APAqtls with Leafcutter
Briana Mittleman
8/15/2018
Last updated: 2018-08-21
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 5ffffe1
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: output/.DS_Store Untracked files: Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed Untracked: analysis/snake.config.notes.Rmd Untracked: data/18486.genecov.txt Untracked: data/APApeaksYL.total.inbrain.bed Untracked: data/Totalpeaks_filtered_clean.bed Untracked: data/YL-SP-18486-T-combined-genecov.txt Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt Untracked: data/bedgraph_peaks/ Untracked: data/bin200.5.T.nuccov.bed Untracked: data/bin200.Anuccov.bed Untracked: data/bin200.nuccov.bed Untracked: data/clean_peaks/ Untracked: data/comb_map_stats.csv Untracked: data/comb_map_stats.xlsx Untracked: data/combined_reads_mapped_three_prime_seq.csv Untracked: data/gencov.test.csv Untracked: data/gencov.test.txt Untracked: data/gencov_zero.test.csv Untracked: data/gencov_zero.test.txt Untracked: data/gene_cov/ Untracked: data/joined Untracked: data/leafcutter/ Untracked: data/merged_combined_YL-SP-threeprimeseq.bg Untracked: data/nuc6up/ Untracked: data/perm_QTL/ Untracked: data/reads_mapped_three_prime_seq.csv Untracked: data/smash.cov.results.bed Untracked: data/smash.cov.results.csv Untracked: data/smash.cov.results.txt Untracked: data/smash_testregion/ Untracked: data/ssFC200.cov.bed Untracked: data/temp.file1 Untracked: data/temp.file2 Untracked: data/temp.gencov.test.txt Untracked: data/temp.gencov_zero.test.txt Untracked: output/picard/ Untracked: output/plots/ Untracked: output/qual.fig2.pdf Unstaged changes: Modified: analysis/28ind.peak.explore.Rmd Modified: analysis/cleanupdtseq.internalpriming.Rmd Modified: analysis/dif.iso.usage.leafcutter.Rmd Modified: analysis/explore.filters.Rmd Modified: analysis/peak.cov.pipeline.Rmd Modified: analysis/pheno.leaf.comb.Rmd Modified: analysis/test.max2.Rmd Modified: code/Snakefile
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 5ffffe1 | brimittleman | 2018-08-21 | BH result plots |
| html | b6e6ed9 | brimittleman | 2018-08-21 | Build site. |
| Rmd | 73516a6 | brimittleman | 2018-08-21 | chr1 results |
| html | d682ab6 | brimittleman | 2018-08-21 | Build site. |
| Rmd | a3c44fb | brimittleman | 2018-08-21 | add code for permute fastqtl |
| html | 5564e25 | brimittleman | 2018-08-20 | Build site. |
| Rmd | 6b1b51c | brimittleman | 2018-08-20 | start qtl analsis, add to index |
I need to run fastQTL to call the apaQTLs.
Imputed snp: /project2/yangili1/tonyzeng/genotyping/imputation_results/ `
module load samtools
#zip file
gzip filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt
module load python
#leafcutter script
python /project2/gilad/briana/threeprimeseq/code/prepare_phenotype_table.py filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz
#source activate three-prime-env
sh filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz_prepare.sh
#run for nuclear as well
gzip filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt
#unload anaconda, load python
python /project2/gilad/briana/threeprimeseq/code/prepare_phenotype_table.py filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz
#load anaconda and env.
sh filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz_prepare.sh
#filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.PCs
#filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.PCs
makeSamplelist.py
#make a sample list
fout = file("/project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/SAMPLE.txt",'w')
for ln in open("/project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/file_id_mapping_nuc.txt", "r"):
bam, sample = ln.split()
line=sample[:-2]
fout.write("NA"+line + "\n")
fout.close()
APAqtl_nominal_nuc.sh
#!/bin/bash
#SBATCH --job-name=APAqtl_nominal_nuc
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=APAqtl_nominal_nuc.out
#SBATCH --error=APAqtl_nominal_nuc.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.qqnorm_chr$i.nominal.out --chunk 1 1 --window 5e4 --include-samples /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/SAMPLE.txt
done
Remove the non matching ind. from the sample list.
Remove 18500, 19092 and 19193, 18497
Try it on the total ones:
APAqtl_nominal_tot.sh
#!/bin/bash
#SBATCH --job-name=APAqtl_nominal_tot
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=APAqtl_nominal_tot.out
#SBATCH --error=APAqtl_nominal_tot.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.qqnorm_chr$i.nominal.out --chunk 1 1 --window 5e4 --include-samples /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/SAMPLE.txt
done
Filter dose files
I need to remove non snps and snps with <.05 from the dosage file.
I will first copy all of the dosage files to my direcory instead of changing tonys.
cp *dose.vcf.gz /project2/gilad/briana/YRI_geno_hg19/I want to write a python script that will read in the files and perform the filters.
I wrote a python script that take in the dose file and a name of an out file. I will write a bash script to wrap this on all of the chrs.
#!/bin/bash
#SBATCH --job-name=filter_dose
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=filter_dose.out
#SBATCH --error=filter_dose.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
module load python
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
python filter_vcf.py chr$i.dose.vcf chr$i.dose.filt.vcf
doneNow I can use these for the fastqtl script instead.
I also updated to only use the first 2 pcs as covariates.
Run permuted version
Permutation pass to calculate correctedp-values for molecular phenotypes.
