Motif Distruption
Briana Mittleman
6/10/2019
Last updated: 2019-06-13
Checks: 6 0
Knit directory: apaQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.3.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190411) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: .Rprofile
Untracked: ._.DS_Store
Untracked: .gitignore
Untracked: _workflowr.yml
Untracked: analysis/._PASdescriptiveplots.Rmd
Untracked: analysis/._cuttoffPercUsage.Rmd
Untracked: analysis/QTLexampleplots.Rmd
Untracked: analysis/cuttoffPercUsage.Rmd
Untracked: analysis/eQTLoverlap.Rmd
Untracked: analysis/oldstuffNotNeeded.Rmd
Untracked: apaQTL.Rproj
Untracked: code/.NascentRNAdtPlotFirstintronicPAS.sh.swp
Untracked: code/._ApaQTL_nominalNonnorm.sh
Untracked: code/._BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/._FC_NucintornUpandDown.sh
Untracked: code/._FC_UTR.sh
Untracked: code/._FC_intornUpandDownsteamPAS.sh
Untracked: code/._FC_newPeaks_olddata.sh
Untracked: code/._HMMpermuteTotal.py
Untracked: code/._HmmPermute.py
Untracked: code/._LC_samplegroups.py
Untracked: code/._NascentRNAdtPlot.sh
Untracked: code/._NascentRNAdtPlot3UTRPAS.sh
Untracked: code/._NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/._NascentRNAdtPlotNucPAS.sh
Untracked: code/._NascentRNAdtPlotTotPAS.sh
Untracked: code/._NascentRNAdtPlotintronicPAS.sh
Untracked: code/._NascnetRNAdtPlotPAS.sh
Untracked: code/._NetSeq_fourthintronDT.sh
Untracked: code/._QTL2bed.py
Untracked: code/._QTL2bed_withstrand.py
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilefiltPAS
Untracked: code/._TESplots100bp.sh
Untracked: code/._TESplots150bp.sh
Untracked: code/._TESplots200bp.sh
Untracked: code/._Untitled
Untracked: code/._ZipandTabPheno.sh
Untracked: code/._aAPAqtl_nominal39ind.sh
Untracked: code/._apaQTLCorrectPvalMakeQQ.R
Untracked: code/._apaQTL_Nominal.sh
Untracked: code/._apaQTL_permuted.sh
Untracked: code/._assignNucIntonpeak2intronlocs.sh
Untracked: code/._assignTotIntronpeak2intronlocs.sh
Untracked: code/._bam2BW_5primemost.sh
Untracked: code/._bed2saf.py
Untracked: code/._bothFracDTplot1stintron.sh
Untracked: code/._bothFracDTplot4thintron.sh
Untracked: code/._bothFrac_FC.sh
Untracked: code/._callPeaksYL.py
Untracked: code/._chooseAnno2SAF.py
Untracked: code/._chooseSignalSite
Untracked: code/._chooseSignalSite.py
Untracked: code/._cluster.json
Untracked: code/._clusterPAS.json
Untracked: code/._clusterfiltPAS.json
Untracked: code/._codingdms2bed.py
Untracked: code/._config.yaml
Untracked: code/._config2.yaml
Untracked: code/._configOLD.yaml
Untracked: code/._convertNominal2SNPLOC.py
Untracked: code/._convertNumeric.py
Untracked: code/._correctNomeqtl.R
Untracked: code/._dag.pdf
Untracked: code/._eQTLgenestestedapa.py
Untracked: code/._encodeRNADTplots.sh
Untracked: code/._extractGenotypes.py
Untracked: code/._extractseqfromqtlfastq.py
Untracked: code/._fc2leafphen.py
Untracked: code/._filter5perc.R
Untracked: code/._filter5percPheno.py
Untracked: code/._filterpeaks.py
Untracked: code/._finalPASbed2SAF.py
Untracked: code/._fix4su304corr.py
Untracked: code/._fix4su604corr.py
Untracked: code/._fix4sukalisto.py
Untracked: code/._fixExandUnexeQTL
Untracked: code/._fixExandUnexeQTL.py
Untracked: code/._fixFChead.py
Untracked: code/._fixFChead_bothfrac.py
Untracked: code/._fixH3k12ac.py
Untracked: code/._fixRNAhead4corr.py
Untracked: code/._fixRNAkalisto.py
Untracked: code/._fixgroupedtranscript.py
Untracked: code/._fixhead_netseqfc.py
Untracked: code/._getAPAfromanyeQTL.py
Untracked: code/._getApapval4eqtl.py
Untracked: code/._getApapval4eqtl_unexp.py
Untracked: code/._getDownstreamIntronNuclear.py
Untracked: code/._getIntronDownstreamPAS.py
Untracked: code/._getIntronUpstreamPAS.py
Untracked: code/._getQTLalleles.py
Untracked: code/._getQTLfastq.sh
Untracked: code/._getUpstreamIntronNuclear.py
Untracked: code/._grouptranscripts.py
Untracked: code/._keep5perMAF.py
Untracked: code/._keepSNP_vcf.sh
Untracked: code/._make5percPeakbed.py
Untracked: code/._makeFileID.py
Untracked: code/._makePheno.py
Untracked: code/._makeSAFbothfrac5perc.py
Untracked: code/._makeSNP2rsidfile.py
Untracked: code/._makeeQTLempirical_unexp.py
Untracked: code/._makeeQTLempiricaldist.py
Untracked: code/._makegencondeTSSfile.py
