P-values and False discoveries under null settings
Chih-Hsuan Wu
Last updated: 2023-11-29
Checks: 5 2
Knit directory: DEanalysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230508) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| ~/Google Drive/My Drive/spatial/10X/DEanalysis | . |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 91b404e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Untracked files:
Untracked: .DS_Store
Untracked: .Rhistory
Untracked: analysis/.DS_Store
Untracked: analysis/.Rapp.history
Untracked: analysis/.Rhistory
Untracked: analysis/Bcells.Rmd
Untracked: analysis/CD14+ Monocytes.Rmd
Untracked: analysis/FD_analysis.Rmd
Untracked: analysis/analysis on Kang.Rmd
Untracked: analysis/data2.Rmd
Untracked: analysis/data_clusters.Rmd
Untracked: analysis/figure/
Untracked: analysis/group12_13.Rmd
Untracked: analysis/group12_19.Rmd
Untracked: analysis/group2_19.Rmd
Untracked: analysis/group8_17-2_19.Rmd
Untracked: analysis/group8_17.Rmd
Untracked: analysis/methods_details.Rmd
Untracked: analysis/new_criteria.Rmd
Untracked: data/.Rhistory
Untracked: data/10X_Kang_DEresult.RData
Untracked: data/10X_inputdata.RData
Untracked: data/10X_inputdata_DEresult.RData
Untracked: data/10X_inputdata_cpm.RData
Untracked: data/10X_inputdata_integrated.RData
Untracked: data/10X_inputdata_lognorm.RData
Untracked: data/10Xdata_annotate.rds
Untracked: data/Bcells.Rmd
Untracked: data/Bcellsce.rds
Untracked: data/data2sce.RData
