Filter Reads Mapping to A’s
Briana Mittleman
6/18/2018
Last updated: 2019-02-15
Checks: 6 0
Knit directory: threeprimeseq/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: data/perm_QTL_trans_noMP_5percov/
Ignored: output/.DS_Store
Untracked files:
Untracked: KalistoAbundance18486.txt
Untracked: analysis/4suDataIGV.Rmd
Untracked: analysis/DirectionapaQTL.Rmd
Untracked: analysis/EvaleQTLs.Rmd
Untracked: analysis/YL_QTL_test.Rmd
Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed
Untracked: analysis/snake.config.notes.Rmd
Untracked: analysis/verifyBAM.Rmd
Untracked: analysis/verifybam_dubs.Rmd
Untracked: code/PeaksToCoverPerReads.py
Untracked: code/strober_pc_pve_heatmap_func.R
Untracked: data/18486.genecov.txt
Untracked: data/APApeaksYL.total.inbrain.bed
Untracked: data/ApaQTLs/
Untracked: data/ChromHmmOverlap/
Untracked: data/DistTXN2Peak_genelocAnno/
Untracked: data/GM12878.chromHMM.bed
Untracked: data/GM12878.chromHMM.txt
Untracked: data/LianoglouLCL/
Untracked: data/LocusZoom/
Untracked: data/NuclearApaQTLs.txt
Untracked: data/PeakCounts/
Untracked: data/PeakCounts_noMP_5perc/
Untracked: data/PeakCounts_noMP_genelocanno/
Untracked: data/PeakUsage/
Untracked: data/PeakUsage_noMP/
Untracked: data/PeakUsage_noMP_GeneLocAnno/
Untracked: data/PeaksUsed/
Untracked: data/PeaksUsed_noMP_5percCov/
Untracked: data/RNAkalisto/
Untracked: data/RefSeq_annotations/
Untracked: data/TotalApaQTLs.txt
Untracked: data/Totalpeaks_filtered_clean.bed
Untracked: data/UnderstandPeaksQC/
Untracked: data/WASP_STAT/
Untracked: data/YL-SP-18486-T-combined-genecov.txt
Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt
Untracked: data/YL_QTL_test/
Untracked: data/apaExamp/
Untracked: data/apaQTL_examp_noMP/
Untracked: data/bedgraph_peaks/
Untracked: data/bin200.5.T.nuccov.bed
Untracked: data/bin200.Anuccov.bed
Untracked: data/bin200.nuccov.bed
Untracked: data/clean_peaks/
Untracked: data/comb_map_stats.csv
Untracked: data/comb_map_stats.xlsx
Untracked: data/comb_map_stats_39ind.csv
Untracked: data/combined_reads_mapped_three_prime_seq.csv
Untracked: data/diff_iso_GeneLocAnno/
Untracked: data/diff_iso_proc/
Untracked: data/diff_iso_trans/
Untracked: data/ensemble_to_genename.txt
Untracked: data/example_gene_peakQuant/
Untracked: data/explainProtVar/
Untracked: data/filtPeakOppstrand_cov_noMP_GeneLocAnno_5perc/
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed
Untracked: data/first50lines_closest.txt
Untracked: data/gencov.test.csv
Untracked: data/gencov.test.txt
Untracked: data/gencov_zero.test.csv
Untracked: data/gencov_zero.test.txt
Untracked: data/gene_cov/
Untracked: data/joined
Untracked: data/leafcutter/
Untracked: data/merged_combined_YL-SP-threeprimeseq.bg
Untracked: data/molPheno_noMP/
Untracked: data/mol_overlap/
Untracked: data/mol_pheno/
Untracked: data/nom_QTL/
Untracked: data/nom_QTL_opp/
Untracked: data/nom_QTL_trans/
Untracked: data/nuc6up/
Untracked: data/nuc_10up/
Untracked: data/other_qtls/
Untracked: data/pQTL_otherphen/
Untracked: data/peakPerRefSeqGene/