Untracked: code/._mergeAllBam.sh
Untracked: code/._mergeBW_norm.sh
Untracked: code/._mergeBamNascent.sh
Untracked: code/._mergeByFracBam.sh
Untracked: code/._mergePeaks.sh
Untracked: code/._mnase1stintron.sh
Untracked: code/._mnaseDT_fourthintron.sh
Untracked: code/._namePeaks.py
Untracked: code/._netseqDTplot1stIntron.sh
Untracked: code/._netseqFC.sh
Untracked: code/._peak2PAS.py
Untracked: code/._peakFC.sh
Untracked: code/._pheno2countonly.R
Untracked: code/._processYRIgen.py
Untracked: code/._qtlRegionseq.sh
Untracked: code/._qtlsPvalOppFrac.py
Untracked: code/._quantassign2parsedpeak.py
Untracked: code/._removeXfromHmm.py
Untracked: code/._removeloc_pheno.py
Untracked: code/._runCorrectNomEqtl.sh
Untracked: code/._runHMMpermuteAPAqtls.sh
Untracked: code/._runHMMpermuteeQTLS.sh
Untracked: code/._runMakeEmpiricaleQTL_unexp.sh
Untracked: code/._runMakeeQTLempirical.sh
Untracked: code/._run_getApaPval4eqtl.sh
Untracked: code/._run_getapafromeQTL.py
Untracked: code/._run_getapafromeQTL.sh
Untracked: code/._run_getapapval4eqtl_unexp.sh
Untracked: code/._run_leafcutterDiffIso.sh
Untracked: code/._run_sepUsagephen.sh
Untracked: code/._run_sepgenobychrom.sh
Untracked: code/._selectNominalPvalues.py
Untracked: code/._sepUsagePhen.py
Untracked: code/._sepgenobychrom.py
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._submit-snakemakePAS.sh
Untracked: code/._submit-snakemakefiltPAS.sh
Untracked: code/._subsetApanoteGene.py
Untracked: code/._subsetUnexplainedeQTLs.py
Untracked: code/._subset_diffisopheno.py
Untracked: code/._subsetpermAPAwithGenelist.py
Untracked: code/._subtrachfiveprimeUTR.sh
Untracked: code/._subtractExons.sh
Untracked: code/._subtractfiveprimeUTR.sh
Untracked: code/._tabixSNPS.sh
Untracked: code/._utrdms2saf.py
Untracked: code/.snakemake/
Untracked: code/APAqtl_nominal.err
Untracked: code/APAqtl_nominal.out
Untracked: code/APAqtl_nominal_39.err
Untracked: code/APAqtl_nominal_39.out
Untracked: code/APAqtl_nominal_nonNorm.err
Untracked: code/APAqtl_nominal_nonNorm.out
Untracked: code/APAqtl_permuted.err
Untracked: code/APAqtl_permuted.out
Untracked: code/ApaQTL_nominalNonnorm.sh
Untracked: code/BothFracDTPlot1stintron.err
Untracked: code/BothFracDTPlot1stintron.out
Untracked: code/BothFracDTPlot4stintron.err
Untracked: code/BothFracDTPlot4stintron.out
Untracked: code/BothFracDTPlotGeneRegions.err
Untracked: code/BothFracDTPlotGeneRegions.out
Untracked: code/BothFracDTPlotGeneRegions_norm.err
Untracked: code/BothFracDTPlotGeneRegions_norm.out
Untracked: code/BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/DistPAS2Sig.py
Untracked: code/EncodeRNADTPlotGeneRegions.err
Untracked: code/EncodeRNADTPlotGeneRegions.out
Untracked: code/FC_NucintornUpandDown.sh
Untracked: code/FC_NucintronPASupandDown.err
Untracked: code/FC_NucintronPASupandDown.out
Untracked: code/FC_UTR.err
Untracked: code/FC_UTR.out
Untracked: code/FC_UTR.sh
Untracked: code/FC_intornUpandDownsteamPAS.sh
Untracked: code/FC_intronPASupandDown.err
Untracked: code/FC_intronPASupandDown.out
Untracked: code/FC_newPAS_olddata.err
Untracked: code/FC_newPAS_olddata.out
Untracked: code/FC_newPeaks_olddata.sh
Untracked: code/HMMpermuteTotal.py
Untracked: code/HmmPermute.p
Untracked: code/HmmPermute.py
Untracked: code/LC_samplegroups.py
Untracked: code/NascentDTPlotGeneRegions.err
Untracked: code/NascentDTPlotGeneRegions.out
Untracked: code/NascentDTPlotPAS.err
Untracked: code/NascentDTPlotPAS.out
Untracked: code/NascentDTPlotPAS_3utr.err
Untracked: code/NascentDTPlotPAS_3utr.out
Untracked: code/NascentDTPlotPAS_firstintron.err
Untracked: code/NascentDTPlotPAS_firstintron.out
Untracked: code/NascentDTPlotPAS_intron.err
Untracked: code/NascentDTPlotPAS_intron.out
Untracked: code/NascentDTPlotPAS_nuc.err
Untracked: code/NascentDTPlotPAS_nuc.out
Untracked: code/NascentDTPlotPAS_tot.err
Untracked: code/NascentDTPlotPAS_tot.out
Untracked: code/NascentRNAdtPlot.sh
Untracked: code/NascentRNAdtPlot3UTRPAS.sh
Untracked: code/NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/NascentRNAdtPlotFirstintronicPAS.sh
Untracked: code/NascentRNAdtPlotNucPAS.sh
Untracked: code/NascentRNAdtPlotTotPAS.sh
Untracked: code/NascentRNAdtPlotintronicPAS.sh
Untracked: code/NascnetRNAdtPlotPAS.sh