Untracked: data/permutation.RData
Untracked: data/vstcounts.Rdata
Unstaged changes:
Modified: analysis/index.Rmd
Modified: code/DE_methods.R
Modified: code/functions_in_rmd.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
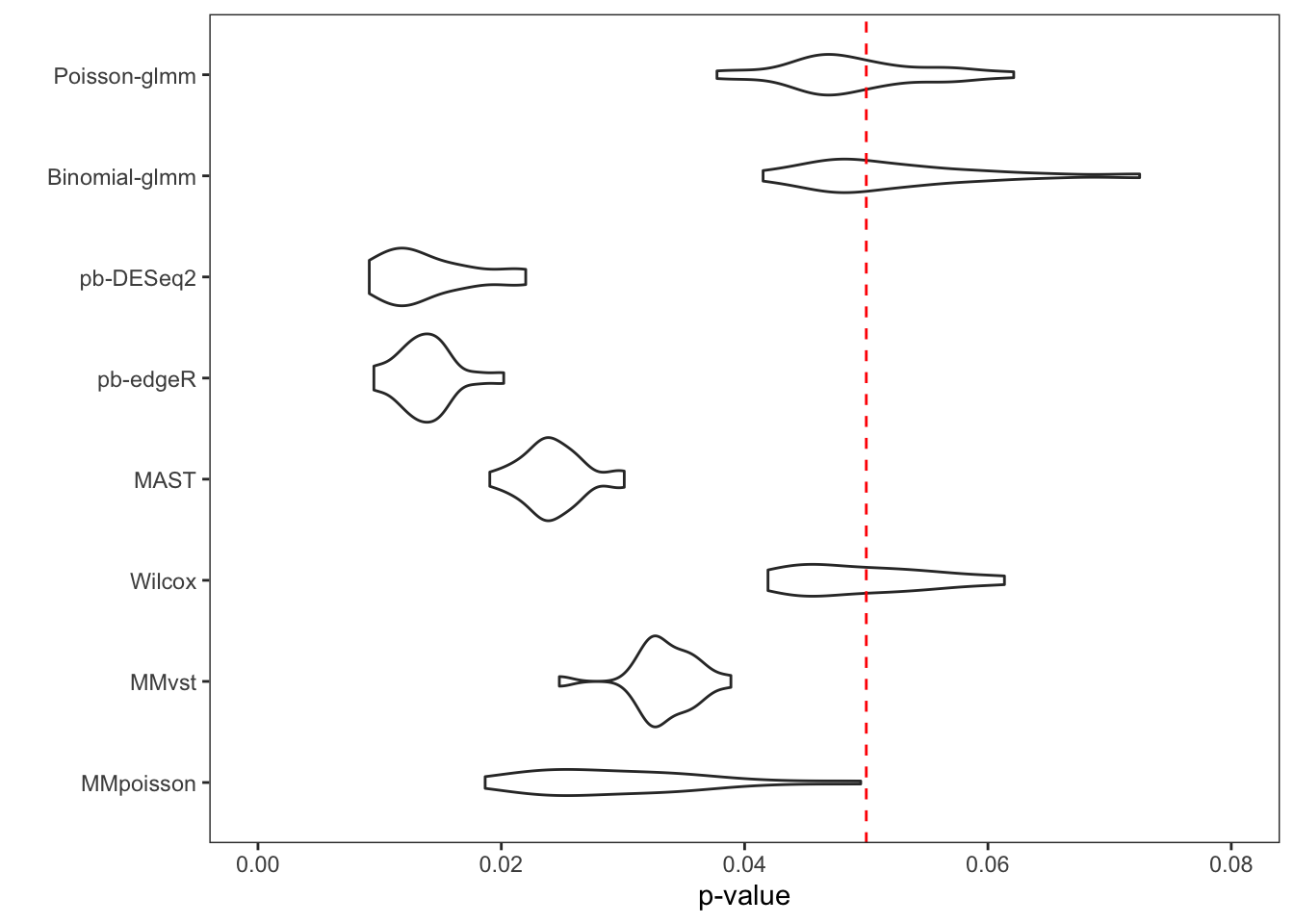
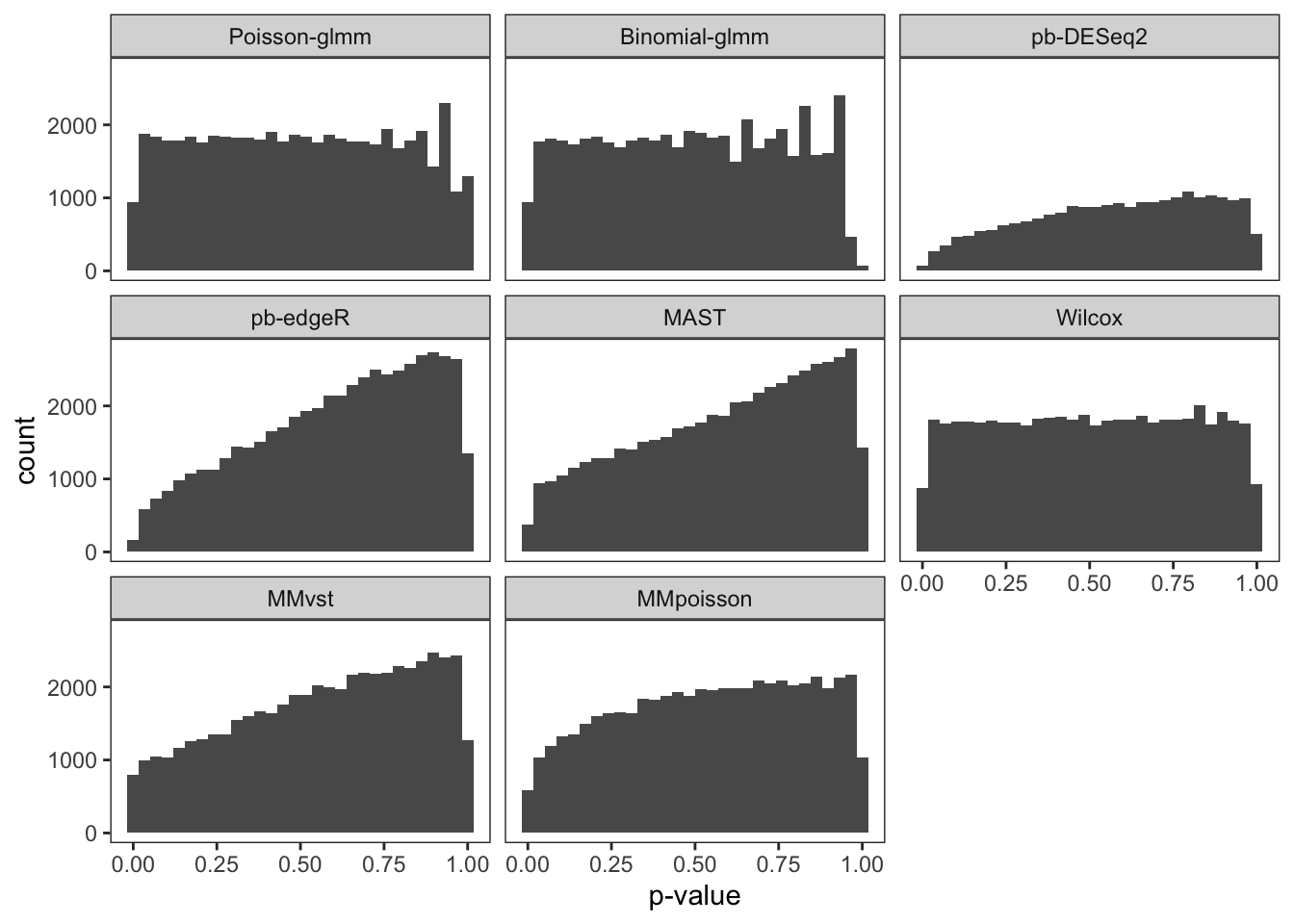
To examine the p-value calibration in real data, we did a permutation on group-of-interest within a null dataset. The cells in the controlled group of B cells were randomly assigned to controlled or stimulated group. We then computed p-values of each gene with different methods. The gene set was restricted to the input genes of Poisson-glmm, and the threshold of Wilcox method was relaxed to prevent filtering out genes. The procedure was repeated 20 times. Each time the proportion of p-value smaller than 0.05 was computed, so as the false discovery DEGs.
From the violin plot below, our glmm methods and Wilcox method are
well-calibrated. However, pseudo-bulk methods, MAST and mixed models
from Muscat are too conservative. Their overall proportion is way less
than 0.05. The histograms of all p-values in these 20 runs are flat for
our glmm methods and Wilcox method, which satisfy the null setting.
However, the p-values of the other methods are overestimated, resulting
conservative results. With either current criteria or our new criteria
to determine DEGs, every method detects at most one false discovery each
run.