Untracked: data/perm_QTL/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/
Untracked: data/perm_QTL_opp/
Untracked: data/perm_QTL_trans/
Untracked: data/perm_QTL_trans_filt/
Untracked: data/protAndAPAAndExplmRes.Rda
Untracked: data/protAndAPAlmRes.Rda
Untracked: data/protAndExpressionlmRes.Rda
Untracked: data/reads_mapped_three_prime_seq.csv
Untracked: data/smash.cov.results.bed
Untracked: data/smash.cov.results.csv
Untracked: data/smash.cov.results.txt
Untracked: data/smash_testregion/
Untracked: data/ssFC200.cov.bed
Untracked: data/temp.file1
Untracked: data/temp.file2
Untracked: data/temp.gencov.test.txt
Untracked: data/temp.gencov_zero.test.txt
Untracked: data/threePrimeSeqMetaData.csv
Untracked: data/threePrimeSeqMetaData55Ind.txt
Untracked: data/threePrimeSeqMetaData55Ind.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.xlsx
Untracked: output/picard/
Untracked: output/plots/
Untracked: output/qual.fig2.pdf
Unstaged changes:
Modified: analysis/28ind.peak.explore.Rmd
Modified: analysis/CompareLianoglouData.Rmd
Modified: analysis/NewPeakPostMP.Rmd
Modified: analysis/apaQTLoverlapGWAS.Rmd
Modified: analysis/cleanupdtseq.internalpriming.Rmd
Modified: analysis/coloc_apaQTLs_protQTLs.Rmd
Modified: analysis/dif.iso.usage.leafcutter.Rmd
Modified: analysis/diff_iso_pipeline.Rmd
Modified: analysis/explainpQTLs.Rmd
Modified: analysis/explore.filters.Rmd
Modified: analysis/flash2mash.Rmd
Modified: analysis/mispriming_approach.Rmd
Modified: analysis/overlapMolQTL.Rmd
Modified: analysis/overlapMolQTL.opposite.Rmd
Modified: analysis/overlap_qtls.Rmd
Modified: analysis/peakOverlap_oppstrand.Rmd
Modified: analysis/peakQCPPlots.Rmd
Modified: analysis/pheno.leaf.comb.Rmd
Modified: analysis/pipeline_55Ind.Rmd
Modified: analysis/swarmPlots_QTLs.Rmd
Modified: analysis/test.max2.Rmd
Modified: analysis/test.smash.Rmd
Modified: analysis/understandPeaks.Rmd
Modified: code/Snakefile
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | ee5c8e3 | Briana Mittleman | 2018-06-26 | Build site. |
| Rmd | e344b95 | Briana Mittleman | 2018-06-26 | final notes |
| html | 61338d7 | Briana Mittleman | 2018-06-20 | Build site. |
| Rmd | a0f23a4 | Briana Mittleman | 2018-06-20 | add stats for all lines and baseline analysis |
| html | d5ac83f | Briana Mittleman | 2018-06-19 | Build site. |
| Rmd | 478db0c | Briana Mittleman | 2018-06-19 | filter 2 samples for 6 As |
| html | 30b415e | Briana Mittleman | 2018-06-19 | Build site. |
| Rmd | 2f53108 | Briana Mittleman | 2018-06-19 | filter A code |
I will use this analysis to develop a filtering method to filter reads that map to genomic locations with PolyA stretches. These reads could be due to priming of the poly dT primer in the protocol rather than actual polyA tails. This will be a problem for our differential APA analysis between total and nuclear RNA if mis primming is more likely to happen in the nuclear fraction. I am adapting a script by Ankeeta Shah to detect misprimming in coelesce seq. The script uses the python package pysam to work with bam files in python like samtools.