Untracked: code/NetSeq_fourthintronDT.sh
Untracked: code/QTL2bed.py
Untracked: code/QTL2bed_withstrand.py
Untracked: code/README.md
Untracked: code/Rplots.pdf
Untracked: code/TESplots100bp.err
Untracked: code/TESplots100bp.out
Untracked: code/TESplots100bp.sh
Untracked: code/TESplots150bp.err
Untracked: code/TESplots150bp.out
Untracked: code/TESplots150bp.sh
Untracked: code/TESplots200bp.err
Untracked: code/TESplots200bp.out
Untracked: code/TESplots200bp.sh
Untracked: code/Untitled
Untracked: code/Upstream100Bases_general.py
Untracked: code/ZipandTabPheno.sh
Untracked: code/aAPAqtl_nominal39ind.sh
Untracked: code/apaQTLCorrectPvalMakeQQ_4pc.R
Untracked: code/apaQTL_Nominal_4pc.sh
Untracked: code/apaQTL_permuted.4pc.sh
Untracked: code/apafacetboxplots.R
Untracked: code/apaqtlfacetboxplots.R
Untracked: code/assignNucIntonpeak2intronlocs.sh
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignTotIntronpeak2intronlocs.sh
Untracked: code/assigntotPeak2Intronicregion.err
Untracked: code/assigntotPeak2Intronicregion.out
Untracked: code/bam2BW_5primemost.sh
Untracked: code/bam2bw.err
Untracked: code/bam2bw.out
Untracked: code/bam2bw_5primemost.err
Untracked: code/bam2bw_5primemost.out
Untracked: code/bothFracDTplot1stintron.sh
Untracked: code/bothFracDTplot4thintron.sh
Untracked: code/bothFrac_FC.err
Untracked: code/bothFrac_FC.out
Untracked: code/bothFrac_FC.sh
Untracked: code/codingdms2bed.py
Untracked: code/convertNominal2SNPLOC.py
Untracked: code/correctNomeqtl.R
Untracked: code/dag.pdf
Untracked: code/dagPAS.pdf
Untracked: code/dagfiltPAS.pdf
Untracked: code/eQTLgenestestedapa.py
Untracked: code/encodeRNADTplots.sh
Untracked: code/extractGenotypes.py
Untracked: code/extractseqfromqtlfastq.py
Untracked: code/fc2leafphen.py
Untracked: code/finalPASbed2SAF.py
Untracked: code/findbuginpeaks.R
Untracked: code/fix4su304corr.py
Untracked: code/fix4su604corr.py
Untracked: code/fix4sukalisto.py
Untracked: code/fixExandUnexeQTL
Untracked: code/fixExandUnexeQTL.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFChead_summary.py
Untracked: code/fixH3k12ac.py
Untracked: code/fixRNAhead4corr.py
Untracked: code/fixRNAkalisto.py
Untracked: code/fixgroupedtranscript.py
Untracked: code/fixhead_netseqfc.py
Untracked: code/genotypesYRI.gen.proc.keep.vcf.log
Untracked: code/genotypesYRI.gen.proc.keep.vcf.recode.vcf
Untracked: code/get100upPAS.py
Untracked: code/getAPAfromanyeQTL.py
Untracked: code/getApapval4eqtl.py
Untracked: code/getApapval4eqtl_unexp.py
Untracked: code/getDownstreamIntronNuclear.py
Untracked: code/getIntronDownstreamPAS.py
Untracked: code/getIntronUpstreamPAS.py
Untracked: code/getQTLalleles.py
Untracked: code/getQTLfastq.sh
Untracked: code/getSeq100up.sh
Untracked: code/getUpstreamIntronNuclear.py
Untracked: code/getseq100up.err
Untracked: code/getseq100up.out
Untracked: code/grouptranscripts.err
Untracked: code/grouptranscripts.out
Untracked: code/grouptranscripts.py
Untracked: code/keep5perMAF.py
Untracked: code/keepSNP_vcf.sh
Untracked: code/log/
Untracked: code/makeSAFbothfrac5perc.py
Untracked: code/makeSNP2rsidfile.py
Untracked: code/makeeQTLempirical_unexp.py
Untracked: code/makeeQTLempiricaldist.py
Untracked: code/makegencondeTSSfile.py
Untracked: code/mergeBW_norm.sh
Untracked: code/mergeBWnorm.err
Untracked: code/mergeBWnorm.out
Untracked: code/mergeBamNacent.err
Untracked: code/mergeBamNacent.out
Untracked: code/mergeBamNascent.sh
Untracked: code/mnase1stintron.sh
Untracked: code/mnaseDTPlot1stintron.err
Untracked: code/mnaseDTPlot1stintron.out
Untracked: code/mnaseDTPlot4thintron.err
Untracked: code/mnaseDTPlot4thintron.out
Untracked: code/mnaseDT_fourthintron.sh
Untracked: code/netDTPlot4thintron.out
Untracked: code/netseqDTplot1stIntron.sh
Untracked: code/netseqFC.err
Untracked: code/netseqFC.out
Untracked: code/netseqFC.sh
Untracked: code/neyDTPlot4thintron.err
Untracked: code/processYRIgen.py
Untracked: code/qtlFacetBoxplots.err
Untracked: code/qtlFacetBoxplots.out
Untracked: code/qtlRegionseq.sh
Untracked: code/qtlsPvalOppFrac.py
Untracked: code/removeXfromHmm.py
Untracked: code/removeloc_pheno.py
Untracked: code/runCorrectNomEqtl.sh
Untracked: code/runCorrectNomeqtl.err
Untracked: code/runCorrectNomeqtl.out
Untracked: code/runHMMpermute.err
