initial_map_qc
Briana Mittleman
5/26/2018
Last updated: 2019-02-15
Checks: 6 0
Knit directory: threeprimeseq/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: data/perm_QTL_trans_noMP_5percov/
Ignored: output/.DS_Store
Untracked files:
Untracked: KalistoAbundance18486.txt
Untracked: analysis/4suDataIGV.Rmd
Untracked: analysis/DirectionapaQTL.Rmd
Untracked: analysis/EvaleQTLs.Rmd
Untracked: analysis/YL_QTL_test.Rmd
Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed
Untracked: analysis/snake.config.notes.Rmd
Untracked: analysis/verifyBAM.Rmd
Untracked: analysis/verifybam_dubs.Rmd
Untracked: code/PeaksToCoverPerReads.py
Untracked: code/strober_pc_pve_heatmap_func.R
Untracked: data/18486.genecov.txt
Untracked: data/APApeaksYL.total.inbrain.bed
Untracked: data/ApaQTLs/
Untracked: data/ChromHmmOverlap/
Untracked: data/DistTXN2Peak_genelocAnno/
Untracked: data/GM12878.chromHMM.bed
Untracked: data/GM12878.chromHMM.txt
Untracked: data/LianoglouLCL/
Untracked: data/LocusZoom/
Untracked: data/NuclearApaQTLs.txt
Untracked: data/PeakCounts/
Untracked: data/PeakCounts_noMP_5perc/
Untracked: data/PeakCounts_noMP_genelocanno/
Untracked: data/PeakUsage/
Untracked: data/PeakUsage_noMP/
Untracked: data/PeakUsage_noMP_GeneLocAnno/
Untracked: data/PeaksUsed/
Untracked: data/PeaksUsed_noMP_5percCov/
Untracked: data/RNAkalisto/
Untracked: data/RefSeq_annotations/
Untracked: data/TotalApaQTLs.txt
Untracked: data/Totalpeaks_filtered_clean.bed
Untracked: data/UnderstandPeaksQC/
Untracked: data/WASP_STAT/
Untracked: data/YL-SP-18486-T-combined-genecov.txt
Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt
Untracked: data/YL_QTL_test/
Untracked: data/apaExamp/
Untracked: data/apaQTL_examp_noMP/
Untracked: data/bedgraph_peaks/
Untracked: data/bin200.5.T.nuccov.bed
Untracked: data/bin200.Anuccov.bed
Untracked: data/bin200.nuccov.bed
Untracked: data/clean_peaks/
Untracked: data/comb_map_stats.csv
Untracked: data/comb_map_stats.xlsx
Untracked: data/comb_map_stats_39ind.csv
Untracked: data/combined_reads_mapped_three_prime_seq.csv
Untracked: data/diff_iso_GeneLocAnno/
Untracked: data/diff_iso_proc/
Untracked: data/diff_iso_trans/
Untracked: data/ensemble_to_genename.txt
Untracked: data/example_gene_peakQuant/
Untracked: data/explainProtVar/
Untracked: data/filtPeakOppstrand_cov_noMP_GeneLocAnno_5perc/
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed
Untracked: data/first50lines_closest.txt
Untracked: data/gencov.test.csv
Untracked: data/gencov.test.txt
Untracked: data/gencov_zero.test.csv
Untracked: data/gencov_zero.test.txt
Untracked: data/gene_cov/
Untracked: data/joined
Untracked: data/leafcutter/
Untracked: data/merged_combined_YL-SP-threeprimeseq.bg
Untracked: data/molPheno_noMP/
Untracked: data/mol_overlap/
Untracked: data/mol_pheno/
Untracked: data/nom_QTL/
Untracked: data/nom_QTL_opp/
Untracked: data/nom_QTL_trans/
Untracked: data/nuc6up/
Untracked: data/nuc_10up/
Untracked: data/other_qtls/
Untracked: data/pQTL_otherphen/
Untracked: data/peakPerRefSeqGene/
Untracked: data/perm_QTL/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/
Untracked: data/perm_QTL_opp/
Untracked: data/perm_QTL_trans/
Untracked: data/perm_QTL_trans_filt/
Untracked: data/protAndAPAAndExplmRes.Rda
Untracked: data/protAndAPAlmRes.Rda
Untracked: data/protAndExpressionlmRes.Rda
Untracked: data/reads_mapped_three_prime_seq.csv
Untracked: data/smash.cov.results.bed
Untracked: data/smash.cov.results.csv
Untracked: data/smash.cov.results.txt
Untracked: data/smash_testregion/
Untracked: data/ssFC200.cov.bed
Untracked: data/temp.file1
Untracked: data/temp.file2
Untracked: data/temp.gencov.test.txt
Untracked: data/temp.gencov_zero.test.txt
