Evaluate Brain Data
Briana Mittleman
7/16/2018
Last updated: 2018-07-16
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 4fede84
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: output/.DS_Store Untracked files: Untracked: analysis/test.smash.Rmd Untracked: data/18486.genecov.txt Untracked: data/APApeaksYL.total.inbrain.bed Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt Untracked: data/bedgraph_peaks/ Untracked: data/bin200.5.T.nuccov.bed Untracked: data/bin200.Anuccov.bed Untracked: data/bin200.nuccov.bed Untracked: data/gene_cov/ Untracked: data/leafcutter/ Untracked: data/nuc6up/ Untracked: data/reads_mapped_three_prime_seq.csv Untracked: data/ssFC200.cov.bed Untracked: output/picard/ Untracked: output/plots/ Untracked: output/qual.fig2.pdf Unstaged changes: Modified: analysis/dif.iso.usage.leafcutter.Rmd Modified: analysis/explore.filters.Rmd Modified: analysis/test.max2.Rmd Modified: code/Snakefile
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 4fede84 | Briana Mittleman | 2018-07-16 | add eval brain analysis |
I downloaded the brain 3’ seq data from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM747470 and I want to use this analysis to see how similar their peaks are to ours eventhough the data is from different cell types.
First I will use the bedtools jaccard function to explore the overlaps. It will give me one stat that is the length(intersection)/length(union) - length(intersection). Here I can have file A brain peaks and file B be our peaks to see the similarity between the sets.
#!/bin/bash
#SBATCH --job-name=jaccard_brain
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=jacard_brain.out
#SBATCH --error=jacard_brain.err
#SBATCH --partition=broadwl
#SBATCH --mem=16G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
bedtools jaccard -a /project2/gilad/briana/threeprimeseq/data/derti_brain/GSM747470_human_brain.sites.clustered.hg19.sort.bed -b /project2/gilad/briana/threeprimeseq/data/peaks/APApeaksYL.total.bed > /project2/gilad/briana/threeprimeseq/data/derti_brain/total.jaccard.txt Results: intersection union-intersection jaccard n_intersections 21371 25414133 0.00084091 21352
The brain set has 89110 peaks and our set has 288350. I will filter ours by score then see if the top 25% have a higher overlap percentage.
library(workflowr)Loading required package: rmarkdownThis is workflowr version 1.0.1
Run ?workflowr for help getting startedlibrary(dplyr)Warning: package 'dplyr' was built under R version 3.4.4
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionlibrary(tidyr)
library(ggplot2)
library(reshape2)Warning: package 'reshape2' was built under R version 3.4.3
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsYL_peaks=read.table("../data/bedgraph_peaks/APApeaks.bed", col.names = c("chr", "start", "end", "count", "strand", "score")) %>% mutate(length=end-start)Warning: package 'bindrcpp' was built under R version 3.4.4I want the counts for the top 25% of the peaks.
quantile(YL_peaks$count) 0% 25% 50% 75% 100%
1.000000e+00 1.343902e+01 2.353933e+01 6.091061e+01 1.604636e+06 I will subset the peaks by having a count > 61.
awk '$4 >= 60 {print}' APApeaksYL.total.bed > APApeaksYL.top25.total.bedI can rerun the jaccard with this and see if it changes, this new file has 72877 peaks.
Results:
intersection union-intersection jaccard n_intersections 13221 6452066 0.00204911 13207
The proportion of overlap increased. Next I can try to make plots where I seperate my peaks by if they have a corresponding one in the brain file then plot the scores. To do this I will first use bedtool intersect to get just my peaks that contain a peak in the brain file. I can then use dplyr to merge them.
Here A is my file and B is the brain file.
#!/bin/bash
#SBATCH --job-name=int_brain
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=int.brain.out
#SBATCH --error=int.brain.err
#SBATCH --partition=broadwl
#SBATCH --mem=16G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
bedtools intersect -wa -a /project2/gilad/briana/threeprimeseq/data/peaks/APApeaksYL.total.bed -b /project2/gilad/briana/threeprimeseq/data/derti_brain/GSM747470_human_brain.sites.clustered.hg19.sort.bed > /project2/gilad/briana/threeprimeseq/data/derti_brain/APApeaksYL.total.inbrain.bed
The resulting file has 21378 peaks.
YL_peaks_overlap=read.table("../data/APApeaksYL.total.inbrain.bed", col.names = c("chr", "start", "end", "count", "strand", "score")) %>% mutate(length=end-start) %>% mutate(in_brain="Y")Now I need to join them.