Untracked: code/runHMMpermute.out
Untracked: code/runHMMpermuteAPAqtls.sh
Untracked: code/runHMMpermuteeQTLS.sh
Untracked: code/runHMMpermuteeQTLs.err
Untracked: code/runHMMpermuteeQTLs.out
Untracked: code/runMakeEmpiricaleQTL_unexp.sh
Untracked: code/runMakeEmpiricaleQTLs.err
Untracked: code/runMakeEmpiricaleQTLs.out
Untracked: code/runMakeEmpiricaleQTLsunex.err
Untracked: code/runMakeEmpiricaleQTLsunex.out
Untracked: code/runMakeeQTLempirical.sh
Untracked: code/run_DistPAS2Sig.err
Untracked: code/run_DistPAS2Sig.out
Untracked: code/run_distPAS2Sig.sh
Untracked: code/run_getAPAfromanyeQTL.err
Untracked: code/run_getAPAfromanyeQTL.out
Untracked: code/run_getApaPval4eQTLs.err
Untracked: code/run_getApaPval4eQTLs.out
Untracked: code/run_getApaPval4eQTLsunexplained.err
Untracked: code/run_getApaPval4eQTLsunexplained.out
Untracked: code/run_getApaPval4eqtl.sh
Untracked: code/run_getapafromeQTL.sh
Untracked: code/run_getapapval4eqtl_unexp.sh
Untracked: code/run_leafcutterDiffIso.sh
Untracked: code/run_leafcutter_ds.err
Untracked: code/run_leafcutter_ds.out
Untracked: code/run_qtlFacetBoxplots.sh
Untracked: code/run_sepUsagephen.sh
Untracked: code/run_sepgenobychrom.err
Untracked: code/run_sepgenobychrom.out
Untracked: code/run_sepgenobychrom.sh
Untracked: code/run_sepusage.err
Untracked: code/run_sepusage.out
Untracked: code/selectNominalPvalues.py
Untracked: code/sepUsagePhen.py
Untracked: code/sepgenobychrom.py
Untracked: code/seqQTLfastq.err
Untracked: code/seqQTLfastq.out
Untracked: code/seqQTLregion.err
Untracked: code/seqQTLregion.out
Untracked: code/snakePASlog.out
Untracked: code/snakefiltPASlog.out
Untracked: code/subsetApanoteGene.py
Untracked: code/subsetUnexplainedeQTLs.py
Untracked: code/subset_diffisopheno.py
Untracked: code/subsetpermAPAwithGenelist.py
Untracked: code/subtract5UTR.err
Untracked: code/subtract5UTR.out
Untracked: code/subtractExons.err
Untracked: code/subtractExons.out
Untracked: code/subtractExons.sh
Untracked: code/subtractfiveprimeUTR.sh
Untracked: code/tabixSNPS.sh
Untracked: code/tabixSNPs.err
Untracked: code/tabixSNPs.out
Untracked: code/transcriptdm2bed.py
Untracked: code/utrdms2saf.py
Untracked: code/vcf_keepsnps.err
Untracked: code/vcf_keepsnps.out
Untracked: code/zipandtabPhen.err
Untracked: code/zipandtabPhen.out
Untracked: data/._.DS_Store
Untracked: data/ApaByEgene/
Untracked: data/CompareOldandNew/
Untracked: data/DTmatrix/
Untracked: data/DiffIso/
Untracked: data/EncodeRNA/
Untracked: data/ExampleQTLPlots/
Untracked: data/GeuvadisRNA/
Untracked: data/HMMqtls/
Untracked: data/Li_eQTLs/
Untracked: data/NascentRNA/
Untracked: data/PAS/
Untracked: data/QTLGenotypes/
Untracked: data/QTLoverlap/
Untracked: data/QTLoverlap_nonNorm/
Untracked: data/README.md
Untracked: data/RNAseq/
Untracked: data/Reads2UTR/
Untracked: data/SignalSiteFiles/
Untracked: data/ThirtyNineIndQtl_nominal/
Untracked: data/apaQTLNominal/
Untracked: data/apaQTLNominal_4pc/
Untracked: data/apaQTLPermuted/
Untracked: data/apaQTLPermuted_4pc/
Untracked: data/apaQTLs/
Untracked: data/assignedPeaks/
Untracked: data/bam/
Untracked: data/bam_clean/
Untracked: data/bam_waspfilt/
Untracked: data/bed_10up/
Untracked: data/bed_clean/
Untracked: data/bed_clean_sort/
Untracked: data/bed_waspfilter/
Untracked: data/bedsort_waspfilter/
Untracked: data/bothFrac_FC/
Untracked: data/bw_norm/
Untracked: data/eQTLs/
Untracked: data/exampleQTLs/
Untracked: data/fastq/
Untracked: data/filterPeaks/
Untracked: data/fourSU/
Untracked: data/h3k27ac/
Untracked: data/highdiffsiggenes.txt
Untracked: data/inclusivePeaks/
Untracked: data/inclusivePeaks_FC/
Untracked: data/intronRNAratio/
Untracked: data/intron_analysis/
Untracked: data/mergedBG/
Untracked: data/mergedBW_byfrac/
Untracked: data/mergedBW_norm/
Untracked: data/mergedBam/
Untracked: data/mergedbyFracBam/
Untracked: data/motifdistrupt/
Untracked: data/netseq/
Untracked: data/nonNorm_pheno/
Untracked: data/nuc_10up/
Untracked: data/nuc_10upclean/
Untracked: data/overlapeQTL_try2/
Untracked: data/overlapeQTLs/
Untracked: data/peakCoverage/
Untracked: data/peaks_5perc/
Untracked: data/phenotype/
Untracked: data/phenotype_5perc/
Untracked: data/sigDiffGenes.txt
Untracked: data/sort/
Untracked: data/sort_clean/