R version 4.2.2 (2022-10-31)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur ... 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] tidyr_1.3.0 MAST_1.24.1
[3] muscat_1.12.1 SeuratObject_4.1.3
[5] Seurat_4.3.0.1 reshape_0.8.9
[7] gridExtra_2.3 pheatmap_1.0.12
[9] SingleCellExperiment_1.20.1 SummarizedExperiment_1.28.0
[11] Biobase_2.58.0 GenomicRanges_1.50.2
[13] GenomeInfoDb_1.34.9 IRanges_2.32.0
[15] S4Vectors_0.36.2 BiocGenerics_0.44.0
[17] MatrixGenerics_1.10.0 matrixStats_1.0.0
[19] ggpubr_0.6.0 dplyr_1.1.2
[21] ggplot2_3.4.2
loaded via a namespace (and not attached):
[1] scattermore_1.2 bit64_4.0.5
[3] knitr_1.27 irlba_2.3.5.1
[5] DelayedArray_0.24.0 data.table_1.14.8
[7] KEGGREST_1.38.0 RCurl_1.98-1.12
[9] doParallel_1.0.17 generics_0.1.3
[11] ScaledMatrix_1.6.0 RhpcBLASctl_0.23-42
[13] cowplot_1.1.1 RSQLite_2.3.1
[15] RANN_2.6.1 future_1.33.0
[17] bit_4.0.5 spatstat.data_3.0-1
[19] httpuv_1.6.11 viridis_0.6.3
[21] xfun_0.39 hms_1.1.3
[23] jquerylib_0.1.4 evaluate_0.21
[25] promises_1.2.0.1 fansi_1.0.4
[27] progress_1.2.2 caTools_1.18.2
[29] igraph_1.5.0 DBI_1.1.3
[31] geneplotter_1.76.0 htmlwidgets_1.6.2
[33] spatstat.geom_3.2-4 purrr_1.0.1
[35] ellipsis_0.3.2 backports_1.4.1
[37] annotate_1.76.0 aod_1.3.2
[39] deldir_1.0-9 sparseMatrixStats_1.10.0
[41] vctrs_0.6.3 ROCR_1.0-11
[43] abind_1.4-5 cachem_1.0.8
[45] withr_2.5.0 progressr_0.13.0
[47] sctransform_0.3.5 prettyunits_1.1.1
[49] goftest_1.2-3 cluster_2.1.4
[51] lazyeval_0.2.2 crayon_1.5.2
[53] spatstat.explore_3.2-1 labeling_0.4.2