APAqtl_perm_tot.sh
#!/bin/bash
#SBATCH --job-name=APAqtl_perm_tot
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=APAqtl_perm_tot.out
#SBATCH --error=APAqtl_perm_tot.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --permute 1000 --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/perm_APAqtl/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.qqnorm_chr$i.perm.out --chunk 1 1 --window 5e4 --include-samples /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/SAMPLE.txt
done
APAqtl_perm_nuc.sh
#!/bin/bash
#SBATCH --job-name=APAqtl_nominal_nuc
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=APAqtl_perm_nuc.out
#SBATCH --error=APAqtl_perm_nuc.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --permute 1000 --vcf /project2/gilad/briana/YRI_geno_hg19/chr$i.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.2PCs --bed /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.qqnorm_chr$i.gz --out /project2/gilad/briana/threeprimeseq/data/perm_APAqtl/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.qqnorm_chr$i.perm.out --chunk 1 1 --window 5e4 --include-samples /project2/gilad/briana/threeprimeseq/data/filt_peak_refGene_cov/SAMPLE.txt
done
The results file has the folowing columns:
- ID of the tested molecular phenotype (in this particular case, the gene ID)
- Number of variants tested in cis for this phenotype
- MLE of the shape1 parameter of the Beta distribution
- MLE of the shape2 parameter of the Beta distribution
- Dummy [To be described later]
- ID of the best variant found for this molecular phenotypes (i.e. with the smallest p-value)
- Distance between the molecular phenotype - variant pair
- The nominal p-value of association that quantifies how significant from 0, the regression coefficient is
- The slope associated with the nominal p-value of association [only in version > v2-184]
- A first permutation p-value directly obtained from the permutations with the direct method. This is basically a corrected version of the nominal p-value that accounts for the fact that multiple variants are tested per molecular phenotype.
- A second permutation p-value obtained via beta approximation. We advice to use this one in any downstream analysis.
I can check the experiments as recomended by the FastQTL site.
d = read.table("permutations.all.chunks.txt.gz", hea=F, stringsAsFactors=F)
colnames(d) = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "ppval", "bpval")
plot(d$ppval, d$bpval, xlab="Direct method", ylab="Beta approximation", main="Check plot")
