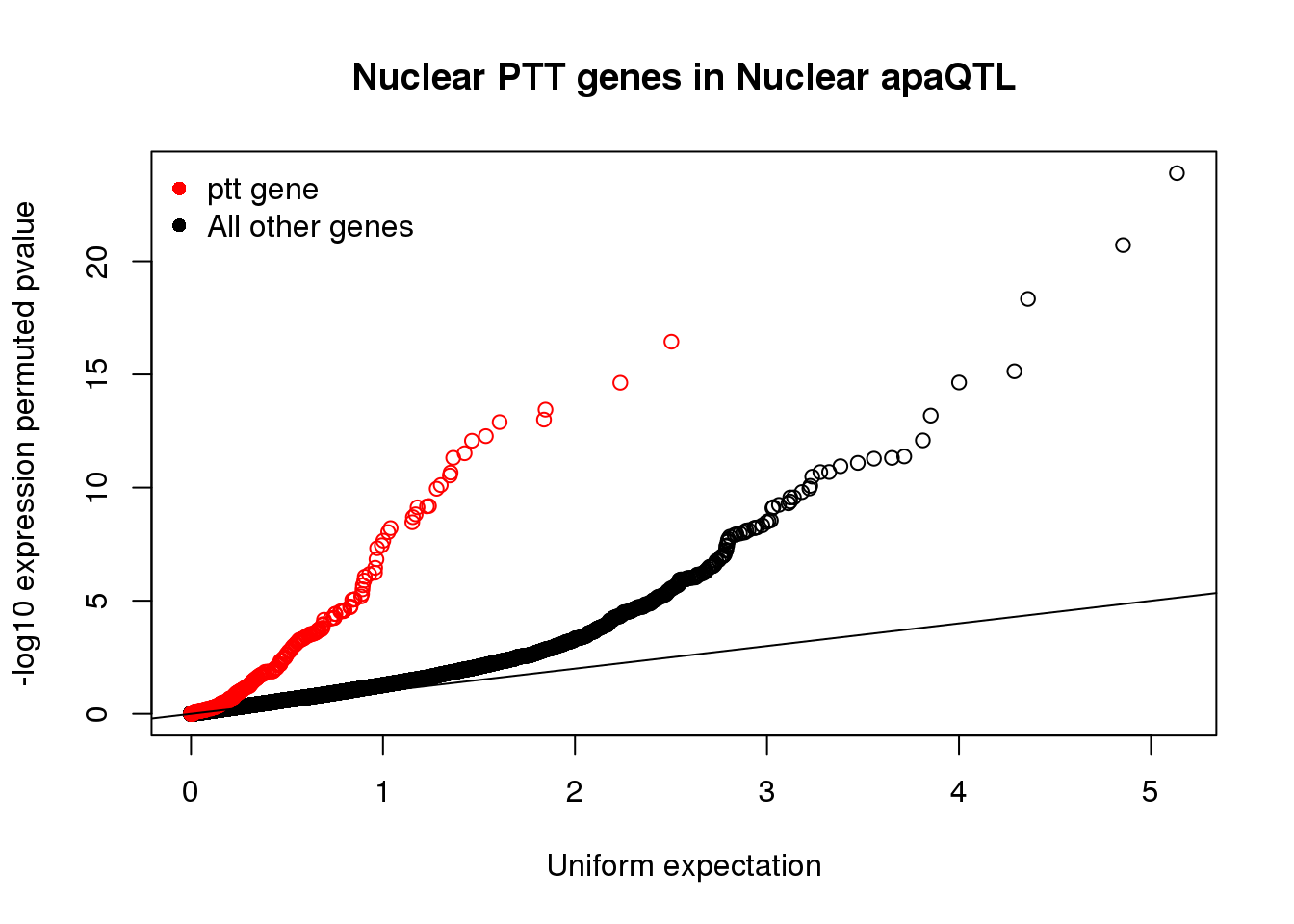
| Version | Author | Date |
|---|---|---|
| 272b0b4 | brimittleman | 2019-07-08 |
These show the sharing is super high and we are getting at similar phenotypes in ptt and apa.