Untracked: data/sort_waspfilter/
Untracked: nohup.out
Untracked: output/._.DS_Store
Untracked: output/._meanCorrelationPhenotypes.svg
Untracked: output/dtPlots/
Untracked: output/fastqc/
Untracked: output/meanCorrelationPhenotypes.svg
Unstaged changes:
Modified: analysis/Readdistagainstfeatures.Rmd
Modified: analysis/index.Rmd
Modified: analysis/nascenttranscription.Rmd
Modified: analysis/nucintronicanalysis.Rmd
Modified: analysis/overlapapaqtlsandeqtls.Rmd
Modified: analysis/rna_netseq_h3k12ac.Rmd
Modified: code/BothFracDTPlotGeneRegions.sh
Modified: code/Snakefile
Deleted: code/Upstream10Bases_general.py
Modified: code/apaQTLCorrectPvalMakeQQ.R
Modified: code/apaQTL_Nominal.sh
Modified: code/apaQTL_permuted.sh
Modified: code/apaQTLsnake.err
Modified: code/bam2bw.sh
Modified: code/bed2saf.py
Modified: code/cluster.json
Modified: code/clusterfiltPAS.json
Modified: code/config.yaml
Modified: code/environment.yaml
Modified: code/makePheno.py
Deleted: code/test.txt
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 02db3a7 | brimittleman | 2019-06-13 | fixbug |
| html | e783f5c | brimittleman | 2019-06-12 | Build site. |
| Rmd | 1f203f7 | brimittleman | 2019-06-12 | add examples |
| html | 5d71c2e | brimittleman | 2019-06-10 | Build site. |
| Rmd | c5fe1c2 | brimittleman | 2019-06-10 | add motif disruption |
In this analysis I will identify apaQTL that modify signal sites for the associated PAS. To do this I will look at the sequences 5bp up and downtream of each QTL snp and look for evidence of AATAAA disruption.
library(tidyverse)── Attaching packages ─────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(BSgenome)Loading required package: BiocGenericsLoading required package: parallel
Attaching package: 'BiocGenerics'The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLBThe following objects are masked from 'package:dplyr':
combine, intersect, setdiff, unionThe following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':
anyDuplicated, append, as.data.frame, basename, cbind,
colMeans, colnames, colSums, dirname, do.call, duplicated,
eval, evalq, Filter, Find, get, grep, grepl, intersect,
is.unsorted, lapply, lengths, Map, mapply, match, mget, order,
paste, pmax, pmax.int, pmin, pmin.int, Position, rank, rbind,
Reduce, rowMeans, rownames, rowSums, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which, which.max,
which.minLoading required package: S4VectorsLoading required package: stats4
Attaching package: 'S4Vectors'The following objects are masked from 'package:dplyr':
first, renameThe following object is masked from 'package:tidyr':
expandThe following object is masked from 'package:base':
expand.gridLoading required package: IRanges
Attaching package: 'IRanges'The following objects are masked from 'package:dplyr':
collapse, desc, sliceThe following object is masked from 'package:purrr':
reduceLoading required package: GenomeInfoDbLoading required package: GenomicRangesLoading required package: BiostringsLoading required package: XVector
Attaching package: 'XVector'The following object is masked from 'package:purrr':
compact
Attaching package: 'Biostrings'The following object is masked from 'package:base':
strsplitLoading required package: rtracklayerlibrary(workflowr)This is workflowr version 1.3.0
Run ?workflowr for help getting startedlibrary(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsGet bedfiles for qtls with the strand:
python QTL2bed_withstrand.py Total
python QTL2bed_withstrand.py NuclearMake bedfile with 5 bases upstream and downstream of snp. Names is gene:peak:loc and the score is the distance between PAS and the snp
totQTLbed=read.table("../data/apaQTLs/Total_apaQTLs4pc_5fdr.WITHSTRAND.bed", header = T, stringsAsFactors = F) %>% mutate(start=as.integer(SNPstart)-4, end=as.integer(SNPend)+6,snpChrint=as.integer(SNPchr) ) %>% select(SNPchr, start, end, name, score, strand)
nucQTLbed=read.table("../data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.WITHSTRAND.bed", header = T, stringsAsFactors = F) %>% mutate(start=as.integer(SNPstart)-4, end=as.integer(SNPend)+6,snpChrint=as.integer(SNPchr) ) %>% select(SNPchr, start, end, name, score, strand)Write these files so I can run bedtools nuc on them.