abline(0, 1, col="red")I will try this first on the resutls from chr1.
nuc.chr1= read.table("../data/perm_QTL/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear.txt.gz.qqnorm_chr1.perm.out",head=F, stringsAsFactors=F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval"))
plot(nuc.chr1$ppval, nuc.chr1$bpval, xlab="Direct method", ylab="Beta approximation", main="Nuclear Check plot")
abline(0, 1, col="red")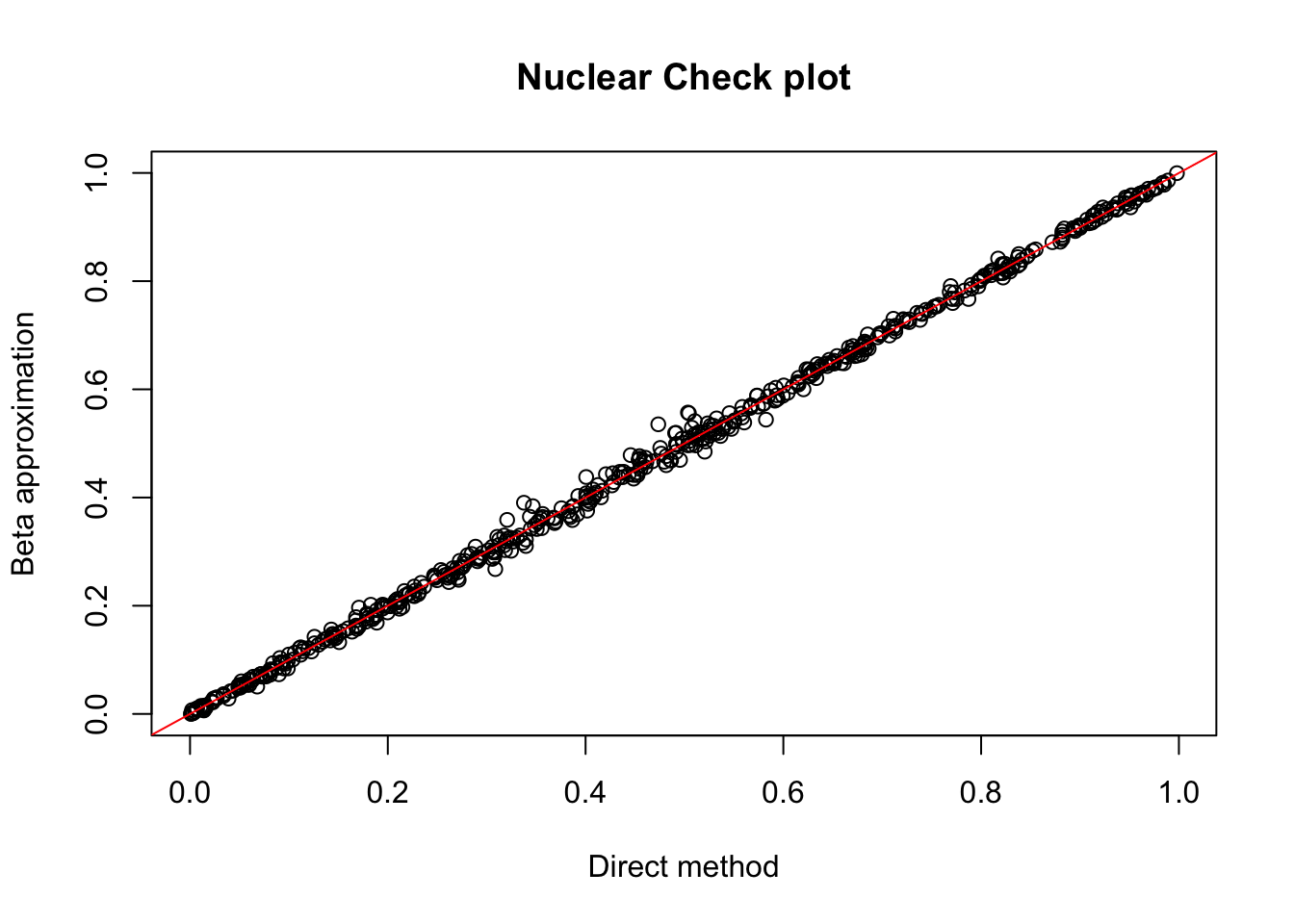
Expand here to see past versions of unnamed-chunk-10-1.png:
| Version | Author | Date |
|---|---|---|
| b6e6ed9 | brimittleman | 2018-08-21 |
tot.chr1=read.table("../data/perm_QTL/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total.txt.gz.qqnorm_chr1.perm.out", head=F, stringsAsFactors = F, col.names= c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval"))
plot(tot.chr1$ppval, tot.chr1$bpval, xlab="Direct method", ylab="Beta approximation", main="Total Check plot")
abline(0, 1, col="red")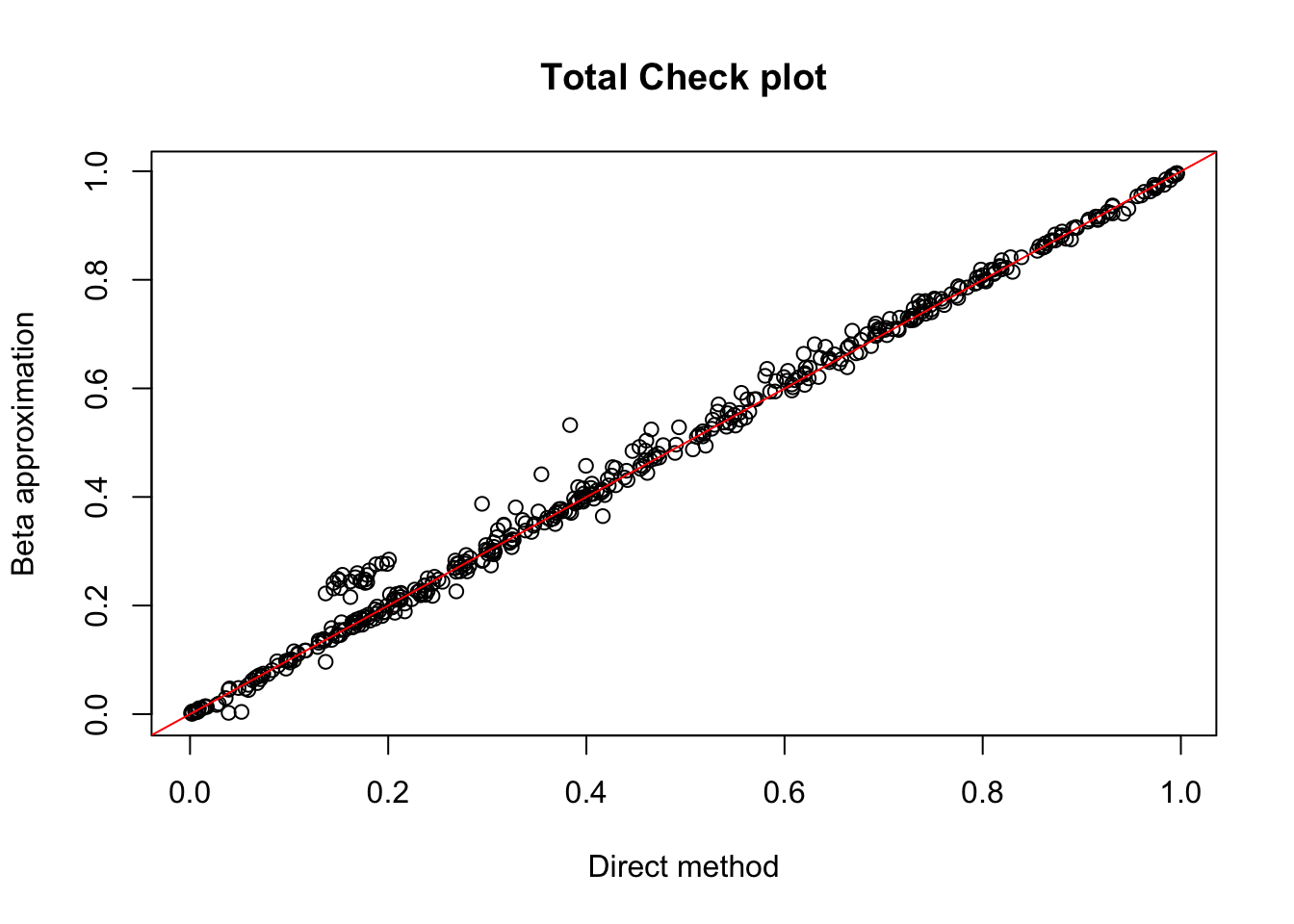
Expand here to see past versions of unnamed-chunk-10-2.png:
| Version | Author | Date |
|---|---|---|
| b6e6ed9 | brimittleman | 2018-08-21 |
Correct for multiple testing:
- Bonferonni