wilcox.test(nucapaPTT$bpval,nucapaNotPTT$bpval,alternative="less")
Wilcoxon rank sum test with continuity correction
data: nucapaPTT$bpval and nucapaNotPTT$bpval
W = 2372300, p-value < 2.2e-16
alternative hypothesis: true location shift is less than 0wilcox.test(nucapaPTT$bpval,nucapaNotPTT$bpval,alternative="less")$p.value[1] 9.869063e-41Is there something interesting about the non overlap genes:
pttOnlyTot=totptt %>% anti_join(totAPA,by="gene")
totapaPTT_pttstatus=totapaPTT %>% separate(pid, into=c("chr", "start", "end", "geneID"),sep=":") %>% separate(geneID, into=c("gene", "pos", "strand", "pas"),sep="_") %>% mutate(APAQTL=ifelse(gene %in% pttOnlyTot$gene, "No", "Yes"))ggplot(totapaPTT_pttstatus,aes(x=APAQTL, y=bpval)) + geom_boxplot()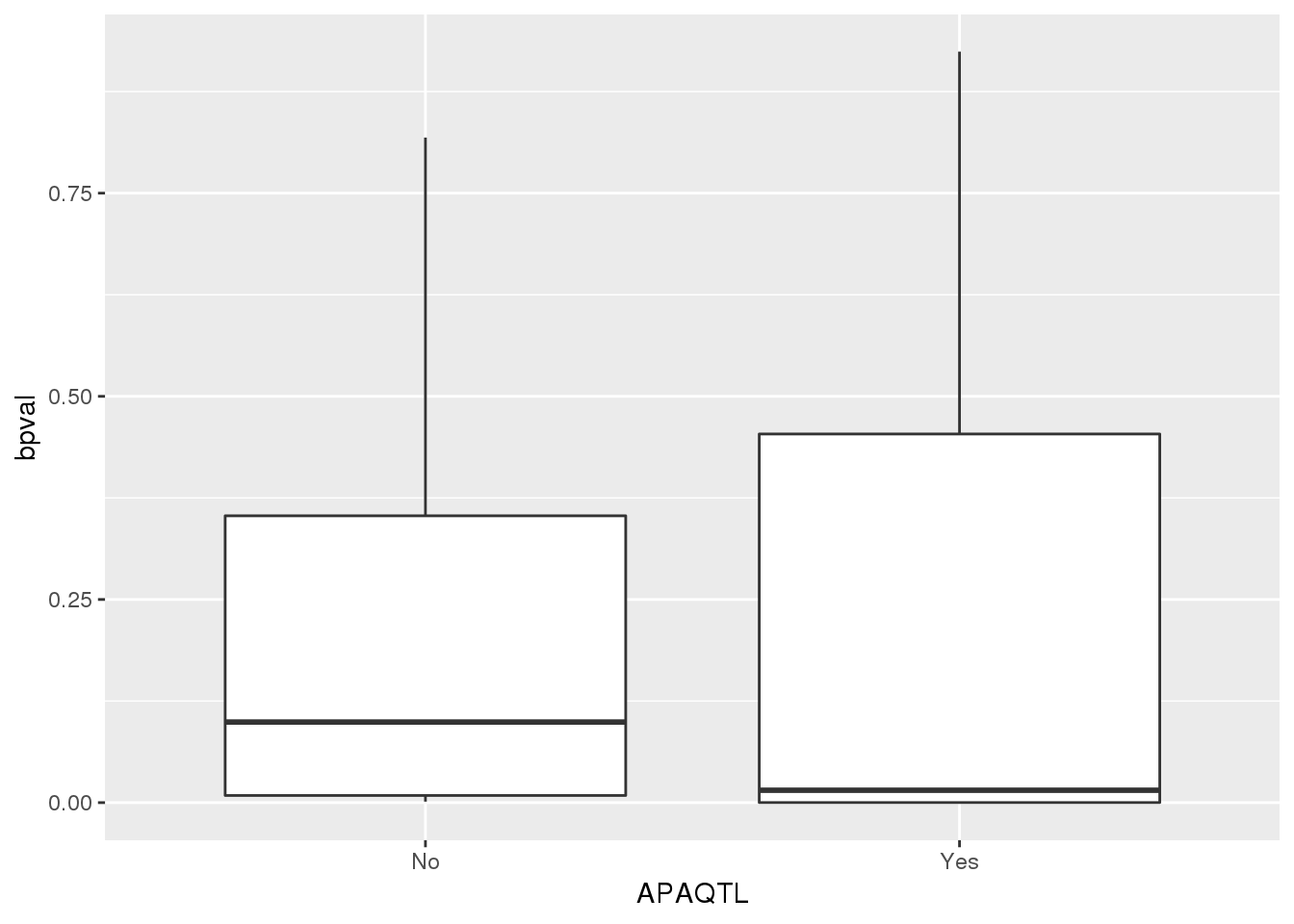
| Version | Author | Date |
|---|---|---|
| 33a700a | brimittleman | 2019-07-25 |
pttOnlyTot$gene[1] "C10orf88" "PHACTR4" "USP37" "ZNF718" pttOnlyNuc=nucptt %>% anti_join(nucAPA,by="gene")
nucapaPTT_pttstatus=nucapaPTT %>% separate(pid, into=c("chr", "start", "end", "geneID"),sep=":") %>% separate(geneID, into=c("gene", "pos", "strand", "pas"),sep="_") %>% mutate(APAQTL=ifelse(gene %in% pttOnlyTot$gene, "No", "Yes"))
