Pipeline for 55 Ind
Briana Mittleman
2/1/2019
Last updated: 2019-02-05
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 7805afd
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: data/.DS_Store Ignored: data/perm_QTL_trans_noMP_5percov/ Ignored: output/.DS_Store Untracked files: Untracked: KalistoAbundance18486.txt Untracked: analysis/4suDataIGV.Rmd Untracked: analysis/DirectionapaQTL.Rmd Untracked: analysis/EvaleQTLs.Rmd Untracked: analysis/IndPeakUsageDiff.Rmd Untracked: analysis/YL_QTL_test.Rmd Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed Untracked: analysis/snake.config.notes.Rmd Untracked: analysis/verifyBAM.Rmd Untracked: analysis/verifybam_dubs.Rmd Untracked: code/PeaksToCoverPerReads.py Untracked: code/strober_pc_pve_heatmap_func.R Untracked: data/18486.genecov.txt Untracked: data/APApeaksYL.total.inbrain.bed Untracked: data/ChromHmmOverlap/ Untracked: data/DistTXN2Peak_genelocAnno/ Untracked: data/GM12878.chromHMM.bed Untracked: data/GM12878.chromHMM.txt Untracked: data/LianoglouLCL/ Untracked: data/LocusZoom/ Untracked: data/NuclearApaQTLs.txt Untracked: data/PeakCounts/ Untracked: data/PeakCounts_noMP_5perc/ Untracked: data/PeakCounts_noMP_genelocanno/ Untracked: data/PeakUsage/ Untracked: data/PeakUsage_noMP/ Untracked: data/PeakUsage_noMP_GeneLocAnno/ Untracked: data/PeaksUsed/ Untracked: data/PeaksUsed_noMP_5percCov/ Untracked: data/RNAkalisto/ Untracked: data/RefSeq_annotations/ Untracked: data/TotalApaQTLs.txt Untracked: data/Totalpeaks_filtered_clean.bed Untracked: data/UnderstandPeaksQC/ Untracked: data/YL-SP-18486-T-combined-genecov.txt Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt Untracked: data/YL_QTL_test/ Untracked: data/apaExamp/ Untracked: data/apaQTL_examp_noMP/ Untracked: data/bedgraph_peaks/ Untracked: data/bin200.5.T.nuccov.bed Untracked: data/bin200.Anuccov.bed Untracked: data/bin200.nuccov.bed Untracked: data/clean_peaks/ Untracked: data/comb_map_stats.csv Untracked: data/comb_map_stats.xlsx Untracked: data/comb_map_stats_39ind.csv Untracked: data/combined_reads_mapped_three_prime_seq.csv Untracked: data/diff_iso_GeneLocAnno/ Untracked: data/diff_iso_proc/ Untracked: data/diff_iso_trans/ Untracked: data/ensemble_to_genename.txt Untracked: data/example_gene_peakQuant/ Untracked: data/explainProtVar/ Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed Untracked: data/first50lines_closest.txt Untracked: data/gencov.test.csv Untracked: data/gencov.test.txt Untracked: data/gencov_zero.test.csv Untracked: data/gencov_zero.test.txt Untracked: data/gene_cov/ Untracked: data/joined Untracked: data/leafcutter/ Untracked: data/merged_combined_YL-SP-threeprimeseq.bg Untracked: data/molPheno_noMP/ Untracked: data/mol_overlap/ Untracked: data/mol_pheno/ Untracked: data/nom_QTL/ Untracked: data/nom_QTL_opp/ Untracked: data/nom_QTL_trans/ Untracked: data/nuc6up/ Untracked: data/nuc_10up/ Untracked: data/other_qtls/ Untracked: data/pQTL_otherphen/ Untracked: data/peakPerRefSeqGene/ Untracked: data/perm_QTL/ Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov/ Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/ Untracked: data/perm_QTL_opp/ Untracked: data/perm_QTL_trans/ Untracked: data/perm_QTL_trans_filt/ Untracked: data/protAndAPAAndExplmRes.Rda Untracked: data/protAndAPAlmRes.Rda Untracked: data/protAndExpressionlmRes.Rda Untracked: data/reads_mapped_three_prime_seq.csv Untracked: data/smash.cov.results.bed Untracked: data/smash.cov.results.csv Untracked: data/smash.cov.results.txt Untracked: data/smash_testregion/ Untracked: data/ssFC200.cov.bed Untracked: data/temp.file1 Untracked: data/temp.file2 Untracked: data/temp.gencov.test.txt Untracked: data/temp.gencov_zero.test.txt Untracked: data/threePrimeSeqMetaData.csv Untracked: data/threePrimeSeqMetaData55Ind.txt Untracked: data/threePrimeSeqMetaData55Ind.xlsx Untracked: output/picard/ Untracked: output/plots/ Untracked: output/qual.fig2.pdf Unstaged changes: Modified: analysis/28ind.peak.explore.Rmd Modified: analysis/CompareLianoglouData.Rmd Modified: analysis/apaQTLoverlapGWAS.Rmd Modified: analysis/cleanupdtseq.internalpriming.Rmd Modified: analysis/coloc_apaQTLs_protQTLs.Rmd Modified: analysis/dif.iso.usage.leafcutter.Rmd Modified: analysis/diff_iso_pipeline.Rmd Modified: analysis/explainpQTLs.Rmd Modified: analysis/explore.filters.Rmd Modified: analysis/flash2mash.Rmd Modified: analysis/mispriming_approach.Rmd Modified: analysis/overlapMolQTL.Rmd Modified: analysis/overlapMolQTL.opposite.Rmd Modified: analysis/overlap_qtls.Rmd Modified: analysis/peakOverlap_oppstrand.Rmd Modified: analysis/peakQCPPlots.Rmd Modified: analysis/pheno.leaf.comb.Rmd Modified: analysis/swarmPlots_QTLs.Rmd Modified: analysis/test.max2.Rmd Modified: analysis/understandPeaks.Rmd Modified: code/Snakefile
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7805afd | Briana Mittleman | 2019-02-05 | new QTL snps |
| html | e869ea5 | Briana Mittleman | 2019-02-05 | Build site. |
| Rmd | a2cd283 | Briana Mittleman | 2019-02-05 | res for full and UTR qtls |
| html | 5fd0c2a | Briana Mittleman | 2019-02-01 | Build site. |