nuc.chr1$bonferroni = p.adjust(nuc.chr1$bpval, method="bonferroni")
plot(-log10(nuc.chr1$bonferroni), main="Nuclear chr1 bonferroni corrected pval")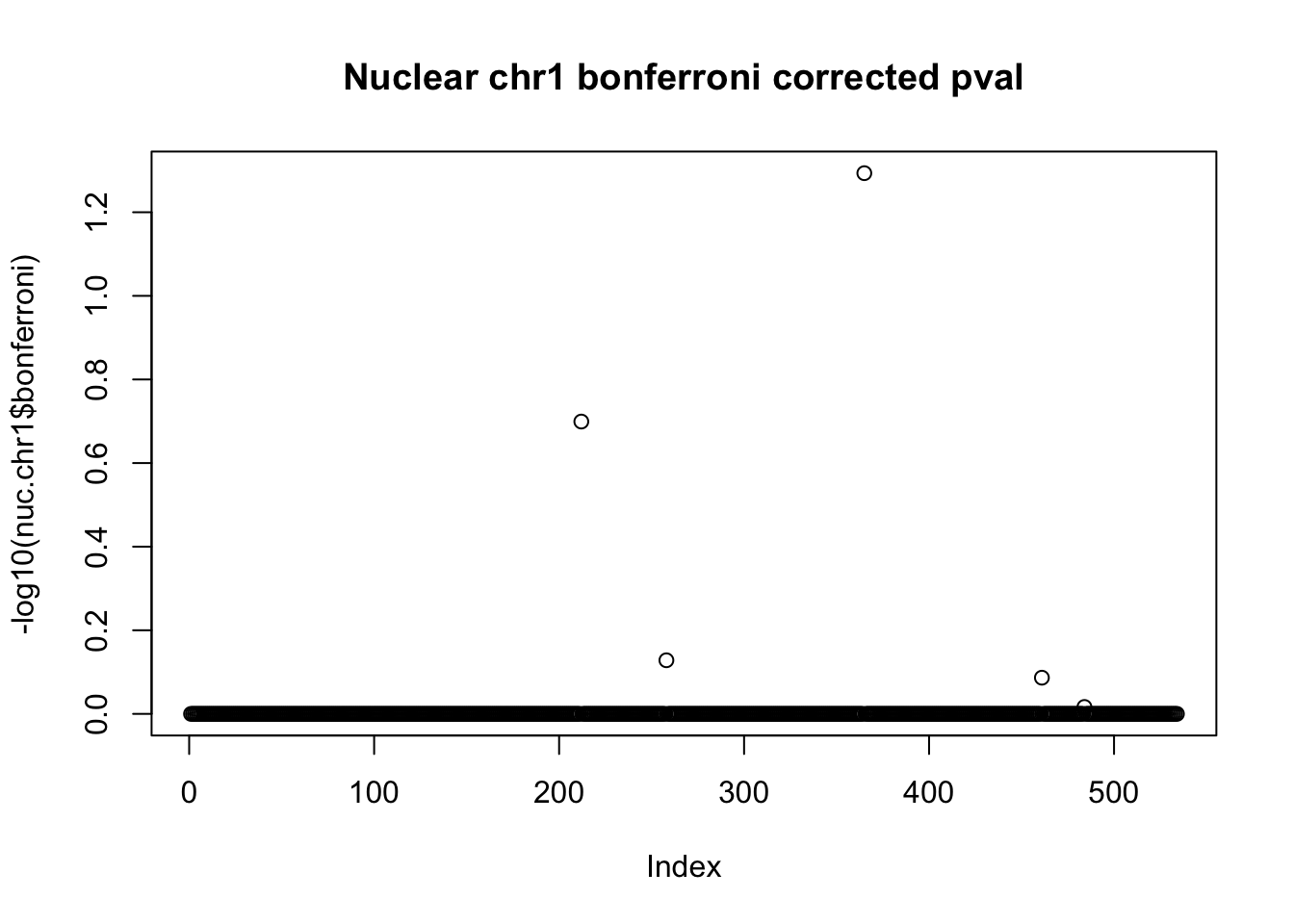
Expand here to see past versions of unnamed-chunk-11-1.png:
| Version | Author | Date |
|---|---|---|
| b6e6ed9 | brimittleman | 2018-08-21 |
tot.chr1$bonferroni = p.adjust(tot.chr1$bpval, method="bonferroni")
plot(-log10(tot.chr1$bonferroni), main="Total chr1 bonferroni corrected pval")
Expand here to see past versions of unnamed-chunk-11-2.png:
| Version | Author | Date |
|---|---|---|
| b6e6ed9 | brimittleman | 2018-08-21 |
< .05 is 1.3 on this plot.
- BH
nuc.chr1$bh=p.adjust(nuc.chr1$bpval, method="fdr")
plot(-log10(nuc.chr1$bh), main="Nuclear chr1 BH corrected pval")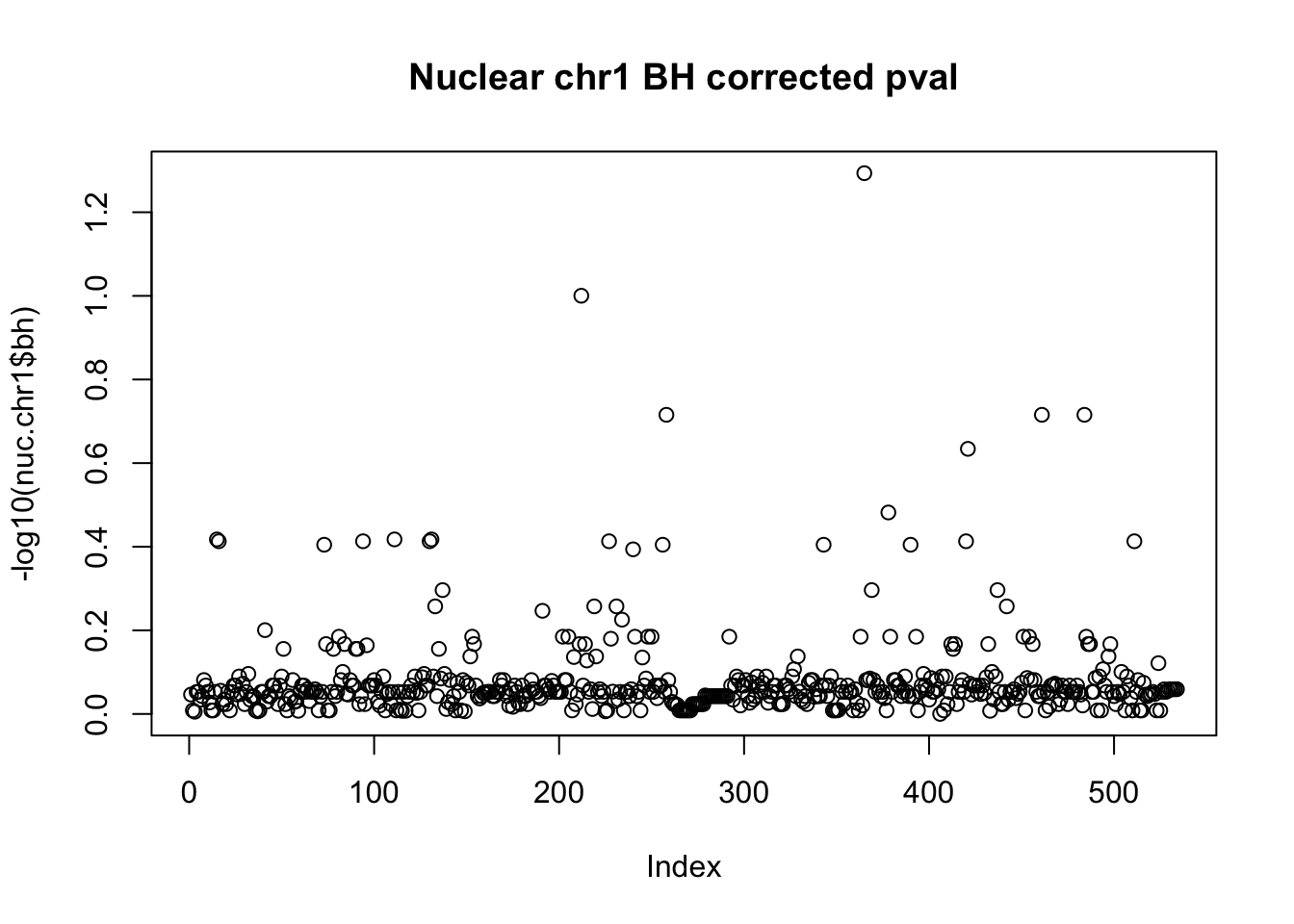
Expand here to see past versions of unnamed-chunk-12-1.png:
| Version | Author | Date |
|---|---|---|
| b6e6ed9 | brimittleman | 2018-08-21 |
tot.chr1$bh=p.adjust(tot.chr1$bpval, method="fdr")
plot(-log10(tot.chr1$bh), main="Total chr1 BH corrected pval")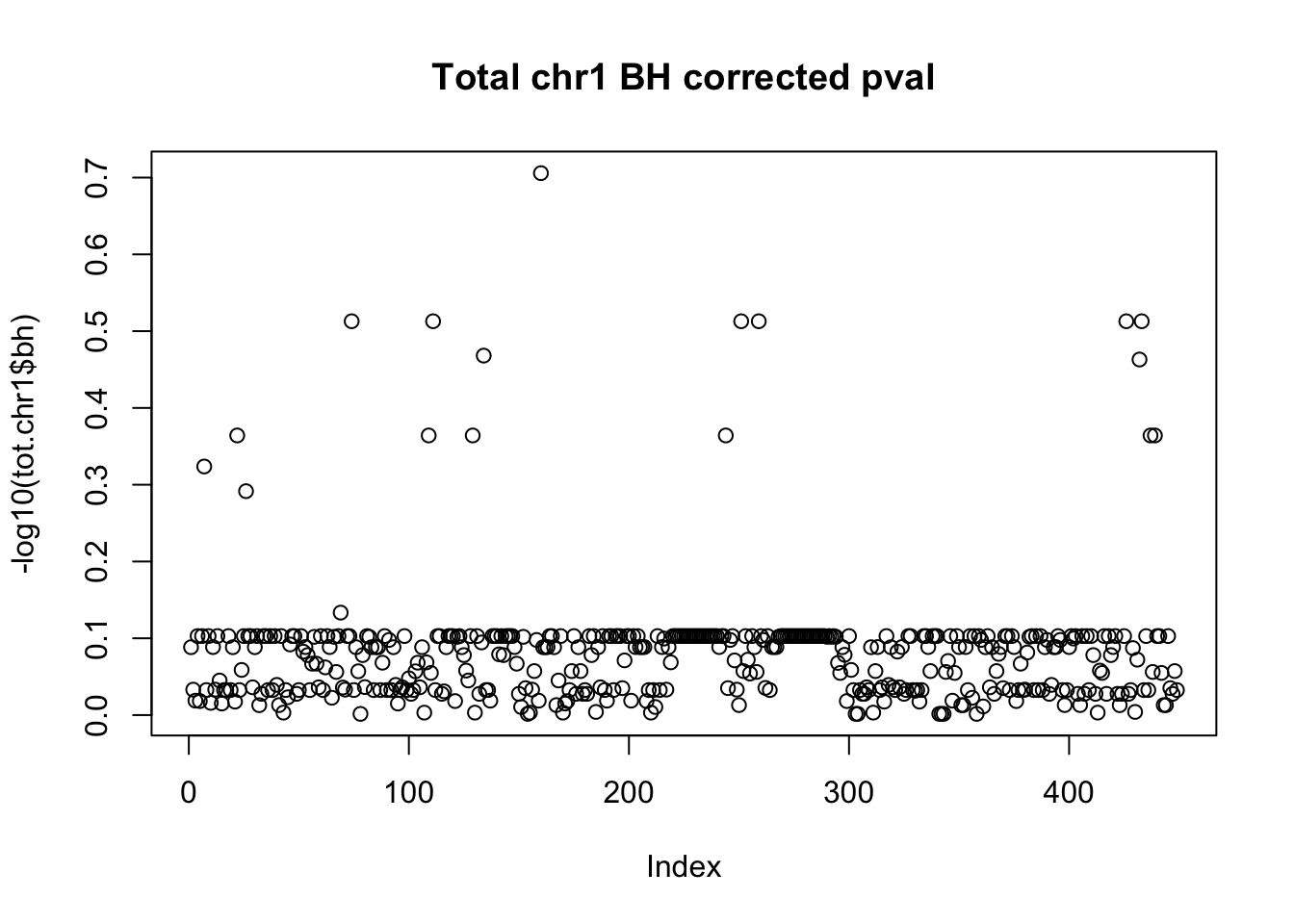
Expand here to see past versions of unnamed-chunk-12-2.png:
| Version | Author | Date |
|---|---|---|
| b6e6ed9 | brimittleman | 2018-08-21 |
10% FDR is 1 on this plot.