Untracked: data/threePrimeSeqMetaData.csv
Untracked: data/threePrimeSeqMetaData55Ind.txt
Untracked: data/threePrimeSeqMetaData55Ind.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.xlsx
Untracked: output/picard/
Untracked: output/plots/
Untracked: output/qual.fig2.pdf
Unstaged changes:
Modified: analysis/28ind.peak.explore.Rmd
Modified: analysis/CompareLianoglouData.Rmd
Modified: analysis/NewPeakPostMP.Rmd
Modified: analysis/apaQTLoverlapGWAS.Rmd
Modified: analysis/cleanupdtseq.internalpriming.Rmd
Modified: analysis/coloc_apaQTLs_protQTLs.Rmd
Modified: analysis/dif.iso.usage.leafcutter.Rmd
Modified: analysis/diff_iso_pipeline.Rmd
Modified: analysis/explainpQTLs.Rmd
Modified: analysis/explore.filters.Rmd
Modified: analysis/flash2mash.Rmd
Modified: analysis/mispriming_approach.Rmd
Modified: analysis/overlapMolQTL.Rmd
Modified: analysis/overlapMolQTL.opposite.Rmd
Modified: analysis/overlap_qtls.Rmd
Modified: analysis/peakOverlap_oppstrand.Rmd
Modified: analysis/peakQCPPlots.Rmd
Modified: analysis/pheno.leaf.comb.Rmd
Modified: analysis/pipeline_55Ind.Rmd
Modified: analysis/swarmPlots_QTLs.Rmd
Modified: analysis/test.max2.Rmd
Modified: analysis/test.smash.Rmd
Modified: analysis/understandPeaks.Rmd
Modified: code/Snakefile
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | df39edc | Briana Mittleman | 2018-06-01 | Build site. |
| Rmd | e4baa3b | Briana Mittleman | 2018-06-01 | compare star and subj |
| html | 7126988 | Briana Mittleman | 2018-05-26 | Build site. |
| Rmd | 252c548 | Briana Mittleman | 2018-05-26 | plot prop mapping with subjunc |
| html | b5cdd59 | Briana Mittleman | 2018-05-26 | Build site. |
| Rmd | 9a49459 | Briana Mittleman | 2018-05-26 | start map qc analysis |
I will use this analysis to look at initial mapping QC for the two mappers I am using.
library(workflowr)This is workflowr version 1.2.0
Run ?workflowr for help getting startedlibrary(ggplot2)
library(tidyr)
library(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithslibrary(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionlibrary(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsaveSubjunc
I created a csv with the number of reads, mapped reads, and proportion of reads mapped per library.
subj_map= read.csv("../data/reads_mapped_three_prime_seq.csv", header=TRUE, stringsAsFactors = FALSE)
subj_map$line=as.factor(subj_map$line)
subj_map$fraction=as.factor(subj_map$fraction)Summaries for each number:
summary(subj_map$reads) Min. 1st Qu. Median Mean 3rd Qu. Max.
5103350 8030068 8776602 8670328 9341566 10931074 summary(subj_map$mapped) Min. 1st Qu. Median Mean 3rd Qu. Max.
3575191 5688940 6268228 6091626 6394260 7788593 summary(subj_map$prop_mapped) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.6658 0.6932 0.7034 0.7025 0.7135 0.7343 Look at this graphically:
subj_melt=melt(subj_map, id.vars=c("line", "fraction"), measure.vars = c("reads", "mapped", "prop_mapped"))subj_prop_mapped= subj_melt %>% filter(variable=="prop_mapped")
subjplot=ggplot(subj_prop_mapped, aes(y=value, x=line, fill=fraction)) + geom_bar(stat="identity",position="dodge") + labs( title="Proportion of reads mapped with Subjunc") + ylab("Proportion mapped") + geom_hline(yintercept = mean(subj_prop_mapped$value)) + annotate("text",4, mean(subj_prop_mapped$value)- .1, vjust = -1, label = "Mean mapping proportion= .702")Star mapping
I added two lines to the csv file with the star map stats for each line.
star_map= read.csv("../data/reads_mapped_three_prime_seq.csv", header=TRUE, stringsAsFactors = FALSE)
star_map$line=as.factor(star_map$line)
star_map$fraction=as.factor(star_map$fraction)Summaries for each number:
summary(star_map$star_mapped) Min. 1st Qu. Median Mean 3rd Qu. Max.