| Rmd | 6ae3bba | Briana Mittleman | 2019-02-01 | add scripts for pipeline |
| html | 3445a3b | Briana Mittleman | 2019-02-01 | Build site. |
| Rmd | 957a0ee | Briana Mittleman | 2019-02-01 | initiate 55 ind pipeline |
library(tidyverse)── Attaching packages ────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.0.0 ✔ purrr 0.2.5
✔ tibble 1.4.2 ✔ dplyr 0.7.6
✔ tidyr 0.8.1 ✔ stringr 1.3.1
✔ readr 1.1.1 ✔ forcats 0.3.0── Conflicts ───────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(workflowr)This is workflowr version 1.1.1
Run ?workflowr for help getting startedFirst I need to move the duplicate files from bed_sort and bam (sort) to a different dir. data/Replicates
YL-SP-18499-N-batch4
YL-SP-18499-T-batch4
YL-SP-18912-N-batch4
YL-SP-18912-T-batch4
YL-SP-19093-N-batch4
YL-SP-19093-T-batch4
YL-SP-19140-N-batch4
YL-SP-19140-T-batch4
Fix these
- 18497-N (18499-N)
- 18497-T (18499-T)
- 18500-N (18501-N)
- 18500-T (18501-N)
QTL analysis pipeline
Mispriming
- Get 10 basepairs upstream: wrap_Upstream10Bases.sh
- Find sequence for these regions: Nuc10BasesUp.sh
- find which are bad run_filterMissprimingInNuc10.sh
- filter bed file run_filterSortBedbyCleanedBed.sh
- sort clean bed file sort_filterSortBedbyCleanedBed.sh
- filter bam files wrap_filterBamforMP.pysam2.sh
- sort and index clean bam SortIndexBam_noMP.sh
- merge clean bam files mergeBamFiles_noMP.sh
- sort and index merged SortIndexMergedBam_noMP.sh
Make Peaks
- create BW mergedBam2Bedgraph.sh
- make it a coverage file run_bgtocov_noMP.sh
- call peaks run_callPeaksYL_noMP.sh
- filter peaks
cat /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP/*.bed > /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP/APApeaks_merged_allchrom_noMP.bed- make SAF file bed2saf_noMP.py
- run feature counts peak_fc_noMP.sh
- filter peaks run_filter_peaks_noMP.sh
- name peaks
x = wc -l /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.bed
122488
seq 1 122488 > peak.num.txt
sort -k1,1 -k2,2n Filtered_APApeaks_merged_allchrom_noMP.bed > Filtered_APApeaks_merged_allchrom_noMP.sort.bed
paste /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.bed peak.num.txt | column -s $'\t' -t > temp
awk '{print $1 "\t" $2 "\t" $3 "\t" $7 "\t" $4 "\t" $5 "\t" $6}' temp > /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.named.bed
#cut the chr
sed 's/^chr//' /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.named.bed > /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR.bed
Assign Gene to peak
- Gene assignments mapnoMPPeaks2GenomeLoc.sh
Get Peak Counts and Usage
- make SAF processGenLocPeakAnno2SAF.py
- feature counts GeneLocAnno_fc_TN_noMP.sh
- fix header fix_head_fc_geneLoc_tot_noMP.py
- fix header fix_head_fc_geneLoc_nuc_noMP.py
- create_fileid_geneLocAnno_total.py (remove top line)
- create_fileid_geneLocAnno_nuclear.py (remove top line)
fout = file("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/file_id_mapping_total_Transcript_head.txt",'w')
infile= open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.fc", "r")
for line, i in enumerate(infile):
if line == 0:
i_list=i.split()
files= i_list[10:-2]
for each in files:
full = each.split("/")[7]
samp= full.split("-")[2:4]
lim="_"
samp_st=lim.join(samp)
outLine= full[:-1] + "\t" + samp_st
fout.write(outLine + "\n")
fout.close()- create_fileid_geneLocAnno_nuclear.py fout = file("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/file_id_mapping_nuclear_Transcript_head.txt",'w')
infile= open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.fc", "r")
for line, i in enumerate(infile):
if line == 0:
i_list=i.split()
files= i_list[10:-2]
for each in files:
full = each.split("/")[7]
samp= full.split("-")[2:4]
lim="_"
samp_st=lim.join(samp)
outLine= full[:-1] + "\t" + samp_st
fout.write(outLine + "\n")
fout.close()- make phenotype run_makePhen_sep_GeneLocAnno_noMP.sh
Change these so that peak IDs are start and end of peak. (code same as in the UTR analysis below)
- convert to usage pheno2CountOnly_genelocAnno.R
- counts to numeric convertCount2Numeric_noMP_GeneLocAnno.py
Make script to filter 5%
- filter_5percUsagePeaks.R
library(tidyverse)
totalPeakUs=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno.fc", header = T, stringsAsFactors = F) %>% separate(chrom, sep = ":", into = c("chr", "start", "end", "id")) %>% separate(id, sep="_", into=c("gene", "strand", "peak"))
nuclearPeakUs=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno.fc", header = T, stringsAsFactors = F) %>% separate(chrom, sep = ":", into = c("chr", "start", "end", "id")) %>% separate(id, sep="_", into=c("gene", "strand", "peak"))
ind=colnames(totalPeakUs)[7:dim(totalPeakUs)[2]]
totalPeakUs_CountNum=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno.CountsOnlyNumeric.txt", col.names = ind)
nuclearPeakUs_CountNum=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno.CountsOnlyNumeric.txt", col.names = ind)
#numeric with anno
totalPeak=as.data.frame(cbind(totalPeakUs[,1:6], totalPeakUs_CountNum))
nuclearPeak=as.data.frame(cbind(nuclearPeakUs[,1:6], nuclearPeakUs_CountNum))
#mean
totalPeakUs_CountNum_mean=rowMeans(totalPeakUs_CountNum)