Extend to all results:
nuc.res= read.table("../data/perm_QTL/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_permQTLresults.out",head=F, stringsAsFactors=F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval"))
plot(nuc.res$ppval, nuc.res$bpval, xlab="Direct method", ylab="Beta approximation", main="Nuclear Check plot")
abline(0, 1, col="red")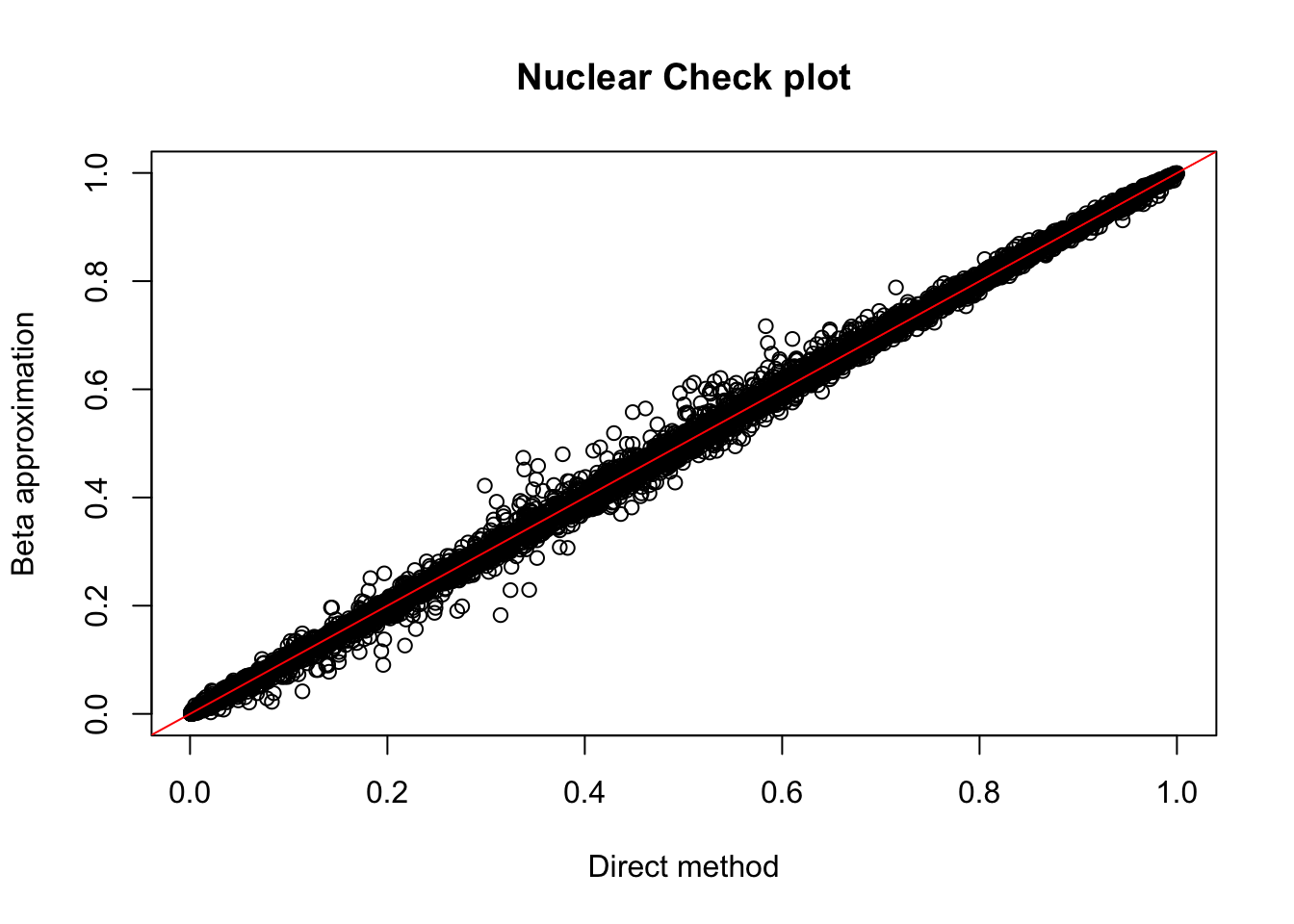
tot.res=read.table("../data/perm_QTL/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_permQTLresults.out", head=F, stringsAsFactors = F, col.names= c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval"))
plot(tot.res$ppval, tot.res$bpval, xlab="Direct method", ylab="Beta approximation", main="Total Check plot")
abline(0, 1, col="red")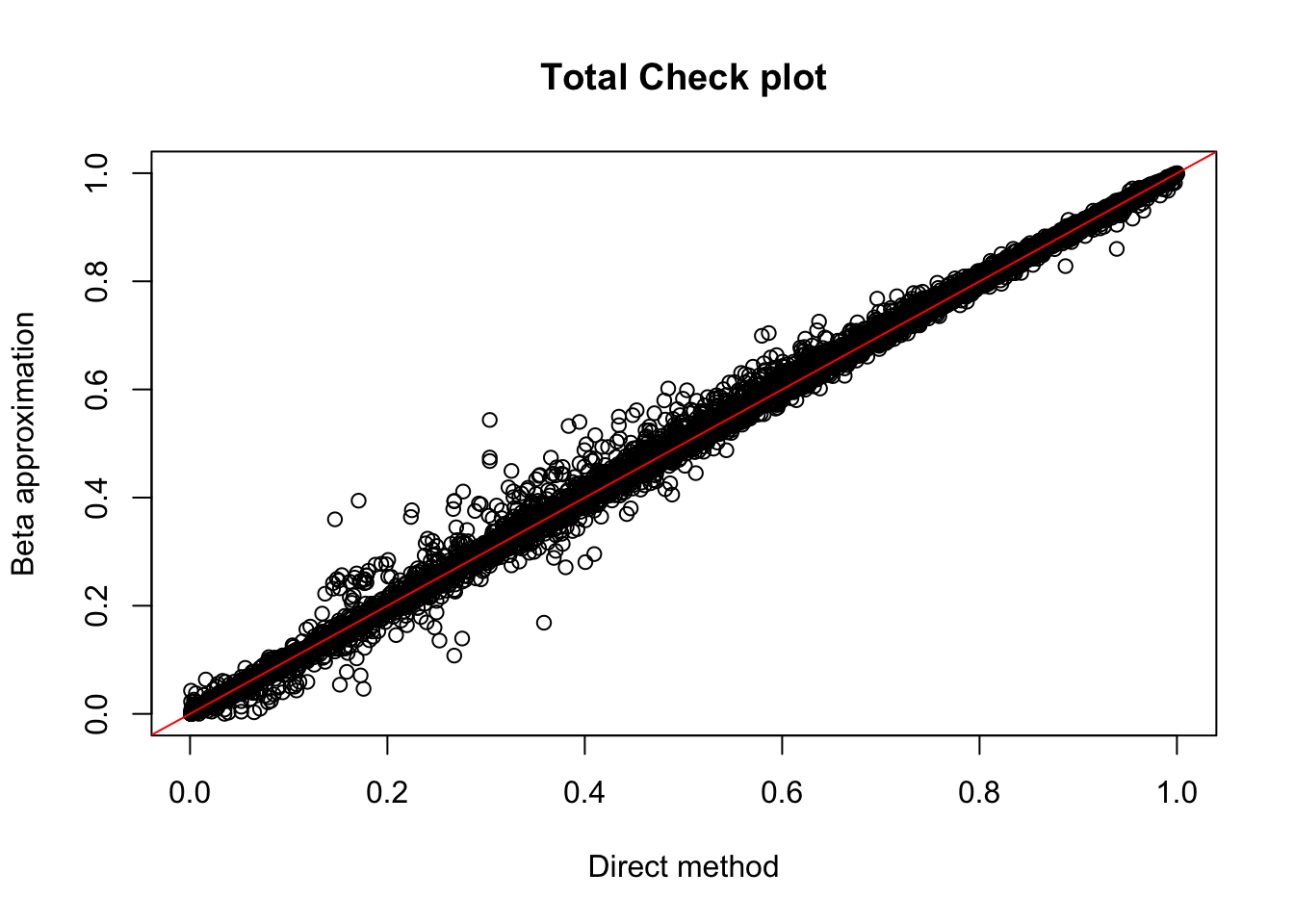
- BH
nuc.res$bh=p.adjust(nuc.res$bpval, method="fdr")
plot(-log10(nuc.res$bh), main="Nuclear BH corrected pval")
abline(h=1, col="red")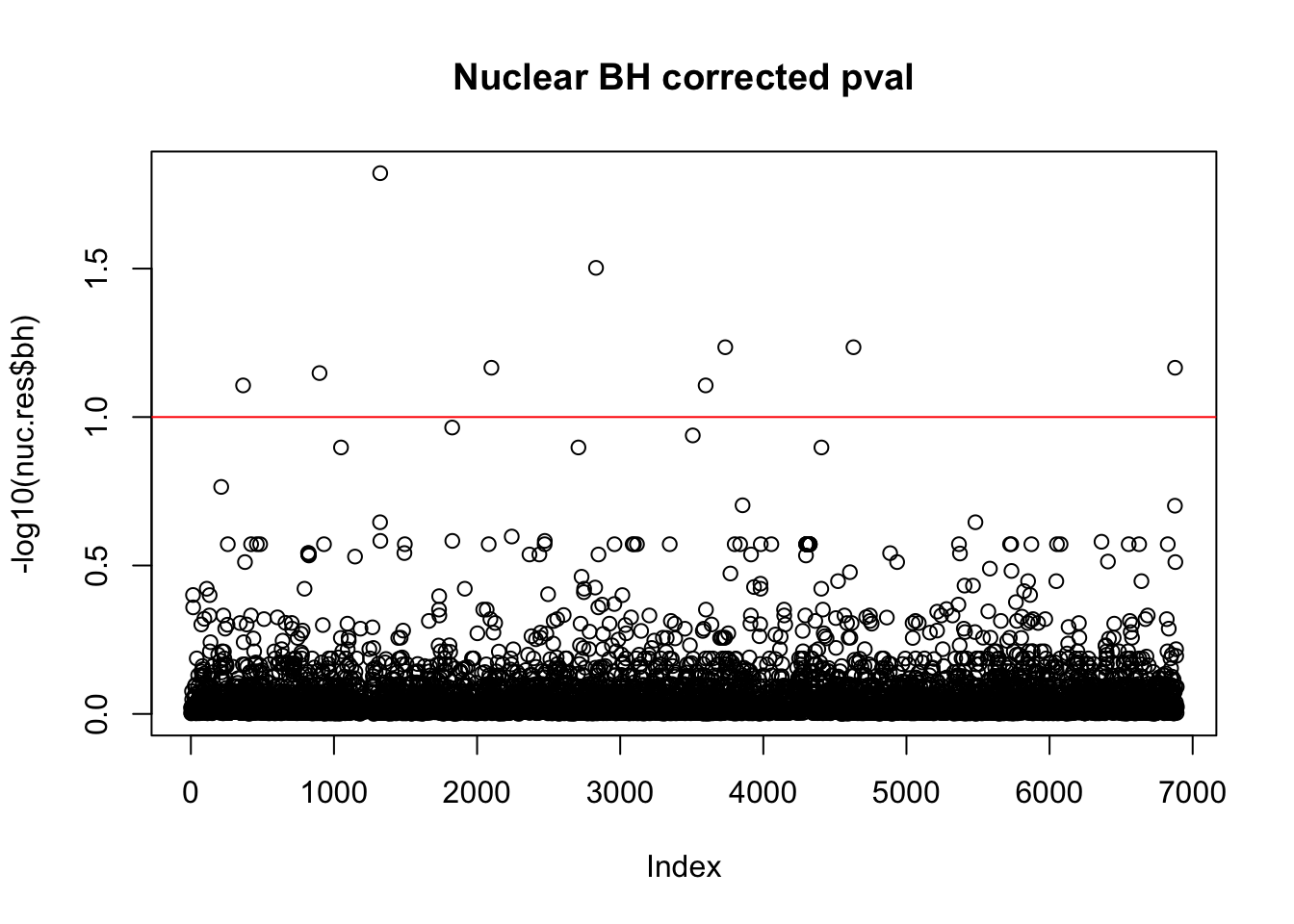
tot.res$bh=p.adjust(tot.res$bpval, method="fdr")
plot(-log10(tot.res$bh), main="Total BH corrected pval")