Coelesce seq script
#!/usr/bin/env python
"""
Usage: python extractReadsWithMismatchesin6FirstNct_noS.py <input_bam> <output_bam>
"""
import sys, pysam, re
iBAM = pysam.Samfile(sys.argv[1], 'r') # reads from the standard input
oBAM = pysam.Samfile(sys.argv[2], 'w', template=iBAM) # output
for line in iBAM:
if (line.is_read2): #for paired end reads, if mate 2
string = line.cigarstring
regex=re.compile('^[0-9]*M') #only look for reads that have M (meaning match or mismatch) at the front of the cigar string
if re.match(regex, string):
md=re.findall(r'\d+', [tag[1] for tag in line.tags if tag[0]=='MD'][0]) #get md tag
if len(md) == 1 : # if there are no mismatches
oBAM.write(line) # write the alignment into the output file
else:
if (not line.is_reverse) and (int(md[0]) >= 6): # if the first mismatch occurs after the 6th nt (from the 5' end)
oBAM.write(line) # write the alignment into the output file
elif (line.is_reverse) and (int(md[-1]) >= 6): # same as above but for reads that align to the reverse strand
oBAM.write(line)
# close files
iBAM.close()
oBAM.close()I need to make the following changes to this script:
Remove first if statement because I do not have paired end reads
get all of the places that have an M in the cigar string. Then look at the one with the longest integer attached. This will correspond to the longest region of the read mapping.
Add a reg exp. to check if mapping region includes 6 A’s.
This should write out a bam with just the reads mapping to 6 A’s.
Update Script
#!/usr/bin/env python
"""
Usage: python filter6As.py <input_bam> <output_bam>
"""
import sys, pysam, re
iBAM = pysam.Samfile(sys.argv[1], 'r') # reads from the standard input
oBAM = pysam.Samfile(sys.argv[2], 'w', template=iBAM) # output
for line in iBAM:
string = line.cigarstring
regex=re.compile('[0-9]*M') #only look for reads that have M (meaning match or mismatch) at the front of the cigar string
test.string="AAAAAA"
if len(re.findall(regex, string))>=1:
#find the logest mapping string
match=re.findall(regex, string)
maxM=0
matchind=0
numM=re.compile('[0-9]*')
for M in range(len(match)):
if re.findall(numM,match[M]) > maxM:
maxM= re.findall(numM,match[M])
matchind=M
longestmatch=match[M]
#query_alignment_sequence
md=re.findall(r'\d+', [tag[1] for tag in line.tags if tag[0]=='MD'][0]) #get md tag
if len(md) == 1 : # if there are no mismatches
oBAM.write(line) # write the alignment into the output file
<!-- else: -->
<!-- if (not line.is_reverse) and (int(md[0]) >= 6): # if the first mismatch occurs after the 6th nt (from the 5' end) -->
<!-- oBAM.write(line) # write the alignment into the output file -->
<!-- elif (line.is_reverse) and (int(md[-1]) >= 6): # same as above but for reads that align to the reverse strand -->
<!-- oBAM.write(line) -->
# close files
iBAM.close()
oBAM.close()Try to not use the cigar string method. Just look at the mapped reads.
#!/usr/bin/env python
"""
Usage: python filter6As.py <input_bam> <output_bam>
"""
import sys, pysam, re
iBAM = pysam.Samfile('/project2/gilad/briana/threeprimeseq/data/sort/YL-SP-19257-T_S25_R1_001-sort.bam', 'r') # reads from the standard input
oBAM = pysam.Samfile('test.bam', 'w', template=iBAM) # output
for line in iBAM:
seq=line.query_alignment_sequence
Aseq=re.compile("AAAAAA")
if len(re.findall(Aseq, seq))>=1:
oBAM.write(line)
iBAM.close()
oBAM.close()What I need to do is combine both of these ideas. I need to test for mismatches using the cigar string, then extract the sequence and test for the multiple As in that section. I could seperate the alligned sequence and the coresponding cigar sequence into a list of tuples. Then I can find the largest mapping section, test for the mismatches and sequence of AAAAAs in this section.
Try on /project2/gilad/briana/threeprimeseq/data/sort/YL-SP-19257-T_S25_R1_001-sort.bam
Alternative method- 6 bases upstream:
An alternative way to think about this is that we expect directly upstream of the read to be 6 A’s. I am going to write a script that changes the bed file to give me the 6 basepairs before the read. This is start -6 to start on the fwd strand and end to encd +6 on rhe reverse strand. I can then use the bedtools nuc tool for these. I will filter the lines that have 100% As on the fwd strand and 100% Ts on the rev strand.