ggplot(nucapaPTT_pttstatus,aes(x=APAQTL, y=bpval)) + geom_boxplot()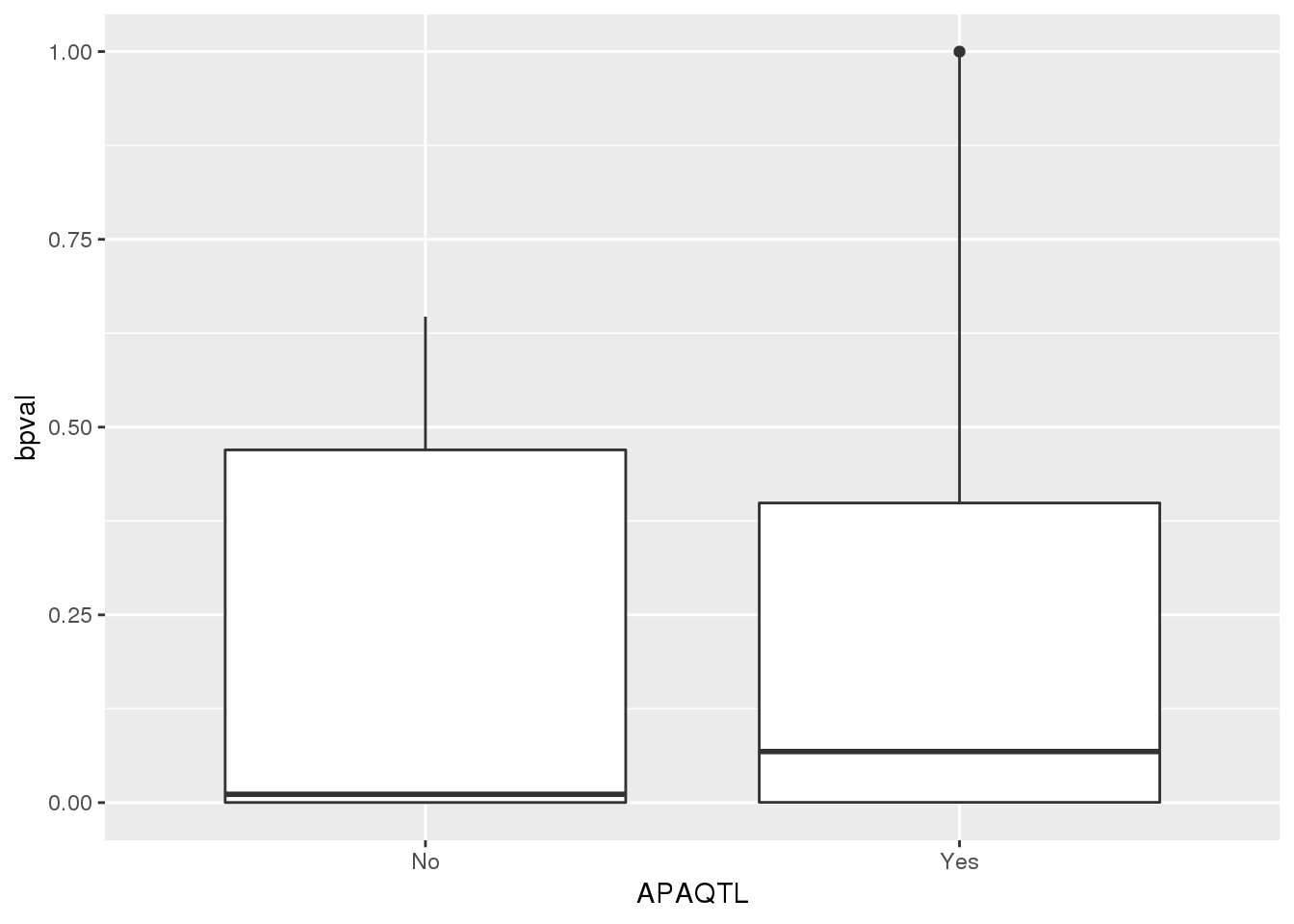
| Version | Author | Date |
|---|---|---|
| 33a700a | brimittleman | 2019-07-25 |
pttOnlyNuc$gene [1] "ATP5C1" "ARHGAP5" "AP4E1" "USP14" "PRKCZ" "MAP3K7CL"
[7] "BCL2L13" "IRAK2" "FYCO1" "IL17RB" "SEC24B" "HSPA9"
[13] "PREP" "ERLIN2" "HACD4" Plot these:
nucpttQTL %>% separate(pid, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene","loc"),sep="_") %>% anti_join(nucAPA,by="gene") %>% select(gene, chr,sid) gene chr sid
1 ATP5C1 10 rs2802460
2 ATP5C1 10 rs2802460
3 ARHGAP5 14 rs114000106
4 ARHGAP5 14 rs114000106
5 AP4E1 15 rs56056949
6 AP4E1 15 rs56056949
7 USP14 18 rs11876726
8 USP14 18 rs11876726
9 PRKCZ 1 rs13302944
10 MAP3K7CL 21 rs9808644
11 BCL2L13 22 rs9604775
12 IRAK2 3 rs6776059
13 FYCO1 3 rs59842697
14 FYCO1 3 rs59842697
15 IL17RB 3 rs2289205
16 IL17RB 3 rs2289205
17 SEC24B 4 rs1509314
18 HSPA9 5 rs41294546
19 HSPA9 5 rs41294546
20 PREP 6 rs76627946
21 PREP 6 rs76627946
22 ERLIN2 8 rs77049216
23 HACD4 9 rs57721805
24 HACD4 9 rs57721805Examples done need to rerun
These are from exploratory analysis
sbatch run_qtlFacetBoxplots.sh Nuclear PPFIA1 11 rs188630869 none
sbatch run_qtlFacetBoxplots.sh Nuclear ZDHHC17 12 rs74792673 none
sbatch run_qtlFacetBoxplots.sh Nuclear ZNF844 19 rs116146966 none