3326506 5426888 5868012 5834521 6314488 7814874 summary(star_map$star_prop_mapped) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.5648 0.6452 0.6558 0.6719 0.7144 0.7827 Look at this graphically:
star_melt=melt(star_map, id.vars=c("line", "fraction"), measure.vars = c("reads", "star_mapped", "star_prop_mapped"))star_prop_mapped= star_melt %>% filter(variable=="star_prop_mapped")
starplot=ggplot(star_prop_mapped, aes(y=value, x=line, fill=fraction)) + geom_bar(stat="identity",position="dodge") + labs( title="Proportion of reads mapped with Star") + ylab("Proportion mapped") + geom_hline(yintercept = mean(star_prop_mapped$value)) + annotate("text",4, mean(star_prop_mapped$value)- .1, vjust = -1, label = "Mean mapping proportion= .672")Compare the plots:
plot_grid(subjplot,starplot)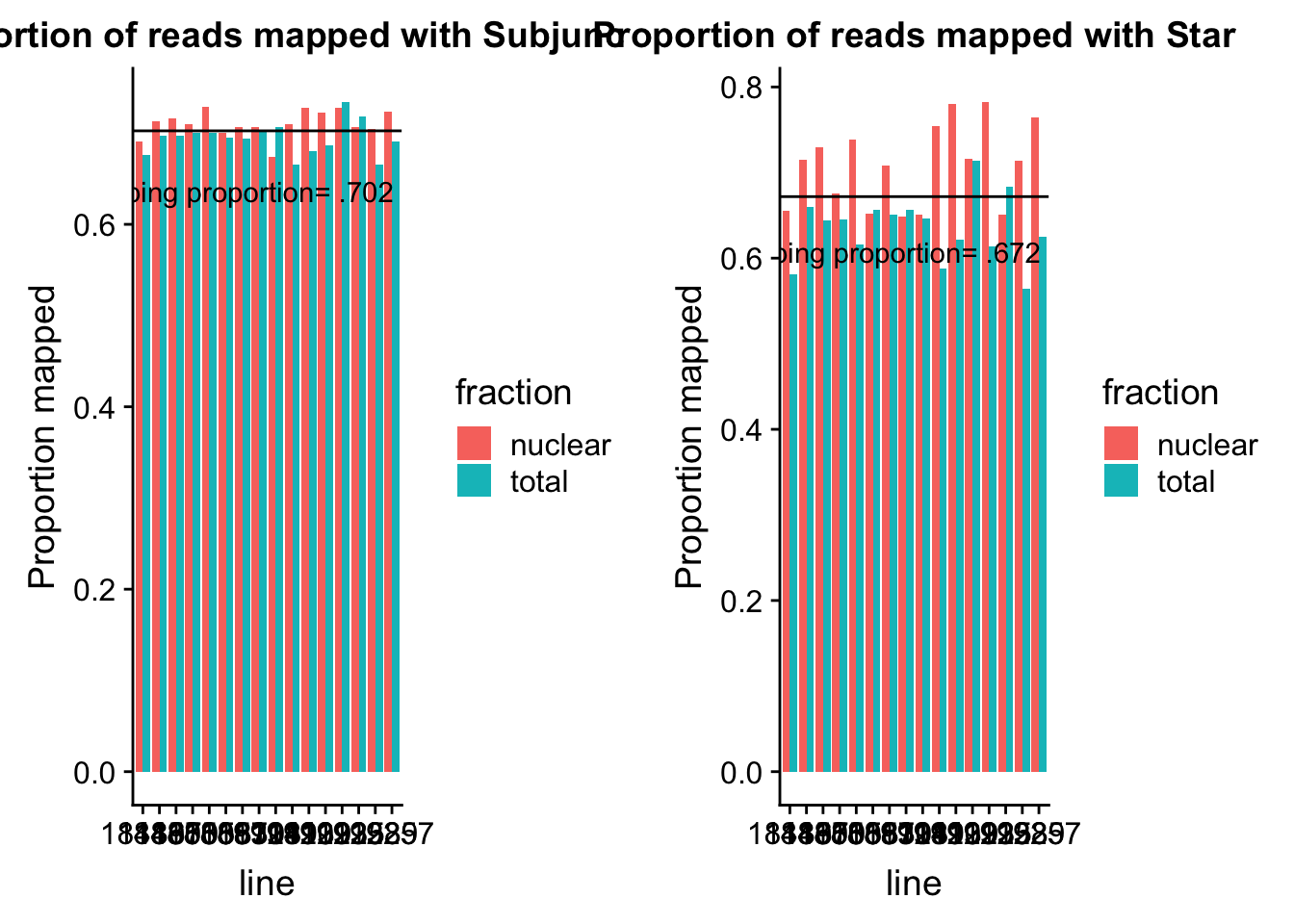
| Version | Author | Date |
|---|---|---|
| df39edc | Briana Mittleman | 2018-06-01 |
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bindrcpp_0.2.2 cowplot_0.9.3 dplyr_0.7.6 reshape2_1.4.3
[5] tidyr_0.8.1 ggplot2_3.0.0 workflowr_1.2.0
loaded via a namespace (and not attached):
[1] Rcpp_0.12.19 compiler_3.5.1 pillar_1.3.0 git2r_0.24.0
[5] plyr_1.8.4 bindr_0.1.1 tools_3.5.1 digest_0.6.17
[9] evaluate_0.13 tibble_1.4.2 gtable_0.2.0 pkgconfig_2.0.2
[13] rlang_0.2.2 yaml_2.2.0 withr_2.1.2 stringr_1.4.0
[17] knitr_1.20 fs_1.2.6 rprojroot_1.3-2 grid_3.5.1
[21] tidyselect_0.2.4 glue_1.3.0 R6_2.3.0 rmarkdown_1.11
[25] purrr_0.2.5 magrittr_1.5 whisker_0.3-2 backports_1.1.2
[29] scales_1.0.0 htmltools_0.3.6 assertthat_0.2.0 colorspace_1.3-2
[33] labeling_0.3 stringi_1.2.4 lazyeval_0.2.1 munsell_0.5.0
[37] crayon_1.3.4