Script to look at positions upstream 6 bases. 6up_bed.sh
#!/bin/bash
#SBATCH --job-name=6upbed
#SBATCH --time=8:00:00
#SBATCH --output=6upbed.out
#SBATCH --error=6upbed.err
#SBATCH --account=pi-yangili1
#SBATCH --partition=broadwl
#SBATCH --mem=20G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#imput sorted bed file
bed=$1
describer=$(echo ${bed} | sed -e 's/.*\YL-SP-//' | sed -e "s/-sort.bed$//")
awk '{if($6== "+") print($1 "\t" $2-6 "\t" $2 "\t" $4 "\t" $5 "\t" $6 ); else print($1 "\t" $3 "\t" $3 + 6 "\t" $4 "\t" $5 "\t" $6)}' $1 | awk '{if($2 <0) print($1 "\t" "0" "\t" $3 "\t" $4 "\t" $5 "\t" $6) ; else print($1 "\t" $2 "\t" $3"\t" $4 "\t" $5 "\t" $6)}' > /project2/gilad/briana/threeprimeseq/data/bed_6up/sixup.${describer}.6up.sort.bed
Write wrapper w_6up.sh:
#!/bin/bash
#SBATCH --job-name=w_6up
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=w_6up.out
#SBATCH --error=w_6up.err
#SBATCH --partition=broadwl
#SBATCH --mem=8G
#SBATCH --mail-type=END
for i in $(ls /project2/gilad/briana/threeprimeseq/data/bed_sort/*.bed); do
sbatch 6up_bed.sh $i
doneThe problem is adding 6 on the end goes outisde the boundaries of the chromosome. I need the lengths of the chromosomes and I need to check for this when I make the file.
I can intersect these files with a bed file with the chromosome lengths then only keep the ones that fully intersect.
The chromosome lengths are in /project2/gilad/briana/genome_anotation_data/chrom_lengths.sort.bed
Intersect with this bed and only keep lines that are 100% in the chrom lengths. a = 6up b= /project2/gilad/briana/genome_anotation_data/chrom_lengths.sort.bed -sorted -wa -f require minimum overlap fraction (1)
#!/bin/bash
#SBATCH --job-name=intchrom
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=intchrom.out
#SBATCH --error=intchrom.err
#SBATCH --partition=broadwl
#SBATCH --mem=20G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#imput 6up sorted bed file
bed=$1
describer=$(echo ${bed} | sed -e 's/.*sixup.//' | sed -e "s/.6up.sort.bed$//")
bedtools intersect -wa -f 1 -a $1 -b /project2/gilad/briana/genome_anotation_data/chrom_lengths2.sort.bed > /project2/gilad/briana/threeprimeseq/data/bed_6upint/sixupint.${describer}.6up.sort.int.bed
wrap function
#!/bin/bash
#SBATCH --job-name=w_int
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=w_int.out
#SBATCH --error=w_int.err
#SBATCH --partition=broadwl
#SBATCH --mem=8G
#SBATCH --mail-type=END
for i in $(ls /project2/gilad/briana/threeprimeseq/data/bed_6up/*.bed); do
sbatch int_chrom.sh $i
doneWrite the nuc script:
#!/bin/bash
#SBATCH --job-name=nuc6up
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=nuc6up.out
#SBATCH --error=nuc6up.err
#SBATCH --partition=broadwl
#SBATCH --mem=20G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#imput 6up sorted bed file
bed=$1
describer=$(echo ${bed} | sed -e 's/.*sixupint.//' | sed -e "s/.6up.sort.int.bed$//")
bedtools nuc -s -fi /project2/gilad/briana/genome_anotation_data/genome/Homo_sapiens.GRCh37.75.dna_sm.all.fa -bed $1 > /project2/gilad/briana/threeprimeseq/data/nuc_6up/sixupnuc.${describer}.bed Wrap this function:
#!/bin/bash
#SBATCH --job-name=w_nuc6
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=w_nuc6.out
#SBATCH --error=w_nuc6.err
#SBATCH --partition=broadwl
#SBATCH --mem=8G
#SBATCH --mail-type=END
for i in $(ls /project2/gilad/briana/threeprimeseq/data/bed_6upint/*.bed); do
sbatch nuc_6up.sh $i
done
Explore results for 2 files
sixupnuc.18486-N_S10_R1_001.bed and sixupnuc.18486-T_S9_R1_001.bed
library(workflowr)This is workflowr version 1.2.0
Run ?workflowr for help getting startedlibrary(ggplot2)
library(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionlibrary(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsavelibrary(tidyr)
library(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsnames=c("chr", "start", "end", "read", "score", "strand", "pct_at", "pct_gc", "numA", "numC", "numG", "numT", "numN", "numOther", "seqlen")
N_18486=read.table("../data/nuc6up/sixupnuc.18486-N_S10_R1_001.bed", col.names = names)
T_18486=read.table("../data/nuc6up/sixupnuc.18486-T_S9_R1_001.bed", col.names = names)Mutate to get the percent A and percent T. I will then use an if statement to keep the + strand reads A percent and the - strand read T percentage.