YL_peaks_join=YL_peaks %>% full_join(YL_peaks_overlap, by = c("chr", "start", "end", "count", "strand", "score", "length"))
YL_peaks_join$in_brain[is.na(YL_peaks_join$in_brain)]="N"
YL_peaks_join_sel=YL_peaks_join %>% select(count, in_brain)Plot these.
ggplot(YL_peaks_join_sel, aes(y=log10(count), x=in_brain, fill=in_brain)) + geom_boxplot() + labs(x="Peak called in brain dataset", y="log10 Score", title="Peak score distribution by inclusion in brain dataset")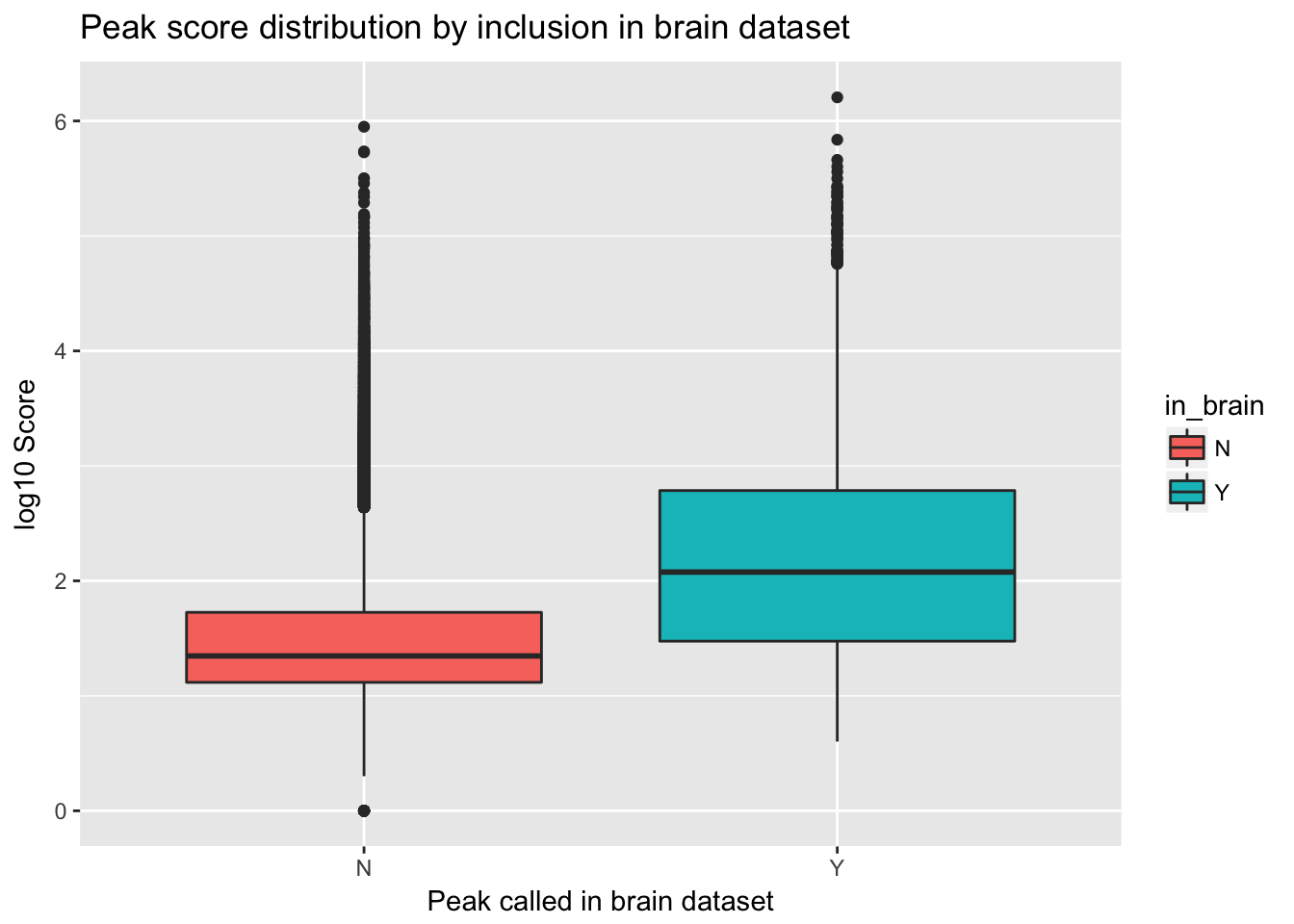