sbatch run_qtlFacetBoxplots.sh Nuclear PRKCZ 1 rs13302944 none
sbatch run_qtlFacetBoxplots.sh Nuclear POU2F1 1 rs6668193 none
sbatch run_qtlFacetBoxplots.sh Nuclear MAP3K7CL 21 rs9808644 none
sbatch run_qtlFacetBoxplots.sh Nuclear BCL2L13 22 rs8141618 none
sbatch run_qtlFacetBoxplots.sh Nuclear LINC00342 2 rs193252777 none
sbatch run_qtlFacetBoxplots.sh Nuclear DZIP3 3 rs112409302 none
sbatch run_qtlFacetBoxplots.sh Nuclear SUB1 5 rs76941541 none
sbatch run_qtlFacetBoxplots.sh Nuclear BLVRA 7 rs1181594 none
sbatch run_qtlFacetBoxplots.sh Nuclear NAPEPLD 7 rs56047178 none
sbatch run_qtlFacetBoxplots.sh Nuclear WNT2 7 rs76105832 none
sbatch run_qtlFacetBoxplots.sh Nuclear HACD4 9 rs57721805 none
totpttQTL %>% separate(pid, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene","loc"),sep="_") %>% anti_join(totAPA,by="gene") %>% select(gene, chr,sid) gene chr sid
1 C10orf88 10 rs7904973
2 C10orf88 10 rs7904973
3 PHACTR4 1 rs76130516
4 USP37 2 rs79468589
5 ZNF718 4 rs6814287
6 ZNF718 4 rs28531086sbatch run_qtlFacetBoxplots.sh Total C10orf88 10 rs7091776 none
sbatch run_qtlFacetBoxplots.sh Total UROS 10 rs11244646 none
sbatch run_qtlFacetBoxplots.sh Total LRRC57 15 rs61489160 none
sbatch run_qtlFacetBoxplots.sh Total ELMOD3 2 rs908302 none