nuclearPeakUs_CountNum_mean=rowMeans(nuclearPeakUs_CountNum)
#append mean to anno
TotalPeakUSMean=as.data.frame(cbind(totalPeakUs[,1:6],totalPeakUs_CountNum_mean))
NuclearPeakUSMean=as.data.frame(cbind(nuclearPeakUs[,1:6],nuclearPeakUs_CountNum_mean))
NuclearPeakUSMean_5perc=NuclearPeakUSMean %>% filter(nuclearPeakUs_CountNum_mean>=.05)
write.table(NuclearPeakUSMean_5perc,file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear_fixed.pheno.5percPeaks.txt", row.names=F, col.names=F, quote = F)
TotalPeakUSMean_5per= TotalPeakUSMean %>% filter(totalPeakUs_CountNum_mean>=.05)
write.table(TotalPeakUSMean_5per,file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total_fixed.pheno.5percPeaks.txt", row.names=F, col.names=F, quote = F)- run_filter_5percUsagePeaks.sh
#!/bin/bash
#SBATCH --job-name=run_filter_5percUsagePeaks
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_filter_5percUsagePeaks.out
#SBATCH --error=run_filter_5percUsagePeaks.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
Rscript filter_5percUsagePeaks.R- filterPheno_bothFraction_GeneLocAnno_5perc.py
QTL analysis
In /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs/
#zip file
gzip filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc
gzip filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc
module load python
#leafcutter script
python /project2/gilad/briana/threeprimeseq/code/prepare_phenotype_table.py filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz
python /project2/gilad/briana/threeprimeseq/code/prepare_phenotype_table.py filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz
#source activate three-prime-env
sh filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz_prepare.sh
sh filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz_prepare.sh
#keep only 2 PCs
head -n 3 filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.PCs > filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.2PCs
head -n 3 filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.PCs > filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.2PCs- Make sample list makeSampleList_newGeneAnno.py
#make a sample list
fout = open("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs/SAMPLE.txt",'w')
for ln in open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/file_id_mapping_total_Transcript_head.txt", "r"):
bam, sample = ln.split()
line=sample[:-2]
fout.write("NA"+line + "\n")
fout.close()(current - need to remove 19092 and 19193)
* APAqtl_nominal_GeneLocAnno_noMP_5percUsage.sh
* APAqtl_perm_GeneLocAnno_noMP_5percUsage.sh (change to 25kb on each side- –window 2.5e4 ) * run_APAqtlpermCorrectQQplot_GeneLocAnno_noMP_5perUs.sh
totQTLs=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total.fixed.pheno_5perc_permResBH.txt", stringsAsFactors = F, header=T)
Sig_TotQTLs= totQTLs %>% filter(-log10(bh)>=1)
nrow(Sig_TotQTLs)[1] 363How many genes are tested:
totQTLs %>% separate(pid, into=c("chr", "start", "end", "id"), sep=":") %>% separate(id, into=c("gene", "strand", "peak"), sep="_") %>% group_by(gene) %>% select(gene) %>% tally() %>% nrow()Warning: Expected 3 pieces. Additional pieces discarded in 3 rows [815,
816, 817].[1] 10633nucQTLs=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear.fixed.pheno_5perc_permResBH.txt", stringsAsFactors = F, header=T)
Sig_NucQTLs= nucQTLs %>% filter(-log10(bh)>=1)
nrow(Sig_NucQTLs)[1] 623How many genes:
nucQTLs %>% separate(pid, into=c("chr", "start", "end", "id"), sep=":") %>% separate(id, into=c("gene", "strand", "peak"), sep="_") %>% group_by(gene) %>% select(gene) %>% tally() %>% nrow()Warning: Expected 3 pieces. Additional pieces discarded in 3 rows [990,
991, 992].[1] 10852QTLs only peaks in the 3’ UTR
I want to subset to only look at the peaks that assign to the 3’ UTRs.
Remake the processGenLocPeakAnno2SAF.py code to only include peaks maping to a 3’ UTR
processGenLocPeakAnno2SAF_3UTRonly.py
inFile="/project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_GeneLoc/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR_geneLoc.bed"
outFile=open("/project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_GeneLoc_3UTR/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR_geneLocParsed_3UTR.SAF" , "w")
outFile.write("GeneID\tChr\tStart\tEnd\tStrand\n")
for ln in open(inFile, "r"):
chrom, start, end, peak, cov, strand, score, anno = ln.split()
if anno==".":
continue
anno_lst=anno.split(",")
if len(anno_lst)==1:
gene=anno_lst[0].split(":")[1]
if anno_lst[0].split(":")[0]=="utr3":
peak_i=int(peak)
start_i=int(start)
end_i=int(end)
ID="peak%d:%s:%d:%d:%s:%s"%(peak_i, chrom, start_i, end_i, strand, gene)
outFile.write("%s\t%s\t%d\t%d\t%s\n"%(ID, chrom, start_i, end_i, strand))
else:
type_dic={}
for each in anno_lst:
type_dic[each.split(":")[0]]=each.split(":")[1]
if "utr3" in type_dic.keys():
gene=type_dic["utr3"]
peak_i=int(peak)
start_i=int(start)
end_i=int(end)
ID="peak%d:%s:%d:%d:%s:%s"%(peak_i, chrom, start_i, end_i, strand, gene)
outFile.write("%s\t%s\t%d\t%d\t%s\n"%(ID, chrom, start_i, end_i, strand))
outFile.close()This results in 28373 peaks.