mkdir ../data/motifdistruptwrite.table(totQTLbed, file="../data/motifdistrupt/TotQTLregion.bed", col.names = F, row.names = F, quote = F, sep="\t")
write.table(nucQTLbed, file="../data/motifdistrupt/NucQTLregion.bed", col.names = F, row.names = F, quote = F, sep="\t")sbatch qtlRegionseq.shEvaluate results:
totSeq=read.table("../data/motifdistrupt/TotQTLregionSequences.bed", header = F, stringsAsFactors = F, col.names =c("chr","start", "end", "name", "Dist", "strand", "pctAT", "pctGC", "numA", "numC", "numG", "numT", "numN", "numoth", "length", "seq") )First plot the distance:
ggplot(totSeq,aes(x=Dist)) + geom_histogram(bins=100)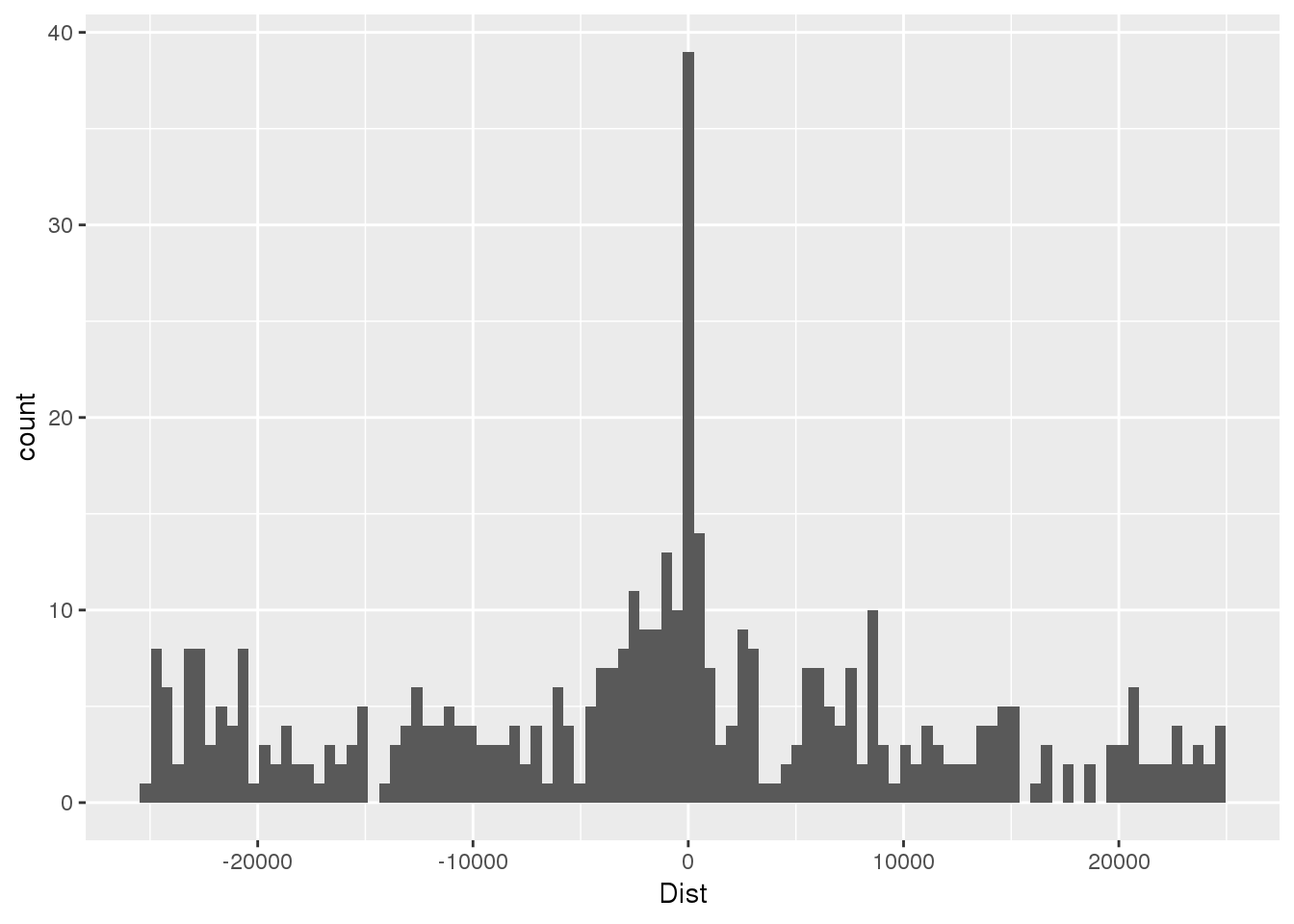
| Version | Author | Date |
|---|---|---|
| e783f5c | brimittleman | 2019-06-12 |
nucSeq=read.table("../data/motifdistrupt/NucQTLregionSequences.bed", header = F, stringsAsFactors = F, col.names =c("chr","start", "end", "name", "Dist", "strand", "pctAT", "pctGC", "numA", "numC", "numG", "numT", "numN", "numoth", "length", "seq") )First plot the distance:
ggplot(nucSeq,aes(x=Dist)) + geom_histogram(bins=100)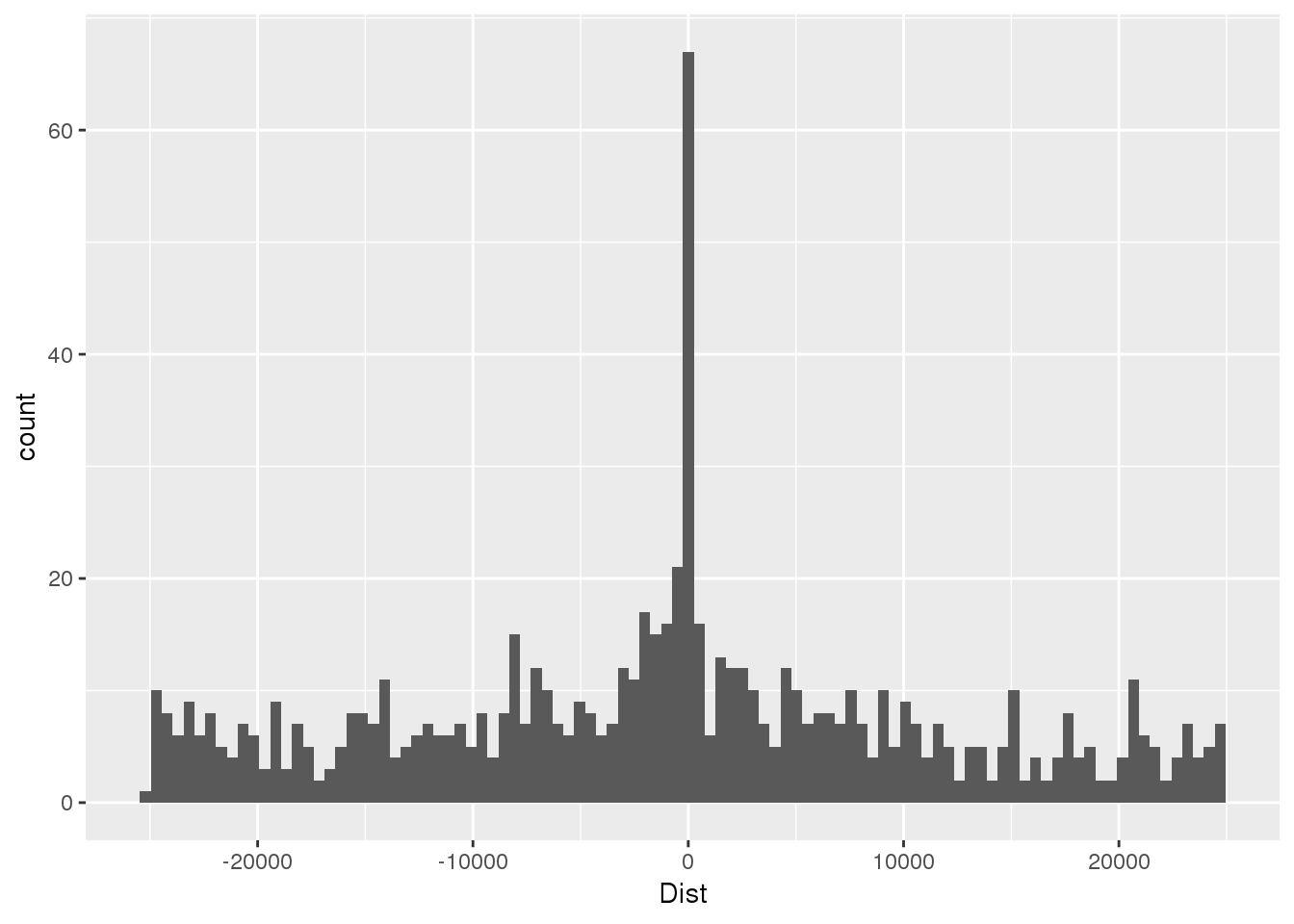
| Version | Author | Date |
|---|---|---|
| e783f5c | brimittleman | 2019-06-12 |
Try with getting the sequences with bedtools getfasta (This reverse compliments the negative strand)
sbatch getQTLfastq.shextract the sequences from these to match with the nuc file above. This is important because this uses the reverse compliment. The snp is the 6th letter.
(fraction is Tot /Nuc)
python extractseqfromqtlfastq.py Tot
python extractseqfromqtlfastq.py NucTotsequp=read.table("../data/motifdistrupt/TotQTLregionSequenceOnly.txt", header = F, stringsAsFactors = F, col.names = "CorrectSeq")
TotSeqComp=as.data.frame(cbind(totSeq,Totsequp)) %>% mutate(sig=ifelse(grepl("AATAAA",CorrectSeq),1, 0))
TotSeqCompSig=TotSeqComp %>% filter(sig==1)
TotSeqCompSig chr start end name Dist strand pctAT
1 15 101610289 101610300 LRRK1:peak47802:utr3 67 + 0.818182
2 19 16438656 16438667 KLF2:peak64649:utr3 454 + 0.909091
3 19 16438656 16438667 KLF2:peak64650:utr3 63 + 0.909091
4 19 43978507 43978518 PHLDB3:peak66481:utr3 -746 - 1.000000