sbatch run_qtlFacetBoxplots.sh Total USP37 2 rs79468589 none
sbatch run_qtlFacetBoxplots.sh Total PLSCR1 3 rs59690244 none
sbatch run_qtlFacetBoxplots.sh Total ZNF718 4 rs6814287 none
sbatch run_qtlFacetBoxplots.sh Total ZSCAN12 6 rs3799500 none
Moved these reslts to a ptt dir in the example qtl dir.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.4.0 forcats_0.3.0 stringr_1.3.1 dplyr_0.8.0.1
[5] purrr_0.3.2 readr_1.3.1 tidyr_0.8.3 tibble_2.1.1
[9] ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.0 cellranger_1.1.0 plyr_1.8.4 compiler_3.5.1
[5] pillar_1.3.1 git2r_0.25.2 highr_0.7 tools_3.5.1
[9] digest_0.6.18 lubridate_1.7.4 jsonlite_1.6 evaluate_0.12
[13] nlme_3.1-137 gtable_0.2.0 lattice_0.20-38 pkgconfig_2.0.2
[17] rlang_0.4.0 cli_1.1.0 rstudioapi_0.10 yaml_2.2.0
[21] haven_1.1.2 withr_2.1.2 xml2_1.2.0 httr_1.3.1
[25] knitr_1.20 hms_0.4.2 generics_0.0.2 fs_1.3.1
[29] rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5 glue_1.3.0
[33] R6_2.3.0 readxl_1.1.0 rmarkdown_1.10 modelr_0.1.2
[37] magrittr_1.5 whisker_0.3-2 backports_1.1.2 scales_1.0.0
[41] htmltools_0.3.6 rvest_0.3.2 assertthat_0.2.0 colorspace_1.3-2
[45] labeling_0.3 stringi_1.2.4 lazyeval_0.2.1 munsell_0.5.0
[49] broom_0.5.1 crayon_1.3.4