N_18486_filt = N_18486 %>% mutate(pc_A=numA/seqlen) %>% mutate(pc_T=numT/seqlen) %>% filter((strand=="+" & pc_A > .8 )|(strand=="-" & pc_T > .8)) %>% select("chr", "start", "end", "read", "score", "strand", "pc_A", "pc_T")
T_18486_filt = T_18486 %>% mutate(pc_A=numA/seqlen) %>% mutate(pc_T=numT/seqlen) %>% filter((strand=="+" & pc_A > .8 )|(strand=="-" & pc_T > .8)) %>% select("chr", "start", "end", "read", "score", "strand", "pc_A", "pc_T")Look at the number of reads matching this:
percN_readex=nrow(N_18486_filt)/nrow(N_18486)
percT_readex=nrow(T_18486_filt)/nrow(T_18486)The percent of reads that would be filtered in the nuclear file is 0.2388687
The percent of reads that would be filtered in the total file is 0.137103
The next step is to write an R script that can process each of the files. I will then wrap this for all of the files.
Expand to all files
#!/bin/rscripts
# usage: ./filter80percA.R infile, outfile
#this script takes the bedtools nuc output for 6bp upstream of the read and filters for 80% A's
#use optparse for management of input arguments I want to be able to imput the 6up nuc file and write out a filter file
library(optparse)
library(dplyr)
library(tidyr)
library(ggplot2)
option_list = list(
make_option(c("-f", "--file"), action="store", default=NA, type='character',
help="input file"),
make_option(c("-o", "--output"), action="store", default=NA, type='character',
help="output file")
)
opt_parser <- OptionParser(option_list=option_list)
opt <- parse_args(opt_parser)
#interrupt execution if no file is supplied
if (is.null(opt$file)){
print_help(opt_parser)
stop("Need input file", call.=FALSE)
}
#import file
names=c("chr", "start", "end", "read", "score", "strand", "pct_at", "pct_gc", "numA", "numC", "numG", "numT", "numN", "numOther", "seqlen")
infile=read.table(file = opt$file, col.names = names)
infile_filt = infile %>% mutate(pc_A=numA/seqlen) %>% mutate(pc_T=numT/seqlen) %>% filter((strand=="+" & pc_A > .8 )|(strand=="-" & pc_T > .8)) %>% select(chr, start, end, read, score, strand, pc_A, pc_T)
write.table(infile_filt, file = opt$output, quote=F, col.names = T, row.names = F, sep="\t")Now I need a bash script that calls this script:
#!/bin/bash
#SBATCH --job-name=run.Rfilt
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=runRfilt.out
#SBATCH --error=runRfilt.err
#SBATCH --partition=broadwl
#SBATCH --mem=20G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
sample=$1
describer=$(echo ${sample} | sed -e 's/.*sixupnuc.//' | sed -e "s/.bed$//")
Rscript filter80percA.R -f $1 -o /project2/gilad/briana/threeprimeseq/data/nuc6A_filt/filtnucA.${describer}.txt
test on /project2/gilad/briana/threeprimeseq/data/nuc_6up/sixupnuc.18486-N_S10_R1_001.bed
Write a wrapper for this:
#!/bin/bash
#SBATCH --job-name=w_rprocess
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=w_rproc.out
#SBATCH --error=w_rproc.err
#SBATCH --partition=broadwl
#SBATCH --mem=8G
#SBATCH --mail-type=END
for i in $(ls /project2/gilad/briana/threeprimeseq/data/nuc_6up/*.bed); do
sbatch run.Rfilt.sh $i
done
Now I can look at the percentage that are filtered out in all lines.