ggplot(YL_peaks_join_sel, aes(x=log10(count), fill=in_brain), bins=50) + geom_density(position="identity", alpha=.5) + labs(x="log10 of Score", title="Distribution of log10 Scores in peaks included in brain dataset")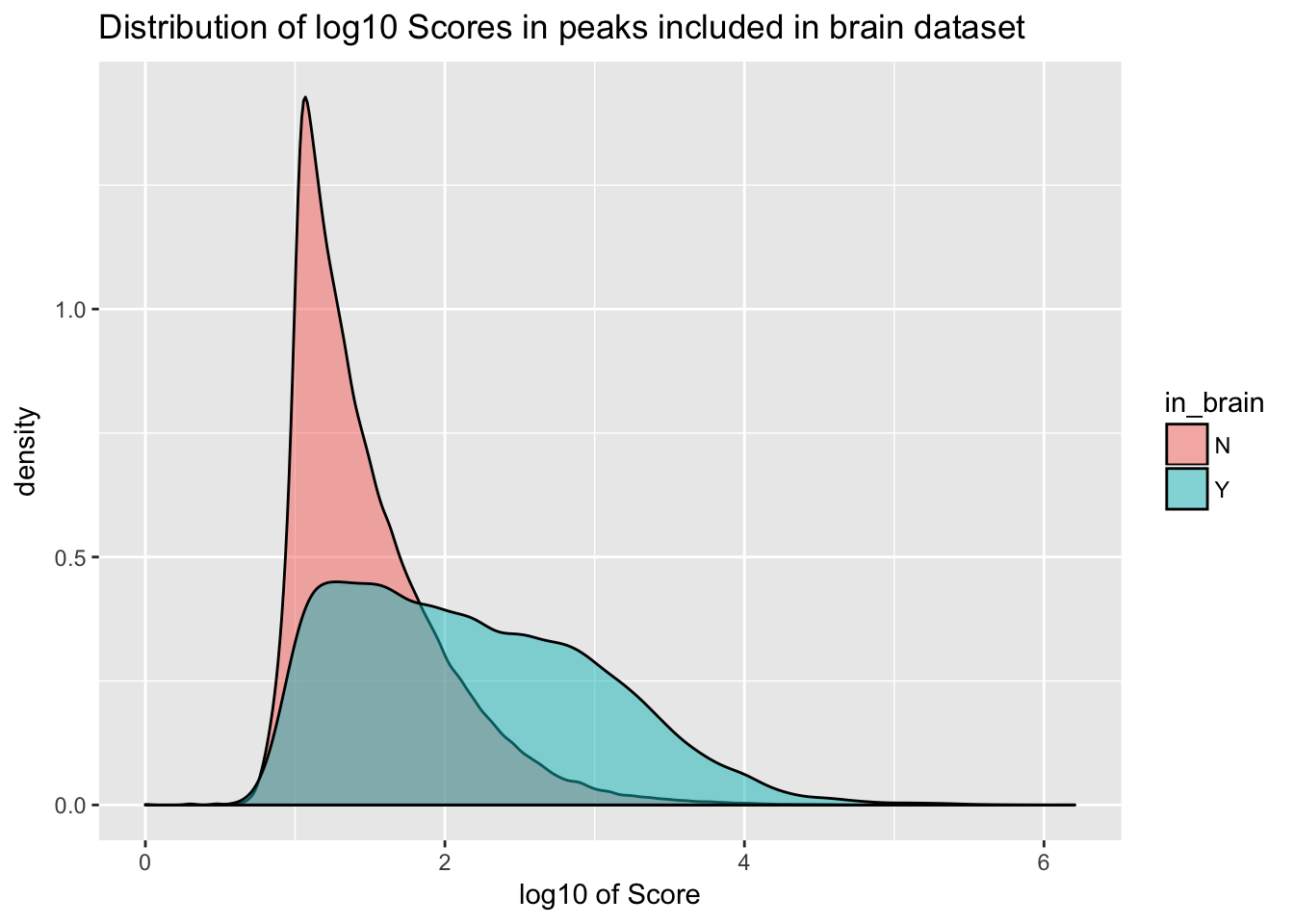
It would be better if the background was just a random subset of the same number. There are 21378 included peaks so I should select a random 21378 to make a background distribution.