- feature counts GeneLocAnno_fc_TN_noMP_3UTR.sh
#!/bin/bash
#SBATCH --job-name=GeneLocAnno_fc_TN_noMP_3UTR
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=GeneLocAnno_fc_TN_noMP_3UTR.out
#SBATCH --error=GeneLocAnno_fc_TN_noMP_3UTR.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
featureCounts -O -a /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_GeneLoc_3UTR/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR_geneLocParsed_3UTR.SAF -F SAF -o /project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fc /project2/gilad/briana/threeprimeseq/data/bam_NoMP_sort/*T-combined-sort.noMP.sort.bam -s 1
featureCounts -O -a /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_GeneLoc_3UTR/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR_geneLocParsed_3UTR.SAF -F SAF -o /project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fc /project2/gilad/briana/threeprimeseq/data/bam_NoMP_sort/*N-combined-sort.noMP.sort.bam -s 1fix_head_fc_geneLoc_tot_noMP_3UTR.py
infile= open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fc", "r")
fout = open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.fc",'w')
for line, i in enumerate(infile):
if line == 1:
i_list=i.split()
libraries=i_list[:6]
for sample in i_list[6:]:
full = sample.split("/")[7]
samp= full.split("-")[2:4]
lim="_"
samp_st=lim.join(samp)
libraries.append(samp_st)
first_line= "\t".join(libraries)
fout.write(first_line + '\n')
else :
fout.write(i)
fout.close()fix header fix_head_fc_geneLoc_nuc_noMP_3UTR.py
infile= open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fc", "r")
fout = open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.fc",'w')
for line, i in enumerate(infile):
if line == 1:
i_list=i.split()
libraries=i_list[:6]
for sample in i_list[6:]:
full = sample.split("/")[7]
samp= full.split("-")[2:4]
lim="_"
samp_st=lim.join(samp)
libraries.append(samp_st)
first_line= "\t".join(libraries)
fout.write(first_line + '\n')
else :
fout.write(i)
fout.close()File IDs:
* /project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/file_id_mapping_nuclear_Transcript_head.txt * c/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/file_id_mapping_total_Transcript_head.txt
- make phenotype run_makePhen_sep_GeneLocAnno_noMP_3UTR.sh
These are also different. The start and end for calling QTLs will be the peak rather than the gene start.
makePhenoRefSeqPeaks_GeneLoc_Total_noMP_3UTR.py
#PYTHON 3
dic_IND = {}
dic_BAM = {}
for ln in open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/file_id_mapping_total_Transcript_head.txt"):
bam, IND = ln.split("\t")
IND = IND.strip()
dic_IND[bam] = IND
if IND not in dic_BAM:
dic_BAM[IND] = []
dic_BAM[IND].append(bam)
#now I have ind dic with keys as the bam and ind as the values
#I also have a bam dic with ind as the keys and bam as the values
inds=list(dic_BAM.keys()) #list of ind libraries
#gene start and end dictionaries:
dic_geneS = {}
dic_geneE = {}
for ln in open("/project2/gilad/briana/genome_anotation_data/RefSeq_annotations/ncbiRefSeq_endAllGenes.sort.bed"):
chrom, start, end, geneID, score, strand = ln.split('\t')
gene= geneID.split(":")[1]
# if "-" in gene:
# gene=gene.split("-")[0]
if gene not in dic_geneS:
dic_geneS[gene]=int(start)
dic_geneE[gene]=int(end)
#list of genes
count_file=open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.fc", "r")
genes=[]
for line , i in enumerate(count_file):
if line > 1:
i_list=i.split()
id=i_list[0]
id_list=id.split(":")
gene=id_list[5]
if gene not in genes:
genes.append(gene)
#make the ind and gene dic
dic_dub={}
for g in genes:
dic_dub[g]={}
for i in inds:
dic_dub[g][i]=0
#populate the dictionary
count_file=open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.fc", "r")
for line, i in enumerate(count_file):
if line > 1:
i_list=i.split()
id=i_list[0]
id_list=id.split(":")
g= id_list[5]
values=list(i_list[6:])
list_list=[]
for ind,val in zip(inds, values):
list_list.append([ind, val])
for num, name in enumerate(list_list):
dic_dub[g][list_list[num][0]] += int(list_list[num][1])
#write the file by acessing the dictionary and putting values in the table ver the value in the dic
fout=open("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.fc","w")
peak=["chrom"]
inds_noL=[]
for each in inds:
indsNA= "NA" + each[:-2]
inds_noL.append(indsNA)