#upload data and melt it
filt_stats=read.csv("../data/nuc6up/sixAup_filterstats.csv", header=T)
filt_stats$Line= as.factor(filt_stats$Line)
filt_stats_melt=melt(filt_stats, id.vars=c("Line", "Fraction")) %>% filter(variable=="perc_filt")
#graph
ggplot(filt_stats_melt, aes(x=Line, fill=Fraction, y=value)) + geom_bar( stat="identity", position="dodge") + labs(title="Filtering out reads with 6 A's upstream of read", y="Percent of reads filtered") + scale_fill_manual(values=c("#D55E00","#0072B2"))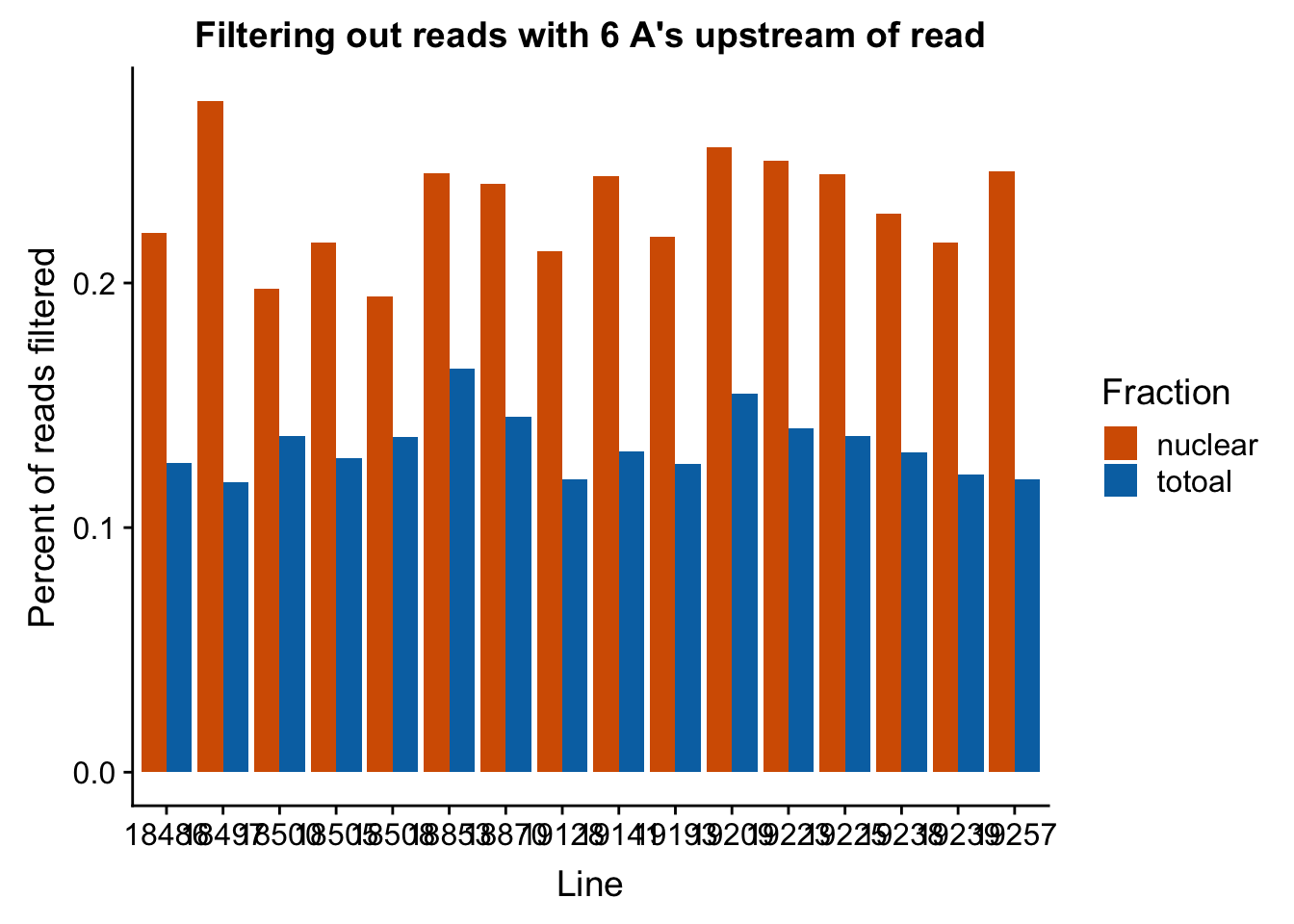
| Version | Author | Date |
|---|---|---|
| 61338d7 | Briana Mittleman | 2018-06-20 |
I want to check how many of reads would be filtered out for Cs and Gs as a baseline. I will use the line I have loaded here.