samp_YLpeaks= sample_n(YL_peaks, 21378)
ggplot() + geom_histogram(data=samp_YLpeaks, aes(log10(count)), bins=100) + geom_histogram(data=YL_peaks_overlap, aes(log10(count)),fill="Red", bins=100) + labs(x="Log10 of Score", title="Scores in Overlapping set compared to scores in random set")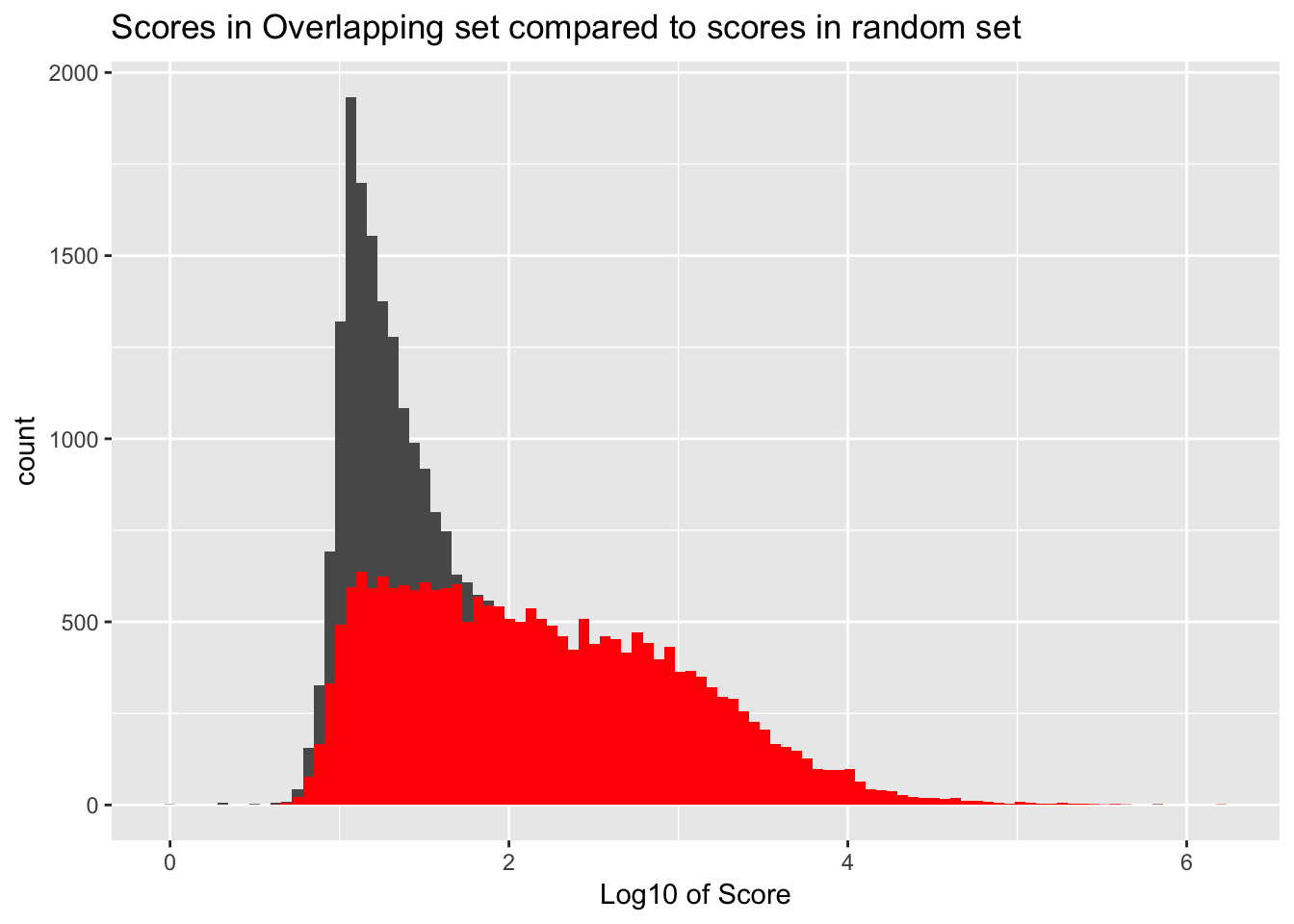
Session information
sessionInfo()R version 3.4.2 (2017-09-28)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bindrcpp_0.2.2 reshape2_1.4.3 ggplot2_2.2.1 tidyr_0.7.2
[5] dplyr_0.7.5 workflowr_1.0.1 rmarkdown_1.8.5
loaded via a namespace (and not attached):
[1] Rcpp_0.12.17 compiler_3.4.2 pillar_1.1.0
[4] git2r_0.21.0 plyr_1.8.4 bindr_0.1.1
[7] R.methodsS3_1.7.1 R.utils_2.6.0 tools_3.4.2
[10] digest_0.6.15 evaluate_0.10.1 tibble_1.4.2
[13] gtable_0.2.0 pkgconfig_2.0.1 rlang_0.2.1
[16] yaml_2.1.19 stringr_1.3.1 knitr_1.18
[19] rprojroot_1.3-2 grid_3.4.2 tidyselect_0.2.4
[22] glue_1.2.0 R6_2.2.2 purrr_0.2.5
[25] magrittr_1.5 whisker_0.3-2 backports_1.1.2
[28] scales_0.5.0 htmltools_0.3.6 assertthat_0.2.0
[31] colorspace_1.3-2 labeling_0.3 stringi_1.2.2
[34] lazyeval_0.2.1 munsell_0.4.3 R.oo_1.22.0
This reproducible R Markdown analysis was created with workflowr 1.0.1