fout.write(" ".join(peak + inds_noL) + '\n' )
count_file=open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.fc", "r")
for line , i in enumerate(count_file):
if line > 1:
i_list=i.split()
id=i_list[0]
id_list=id.split(":")
gene=int(id_list[5])
#start=dic_geneS[id_list[5]]
start=int(id_list[2])
#end=dic_geneE[id_list[5]]
end=id_list[3]
buff=[]
buff.append("chr%s:%d:%d:%s_%s_%s"%(id_list[1], start, end, id_list[5], id_list[4], id_list[0]))
for x,y in zip(i_list[6:], inds):
b=int(dic_dub[gene][y])
t=int(x)
buff.append("%d/%d"%(t,b))
fout.write(" ".join(buff)+ '\n')
fout.close()makePhenoRefSeqPeaks_GeneLoc_Nuclear_noMP_3UTR.py
#PYTHON 3
dic_IND = {}
dic_BAM = {}
for ln in open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno/file_id_mapping_nuclear_Transcript_head.txt"):
bam, IND = ln.split("\t")
IND = IND.strip()
dic_IND[bam] = IND
if IND not in dic_BAM:
dic_BAM[IND] = []
dic_BAM[IND].append(bam)
#now I have ind dic with keys as the bam and ind as the values
#I also have a bam dic with ind as the keys and bam as the values
inds=list(dic_BAM.keys()) #list of ind libraries
#gene start and end dictionaries:
dic_geneS = {}
dic_geneE = {}
for ln in open("/project2/gilad/briana/genome_anotation_data/RefSeq_annotations/ncbiRefSeq_endAllGenes.sort.bed"):
chrom, start, end, geneID, score, strand = ln.split('\t')
gene= geneID.split(":")[1]
# if "-" in gene:
# gene=gene.split("-")[0]
if gene not in dic_geneS:
dic_geneS[gene]=int(start)
dic_geneE[gene]=int(end)
#list of genes
count_file=open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.fc", "r")
genes=[]
for line , i in enumerate(count_file):
if line > 1:
i_list=i.split()
id=i_list[0]
id_list=id.split(":")
gene=id_list[5]
if gene not in genes:
genes.append(gene)
#make the ind and gene dic
dic_dub={}
for g in genes:
dic_dub[g]={}
for i in inds:
dic_dub[g][i]=0
#populate the dictionary
count_file=open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.fc", "r")
for line, i in enumerate(count_file):
if line > 1:
i_list=i.split()
id=i_list[0]
id_list=id.split(":")
g= id_list[5]
values=list(i_list[6:])
list_list=[]
for ind,val in zip(inds, values):
list_list.append([ind, val])
for num, name in enumerate(list_list):
dic_dub[g][list_list[num][0]] += int(list_list[num][1])
#write the file by acessing the dictionary and putting values in the table ver the value in the dic
fout=open("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.fc","w")
peak=["chrom"]
inds_noL=[]
for each in inds:
indsNA= "NA" + each[:-2]
inds_noL.append(indsNA)
fout.write(" ".join(peak + inds_noL) + '\n' )
count_file=open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.fc", "r")
for line , i in enumerate(count_file):
if line > 1:
i_list=i.split()
id=i_list[0]
id_list=id.split(":")
gene=id_list[5]
#start=dic_geneS[id_list[5]]
start=int(id_list[2])
#end=dic_geneE[id_list[5]]
end=int(id_list[3])
buff=[]
buff.append("chr%s:%d:%d:%s_%s_%s"%(id_list[1], start, end, id_list[5], id_list[4], id_list[0]))
for x,y in zip(i_list[6:], inds):
b=int(dic_dub[gene][y])
t=int(x)
buff.append("%d/%d"%(t,b))
fout.write(" ".join(buff)+ '\n')
fout.close()- convert to usage pheno2CountOnly_genelocAnno_3UTR.R
library(reshape2)
library(tidyverse)
totalPeakUs=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.fc", header = T, stringsAsFactors = F) %>% separate(chrom, sep = ":", into = c("chr", "start", "end", "id")) %>% separate(id, sep="_", into=c("gene", "strand", "peak"))
nuclearPeakUs=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.fc", header = T, stringsAsFactors = F) %>% separate(chrom, sep = ":", into = c("chr", "start", "end", "id")) %>% separate(id, sep="_", into=c("gene", "strand", "peak"))
write.table(totalPeakUs[,7:dim(totalPeakUs)[2]], file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.CountsOnly",quote=FALSE, col.names = F, row.names = F)
write.table(nuclearPeakUs[,7:dim(nuclearPeakUs)[2]], file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.CountsOnly",quote=FALSE, col.names = F, row.names = F)- counts to numeric convertCount2Numeric_noMP_GeneLocAnno_3UTR.py def convert(infile, outfile):
final=open(outfile, "w")
for ln in open(infile, "r"):
line_list=ln.split()
new_list=[]
for i in line_list:
num, dem = i.split("/")
if dem == "0":
perc = "0.00"
else:
perc = int(num)/int(dem)
perc=round(perc,2)