N_18486_filtC = N_18486 %>% mutate(pc_C=numC/seqlen) %>% mutate(pc_G=numG/seqlen) %>% filter((strand=="+" & pc_C > .8 )|(strand=="-" & pc_G > .8)) %>% select("chr", "start", "end", "read", "score", "strand", "pc_C", "pc_G")
T_18486_filtC = T_18486 %>% mutate(pc_C=numC/seqlen) %>% mutate(pc_G=numG/seqlen) %>% filter((strand=="+" & pc_C > .8 )|(strand=="-" & pc_G > .8)) %>% select("chr", "start", "end", "read", "score", "strand", "pc_C", "pc_G")
percNC_readex=nrow(N_18486_filtC)/nrow(N_18486)
percTC_readex=nrow(T_18486_filtC)/nrow(T_18486)If we look for stretches of Cs we get:
The percent of reads that would be filtered in the nuclear file is 4.894273710^{-4}
The percent of reads that would be filtered in the total file is 7.527303710^{-4}
We could change it because Nuc flips the ref genome
N_18486_filt_s = N_18486 %>% mutate(pc_A=numA/seqlen) %>% mutate(pc_T=numT/seqlen) %>% filter((strand=="+" & pc_T > .8 )) %>% select("chr", "start", "end", "read", "score", "strand", "pc_A", "pc_T")
T_18486_filt_s = T_18486 %>% mutate(pc_A=numA/seqlen) %>% mutate(pc_T=numT/seqlen) %>% filter((strand=="+" & pc_T > .8 )) %>% select("chr", "start", "end", "read", "score", "strand", "pc_A", "pc_T")
percN_readexS=nrow(N_18486_filt_s)/nrow(N_18486)
percT_readexS=nrow(T_18486_filt_s)/nrow(T_18486)Notes:
nuc profiles the sequence accorrding to strand. I dont need to look at the opposite metric for the negative strand
we care about %T because this is the read from the sequencing primer
The next step for this analysis is to create a snakepipeline with parameters I can control for the number of bases upstream we care about and the percent of T’s we filter on.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bindrcpp_0.2.2 reshape2_1.4.3 tidyr_0.8.1 cowplot_0.9.3
[5] dplyr_0.7.6 ggplot2_3.0.0 workflowr_1.2.0
loaded via a namespace (and not attached):
[1] Rcpp_0.12.19 compiler_3.5.1 pillar_1.3.0 git2r_0.24.0
[5] plyr_1.8.4 bindr_0.1.1 tools_3.5.1 digest_0.6.17
[9] evaluate_0.13 tibble_1.4.2 gtable_0.2.0 pkgconfig_2.0.2
[13] rlang_0.2.2 yaml_2.2.0 withr_2.1.2 stringr_1.4.0
[17] knitr_1.20 fs_1.2.6 rprojroot_1.3-2 grid_3.5.1
[21] tidyselect_0.2.4 glue_1.3.0 R6_2.3.0 rmarkdown_1.11
[25] purrr_0.2.5 magrittr_1.5 whisker_0.3-2 backports_1.1.2
[29] scales_1.0.0 htmltools_0.3.6 assertthat_0.2.0 colorspace_1.3-2
[33] labeling_0.3 stringi_1.2.4 lazyeval_0.2.1 munsell_0.5.0
[37] crayon_1.3.4