5 2 131132114 131132125 PTPN18:peak74525:utr3 -776 + 0.818182
6 2 197855147 197855158 ANKRD44:peak77452:intron 20 - 1.000000
pctGC numA numC numG numT numN numoth length seq CorrectSeq
1 0.181818 8 1 1 1 0 0 11 AAAATAAACAG AAAATAAACAG
2 0.090909 8 1 0 2 0 0 11 AAAATAAAACT AAAATAAAACT
3 0.090909 8 1 0 2 0 0 11 AAAATAAAACT AAAATAAAACT
4 0.000000 1 0 0 10 0 0 11 ttttatttttt AAAAAATAAAA
5 0.181818 8 0 2 1 0 0 11 AGAAATAAAAG AGAAATAAAAG
6 0.000000 5 0 0 6 0 0 11 TTTATTTAAAA TTTTAAATAAA
sig
1 1
2 1
3 1
4 1
5 1
6 1Nucsequp=read.table("../data/motifdistrupt/NucQTLregionSequenceOnly.txt", header = F, stringsAsFactors = F, col.names = "CorrectSeq")
NucSeqComp=as.data.frame(cbind(nucSeq,Nucsequp)) %>% mutate(sig=ifelse(grepl("AATAAA",CorrectSeq),1, 0))
NucSeqCompSig=NucSeqComp %>% filter(sig==1)
NucSeqCompSig chr start end name Dist strand pctAT
1 15 101610289 101610300 LRRK1:peak47802:utr3 67 + 0.818182
2 15 101610289 101610300 LRRK1:peak47806:utr3 -2713 + 0.818182
3 19 16438656 16438667 KLF2:peak64649:utr3 454 + 0.909091
4 19 16438656 16438667 KLF2:peak64650:utr3 63 + 0.909091
5 19 43978507 43978518 PHLDB3:peak66481:utr3 -746 - 1.000000
6 19 58433644 58433655 ZNF418:peak68038:utr3 18 - 0.818182
7 2 33775278 33775289 RASGRP3:peak69728:utr3 -12759 + 1.000000
8 4 84367754 84367765 MRPS18C:peak99427:utr3 -14730 + 0.727273
9 6 167409236 167409247 MIR3939:peak119106:end 1476 - 1.000000
10 6 167409236 167409247 MIR3939:peak119107:end -1662 - 1.000000
pctGC numA numC numG numT numN numoth length seq CorrectSeq
1 0.181818 8 1 1 1 0 0 11 AAAATAAACAG AAAATAAACAG
2 0.181818 8 1 1 1 0 0 11 AAAATAAACAG AAAATAAACAG
3 0.090909 8 1 0 2 0 0 11 AAAATAAAACT AAAATAAAACT
4 0.090909 8 1 0 2 0 0 11 AAAATAAAACT AAAATAAAACT
5 0.000000 1 0 0 10 0 0 11 ttttatttttt AAAAAATAAAA
6 0.181818 3 2 0 6 0 0 11 cttttattaac GTTAATAAAAG
7 0.000000 8 0 0 3 0 0 11 ataataaataa ATAATAAATAA
8 0.272727 7 2 1 1 0 0 11 agccAATAAAA AGCCAATAAAA
9 0.000000 2 0 0 9 0 0 11 tattttttatt AATAAAAAATA
10 0.000000 2 0 0 9 0 0 11 tattttttatt AATAAAAAATA
sig
1 1
2 1
3 1
4 1
5 1
6 1
7 1
8 1
9 1
10 1These are pretty far from the peak and probably not the mechanism for these.