perc= str(perc)
new_list.append(perc)
final.write("\t".join(new_list)+ '\n')
final.close()
convert("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.CountsOnly","/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.CountsOnlyNumeric.txt")
convert("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.CountsOnly","/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.CountsOnlyNumeric.txt")filter_5percUsagePeaks_3UTR.R
library(tidyverse)
totalPeakUs=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.fc", header = T, stringsAsFactors = F) %>% separate(chrom, sep = ":", into = c("chr", "start", "end", "id")) %>% separate(id, sep="_", into=c("gene", "strand", "peak"))
nuclearPeakUs=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.fc", header = T, stringsAsFactors = F) %>% separate(chrom, sep = ":", into = c("chr", "start", "end", "id")) %>% separate(id, sep="_", into=c("gene", "strand", "peak"))
ind=colnames(totalPeakUs)[7:dim(totalPeakUs)[2]]
totalPeakUs_CountNum=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.CountsOnlyNumeric.txt", col.names = ind)
nuclearPeakUs_CountNum=read.table("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.CountsOnlyNumeric.txt", col.names = ind)
#numeric with anno
totalPeak=as.data.frame(cbind(totalPeakUs[,1:6], totalPeakUs_CountNum))
nuclearPeak=as.data.frame(cbind(nuclearPeakUs[,1:6], nuclearPeakUs_CountNum))
#mean
totalPeakUs_CountNum_mean=rowMeans(totalPeakUs_CountNum)
nuclearPeakUs_CountNum_mean=rowMeans(nuclearPeakUs_CountNum)
#append mean to anno
TotalPeakUSMean=as.data.frame(cbind(totalPeakUs[,1:6],totalPeakUs_CountNum_mean))
NuclearPeakUSMean=as.data.frame(cbind(nuclearPeakUs[,1:6],nuclearPeakUs_CountNum_mean))
NuclearPeakUSMean_5perc=NuclearPeakUSMean %>% filter(nuclearPeakUs_CountNum_mean>=.05)
write.table(NuclearPeakUSMean_5perc,file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear.3UTR_fixed.pheno.5percPeaks.txt", row.names=F, col.names=F, quote = F)
TotalPeakUSMean_5per= TotalPeakUSMean %>% filter(totalPeakUs_CountNum_mean>=.05)
write.table(TotalPeakUSMean_5per,file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total.3UTR_fixed.pheno.5percPeaks.txt", row.names=F, col.names=F, quote = F)- run_filter_5percUsagePeaks_3UTR.sh
#!/bin/bash
#SBATCH --job-name=run_filter_5percUsagePeaks_3UTR
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_filter_5percUsagePeaks_3UTR.out
#SBATCH --error=run_filter_5percUsagePeaks_3UTR.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
Rscript filter_5percUsagePeaks_3UTR.RfilterPheno_bothFraction_GeneLocAnno_5perc_3UTR.py
#python
totalokPeaks5perc_file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total.3UTR_fixed.pheno.5percPeaks.txt"
totalokPeaks5perc={}
for ln in open(totalokPeaks5perc_file,"r"):
peakname=ln.split()[5]
totalokPeaks5perc[peakname]=""
nuclearokPeaks5perc_file="/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear.3UTR_fixed.pheno.5percPeaks.txt"
nuclearokPeaks5perc={}
for ln in open(nuclearokPeaks5perc_file,"r"):
peakname=ln.split()[5]
nuclearokPeaks5perc[peakname]=""
totalPhenoBefore=open("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno.fc","r")
totalPhenoAfter=open("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc", "w")
for num, ln in enumerate(totalPhenoBefore):
if num ==0:
totalPhenoAfter.write(ln)
else:
id=ln.split()[0].split(":")[3].split("_")[2]
if id in totalokPeaks5perc.keys():
totalPhenoAfter.write(ln)
totalPhenoAfter.close()
nuclearPhenoBefore=open("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno.fc","r")
nuclearPhenoAfter=open("/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc", "w")
for num, ln in enumerate(nuclearPhenoBefore):
if num ==0:
nuclearPhenoAfter.write(ln)
else:
id=ln.split()[0].split(":")[3].split("_")[2]
if id in nuclearokPeaks5perc.keys():
nuclearPhenoAfter.write(ln)
nuclearPhenoAfter.close() In /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/
#zip file
gzip filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc
gzip filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc
module load python
#leafcutter script
python /project2/gilad/briana/threeprimeseq/code/prepare_phenotype_table.py filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz
python /project2/gilad/briana/threeprimeseq/code/prepare_phenotype_table.py filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz
#source activate three-prime-env
sh filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz_prepare.sh
sh filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz_prepare.sh
#keep only 2 PCs
head -n 3 filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.PCs > filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.2PCs
head -n 3 filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.PCs > filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.2PCsQTL scripts:
Same smaple list
- APAqtl_nominal_GeneLocAnno_noMP_5percUsage_3UTR.sh
#!/bin/bash
#SBATCH --job-name=APAqtl_nominal_GeneLocAnno_noMP_5percUsage
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=APAqtl_nominal_GeneLocAnno_noMP_5percUsage.out
#SBATCH --error=APAqtl_nominal_GeneLocAnno_noMP_5percUsage.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.nominal.out --chunk 1 1 --window 5e5 --include-samples /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs/SAMPLE.txt
done
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.nominal.out --chunk 1 1 --window 5e5 --include-samples /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs/SAMPLE.txt
done- APAqtl_perm_GeneLocAnno_noMP_5percUsage_3UTR.sh
#!/bin/bash
#SBATCH --job-name=APAqtl_perm_GeneLocAnno_noMP_5percUsage
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=APAqtl_perm_GeneLocAnno_noMP_5percUsage.out
#SBATCH --error=APAqtl_perm_GeneLocAnno_noMP_5percUsage.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --permute 1000 --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.perm.out --chunk 1 1 --window 5e5 --include-samples /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs/SAMPLE.txt
done
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --permute 1000 --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc.fc.gz.qqnorm_chr$i.perm.out --chunk 1 1 --window 5e5 --include-samples /project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno_5percUs/SAMPLE.txt
done- run_APAqtlpermCorrectQQplot_GeneLocAnno_noMP_5perUs_3UTR.sh
APAqtlpermCorrectQQplot_GeneLocAnno_noMP_5perUs_3UTR.R
library(dplyr)
##total results
tot.perm= read.table("/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc_permRes.txt",head=F, stringsAsFactors=F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval"))
#BH correction
tot.perm$bh=p.adjust(tot.perm$bpval, method="fdr")
#plot qqplot
png("/project2/gilad/briana/threeprimeseq/output/plots/qqplot_total_APAperm_GeneLocAnno_noMP_5percCov_3UTR.png")
qqplot_total= qqplot(-log10(runif(nrow(tot.perm))), -log10(tot.perm$bpval),ylab="-log10 Total permuted pvalue", xlab="Uniform expectation", main="Total permuted pvalues for all snps\n Gene Loc Anno")
abline(0,1)
dev.off()
#write df with BH
write.table(tot.perm, file = "/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc_permResBH.txt", col.names = T, row.names = F, quote = F)
##nuclear results
nuc.perm= read.table("/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc_permRes.txt",head=F, stringsAsFactors=F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval"))
nuc.perm$bh=p.adjust(nuc.perm$bpval, method="fdr")
#plot qqplot
png("/project2/gilad/briana/threeprimeseq/output/plots/qqplot_nuclear_APAperm_GeneLocAnno_noMP_5percCov_3UTR.png")
qqplot(-log10(runif(nrow(nuc.perm))), -log10(nuc.perm$bpval),ylab="-log10 Nuclear permuted pvalue", xlab="Uniform expectation", main="Nuclear permuted pvalues for all snps \n Gene Loc Anno")
abline(0,1)
dev.off()
# write df with BH
write.table(nuc.perm, file = "/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc_permResBH.txt", col.names = T, row.names = F, quote = F)totQTLs_UTR=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.3UTR.fixed.pheno_5perc_permResBH.txt", stringsAsFactors = F, header=T)
Sig_TotQTLs_UTR= totQTLs_UTR %>% filter(-log10(bh)>=1)
nrow(Sig_TotQTLs_UTR)[1] 114How many genes are tested:
totQTLs_UTR %>% separate(pid, into=c("chr", "start", "end", "id"), sep=":") %>% separate(id, into=c("gene", "strand", "peak"), sep="_") %>% group_by(gene) %>% select(gene) %>% tally() %>% nrow()[1] 6313nucQTLs_UTR=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.3UTR.fixed.pheno_5perc_permResBH.txt", stringsAsFactors = F, header=T)
Sig_NucQTLs_UTR= nucQTLs_UTR %>% filter(-log10(bh)>=1)