abline(h=1, col="red")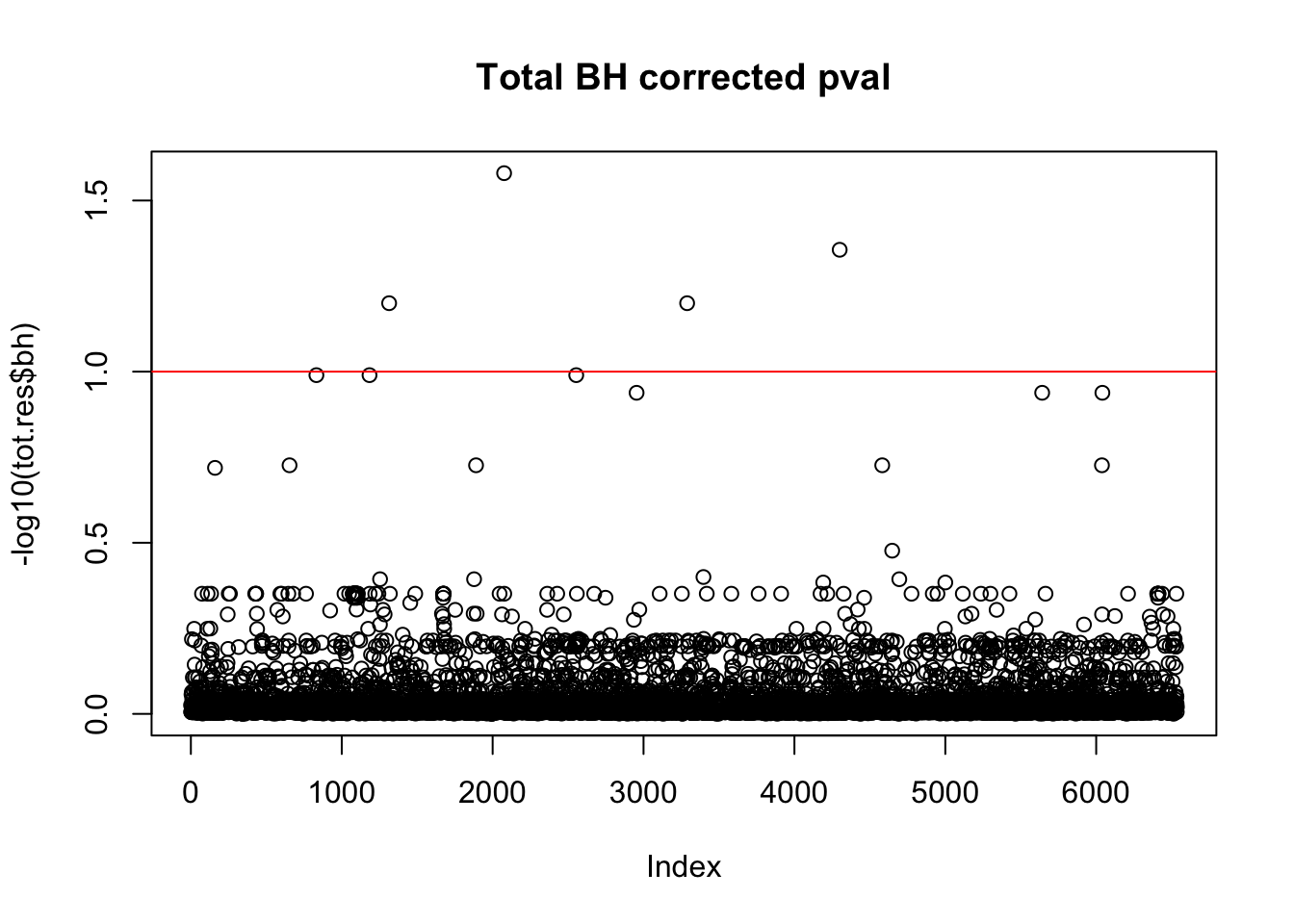
Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] workflowr_1.1.1 Rcpp_0.12.18 digest_0.6.15
[4] rprojroot_1.3-2 R.methodsS3_1.7.1 backports_1.1.2
[7] git2r_0.23.0 magrittr_1.5 evaluate_0.11
[10] stringi_1.2.4 whisker_0.3-2 R.oo_1.22.0
[13] R.utils_2.6.0 rmarkdown_1.10 tools_3.5.1
[16] stringr_1.3.1 yaml_2.1.19 compiler_3.5.1
[19] htmltools_0.3.6 knitr_1.20
This reproducible R Markdown analysis was created with workflowr 1.1.1