I can look at this another way by subsetting to those close to the peak.
TotSeqComp_Close=TotSeqComp %>% filter(abs(Dist)<200) %>% select(name,Dist,CorrectSeq,sig)
NucSeqComp_Close=NucSeqComp %>% filter(abs(Dist)<200) %>% select(name,Dist,CorrectSeq,sig)Look at examples:
Nuclear:
Disrupt: - ZNF418 rs75991626 T C (break signal site for peak68038), also associated with increased usage of the downstream UTR pas.
LRRK1:peak47802:utr3 rs15342 T-C disrupt signal site for peak47802
KLF2:peak64650:utr3 rs11086029 T- A disrupt signal site for peak64650, increased usage of upstream pas still in UTR
ANKRD44:peak77452:intron rs715185 T-C disrupt signal site for ANKRD44
Creating site: - LOC102725022- peak43230 rs4566122 G->A creates a signal site for PAS
Total:
Disrupt: - ZNF418 rs75991626 T C (break signal site for peak68038), also associated with increased usage of the downstream UTR pas.
ANKRD44:peak77452:intron rs715185 T-C disrupt signal site for ANKRD44
KLF2:peak64650:utr3 rs11086029 T- A disrupt signal site for peak64650, increased usage of upstream pas still in UTR
Make boxplots for these:
I wrote the code for these in the example plots analysis
Fraction=$1 gene=$2 chrom=$3 snp=$4 peakID=$5
sbatch run_qtlFacetBoxplots.sh Nuclear ZNF418 19 rs75991626 peak68038
This is not the best way to look at this. It may be a snp in LD. Also this is the distance to the peak not the PAS.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] reshape2_1.4.3 workflowr_1.3.0 BSgenome_1.50.0
[4] rtracklayer_1.42.0 Biostrings_2.50.1 XVector_0.22.0
[7] GenomicRanges_1.34.0 GenomeInfoDb_1.18.1 IRanges_2.16.0
[10] S4Vectors_0.20.1 BiocGenerics_0.28.0 forcats_0.3.0
[13] stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2
[16] readr_1.3.1 tidyr_0.8.3 tibble_2.1.1
[19] ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Biobase_2.42.0 httr_1.3.1
[3] jsonlite_1.6 modelr_0.1.2
[5] assertthat_0.2.0 GenomeInfoDbData_1.2.0
[7] cellranger_1.1.0 Rsamtools_1.34.0
[9] yaml_2.2.0 pillar_1.3.1
[11] backports_1.1.2 lattice_0.20-38
[13] glue_1.3.0 digest_0.6.18
[15] rvest_0.3.2 colorspace_1.3-2
[17] htmltools_0.3.6 Matrix_1.2-15
[19] plyr_1.8.4 XML_3.98-1.16
[21] pkgconfig_2.0.2 broom_0.5.1
[23] haven_1.1.2 zlibbioc_1.28.0
[25] scales_1.0.0 whisker_0.3-2
[27] BiocParallel_1.16.0 git2r_0.25.2
[29] generics_0.0.2 withr_2.1.2
[31] SummarizedExperiment_1.12.0 lazyeval_0.2.1
[33] cli_1.0.1 magrittr_1.5
[35] crayon_1.3.4 readxl_1.1.0
[37] evaluate_0.12 fs_1.2.6
[39] nlme_3.1-137 xml2_1.2.0
[41] tools_3.5.1 hms_0.4.2
[43] matrixStats_0.54.0 munsell_0.5.0
[45] DelayedArray_0.8.0 compiler_3.5.1
[47] rlang_0.3.1 grid_3.5.1
[49] RCurl_1.95-4.11 rstudioapi_0.10
[51] labeling_0.3 bitops_1.0-6
[53] rmarkdown_1.10 gtable_0.2.0
[55] R6_2.3.0 GenomicAlignments_1.18.0
[57] lubridate_1.7.4 knitr_1.20
[59] rprojroot_1.3-2 stringi_1.2.4
[61] Rcpp_1.0.0 tidyselect_0.2.5