nrow(Sig_NucQTLs_UTR)[1] 108How many genes:
nucQTLs_UTR %>% separate(pid, into=c("chr", "start", "end", "id"), sep=":") %>% separate(id, into=c("gene", "strand", "peak"), sep="_") %>% group_by(gene) %>% select(gene) %>% tally() %>% nrow()[1] 6349Diff Isoform Pipeline:
Pipeline comes from New gene Annodation Dif Iso
* filternamePeaks5percCov_GeneLocAnno.py * bothFrac_processed_GeneLocAnno_FC.sh * fix_head_fc_procBothFrac_GeneLocAnno.py * fc2leafphen_processed_GeneLocAnno.py
* subset_diffisopheno_processed_GeneLocAnno.py/ run_subset_diffisopheno_processed_GeneLocAnno.sh * makeLCSampleList_processed_GeneLocAnno.py
* run_leafcutter_ds_bychrom_processed_GeneLocAnno.sh
awk '{if(NR>1)print}' /project2/gilad/briana/threeprimeseq/data/diff_iso_processed_GeneLocAnno/TN_diff_isoform_GeneLocAnno_chr*.txt_effect_sizes.txt > /project2/gilad/briana/threeprimeseq/data/diff_iso_processed_GeneLocAnno/TN_diff_isoform_GeneLocAnno_AllChrom.txt_effect_sizes.txt
awk '{if(NR>1)print}' /project2/gilad/briana/threeprimeseq/data/diff_iso_processed_GeneLocAnno/TN_diff_isoform_GeneLocAnno_chr*cluster_significance.txt > /project2/gilad/briana/threeprimeseq/data/diff_iso_processed_GeneLocAnno/TN_diff_isoform_GeneLocAnno_AllChrom.txt_cluster_significance.txtdiffIso=read.table("../data/diff_iso_GeneLocAnno/TN_diff_isoform_GeneLocAnno_AllChrom.txt_cluster_significance.txt", header = F,col.names = c("status", "loglr", "df", "p", "cluster", "p.adjust"),stringsAsFactors = F,sep="\t") %>% filter(status == "Success")
diffIso$p.adjust=as.numeric(as.character(diffIso$p.adjust))
qqplot(-log10(runif(nrow(diffIso))), -log10(diffIso$p.adjust),ylab="-log10 Total Adjusted Leafcutter pvalue", xlab="-log 10 Uniform expectation", main="Leafcutter differencial isoform analysis between fractions")
abline(0,1)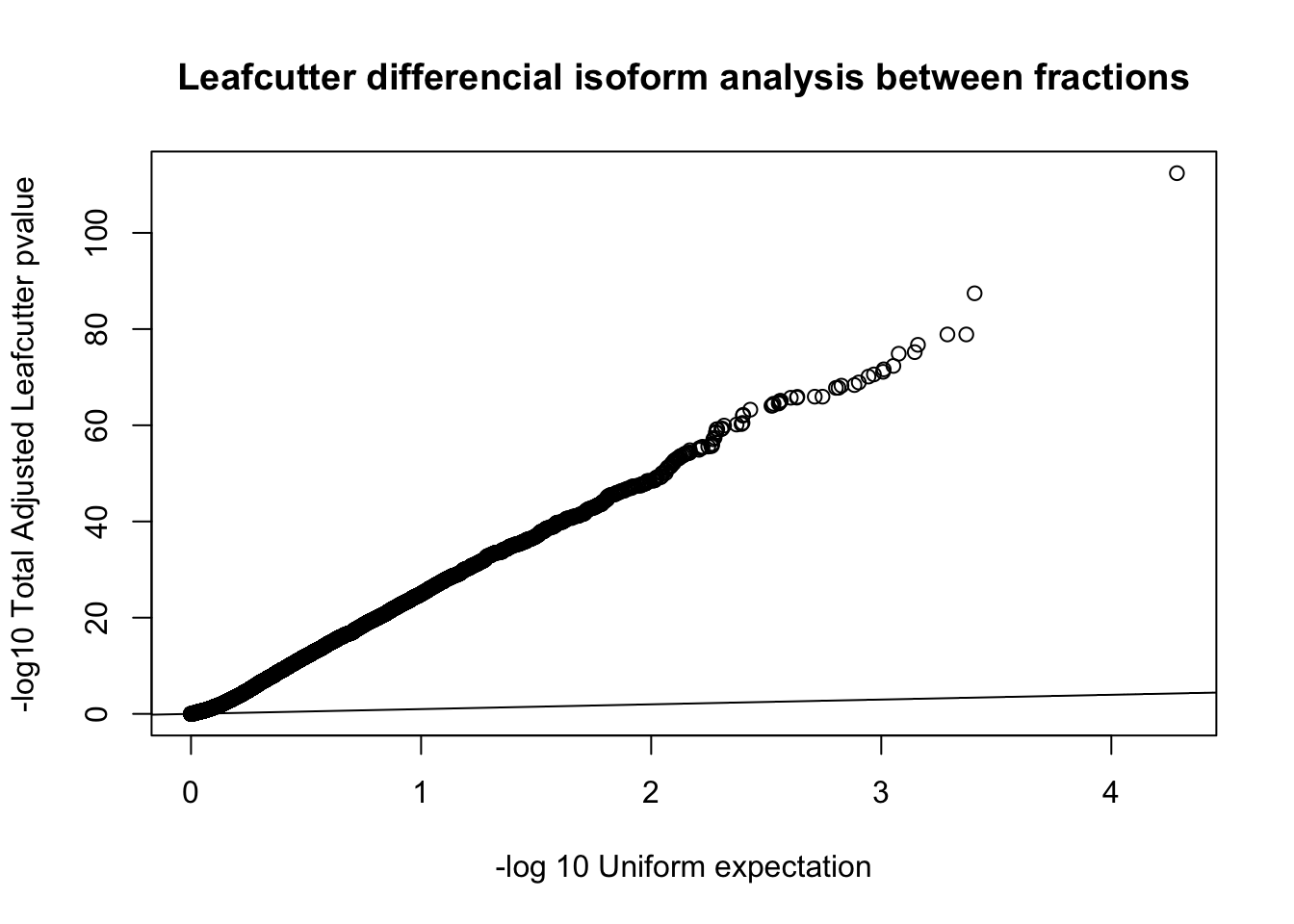
effectsize=read.table("../data/diff_iso_GeneLocAnno/TN_diff_isoform_GeneLocAnno_AllChrom.txt_effect_sizes.txt", stringsAsFactors = F, col.names=c('intron', 'logef' ,'Nuclear', 'Total','deltapsi'))
effectsize$deltapsi=as.numeric(as.character(effectsize$deltapsi))Warning: NAs introduced by coercionfilterHighPSI=effectsize %>% filter(abs(deltapsi)>.4) %>% arrange(deltapsi)
slice(filterHighPSI, 1:10) intron logef Nuclear
1 chr16:81123538:81123627:GCSH -1.79087571426241 0.724883044705174
2 chr3:25705115:25705187:MIR4442 -1.32432047353345 0.660590394608791
3 chr6:84007319:84007404:ME1 -1.68685519425574 0.671530253986571
4 chr19:36806427:36806515:LINC00665 -1.17617088053315 0.809837765186814
5 chr14:47718556:47718593:MDGA2 -1.1825787812967 0.659822642518761
6 chr14:51976014:51976057:FRMD6 -1.56759469994778 0.636611882071359
7 chr20:58647920:58648008:C20orf197 -1.09984402063945 0.691727788494324
8 chr6:37812035:37812094:ZFAND3 -1.61085385492872 0.617570611488878
9 chr8:104953022:104953097:RIMS2 -1.39009184488063 0.611545558430189
10 chr7:147596894:147596932:CNTNAP2 -0.827276301229926 0.72731101101958
Total deltapsi
1 0.151199900846243 -0.5736831
2 0.121029754316441 -0.5395606
3 0.134625920458626 -0.5369043
4 0.28835529406143 -0.5214825
5 0.154119006269487 -0.5057036
6 0.139375449680577 -0.4972364
7 0.199171791192942 -0.4925560
8 0.127058493301525 -0.4905121
9 0.13467815341195 -0.4768674
10 0.259840841892183 -0.4674702Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bindrcpp_0.2.2 workflowr_1.1.1 forcats_0.3.0 stringr_1.3.1
[5] dplyr_0.7.6 purrr_0.2.5 readr_1.1.1 tidyr_0.8.1
[9] tibble_1.4.2 ggplot2_3.0.0 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_0.12.19 cellranger_1.1.0 plyr_1.8.4
[4] compiler_3.5.1 pillar_1.3.0 git2r_0.23.0
[7] bindr_0.1.1 R.methodsS3_1.7.1 R.utils_2.7.0
[10] tools_3.5.1 digest_0.6.17 lubridate_1.7.4
[13] jsonlite_1.5 evaluate_0.11 nlme_3.1-137
[16] gtable_0.2.0 lattice_0.20-35 pkgconfig_2.0.2
[19] rlang_0.2.2 cli_1.0.1 rstudioapi_0.8
[22] yaml_2.2.0 haven_1.1.2 withr_2.1.2
[25] xml2_1.2.0 httr_1.3.1 knitr_1.20
[28] hms_0.4.2 rprojroot_1.3-2 grid_3.5.1
[31] tidyselect_0.2.4 glue_1.3.0 R6_2.3.0
[34] readxl_1.1.0 rmarkdown_1.10 modelr_0.1.2
[37] magrittr_1.5 whisker_0.3-2 backports_1.1.2
[40] scales_1.0.0 htmltools_0.3.6 rvest_0.3.2
[43] assertthat_0.2.0 colorspace_1.3-2 stringi_1.2.4
[46] lazyeval_0.2.1 munsell_0.5.0 broom_0.5.0
[49] crayon_1.3.4 R.oo_1.22.0
This reproducible R Markdown analysis was created with workflowr 1.1.1