[55] edgeR_3.40.2 pkgconfig_2.0.3
[57] nlme_3.1-162 vipor_0.4.5
[59] blme_1.0-5 rlang_1.1.1
[61] globals_0.16.2 lifecycle_1.0.3
[63] miniUI_0.1.1.1 rsvd_1.0.5
[65] rprojroot_2.0.3 polyclip_1.10-4
[67] lmtest_0.9-40 Matrix_1.5-4.1
[69] carData_3.0-5 boot_1.3-28.1
[71] zoo_1.8-12 beeswarm_0.4.0
[73] ggridges_0.5.4 GlobalOptions_0.1.2
[75] png_0.1-8 viridisLite_0.4.2
[77] rjson_0.2.21 bitops_1.0-7
[79] KernSmooth_2.23-22 Biostrings_2.66.0
[81] blob_1.2.4 DelayedMatrixStats_1.20.0
[83] workflowr_1.7.0 shape_1.4.6
[85] stringr_1.5.0 parallelly_1.36.0
[87] spatstat.random_3.1-5 remaCor_0.0.16
[89] rstatix_0.7.2 ggsignif_0.6.4
[91] beachmat_2.14.2 scales_1.2.1
[93] memoise_2.0.1 magrittr_2.0.3
[95] plyr_1.8.8 ica_1.0-3
[97] gplots_3.1.3 zlibbioc_1.44.0
[99] compiler_4.2.2 RColorBrewer_1.1-3
[101] clue_0.3-64 lme4_1.1-34
[103] DESeq2_1.38.3 fitdistrplus_1.1-11
[105] cli_3.6.1 XVector_0.38.0
[107] lmerTest_3.1-3 listenv_0.9.0
[109] patchwork_1.1.2 pbapply_1.7-2
[111] TMB_1.9.5 MASS_7.3-60
[113] mgcv_1.9-0 tidyselect_1.2.0
[115] stringi_1.7.12 highr_0.10
[117] yaml_2.3.7 BiocSingular_1.14.0
[119] locfit_1.5-9.8 ggrepel_0.9.3
[121] grid_4.2.2 sass_0.4.7
[123] tools_4.2.2 future.apply_1.11.0
[125] parallel_4.2.2 circlize_0.4.15
[127] rstudioapi_0.15.0 foreach_1.5.2
[129] git2r_0.32.0 EnvStats_2.8.0
[131] farver_2.1.1 Rtsne_0.16
[133] digest_0.6.33 shiny_1.7.4.1
[135] Rcpp_1.0.11 car_3.1-2
[137] broom_1.0.5 scuttle_1.8.4
[139] later_1.3.1 RcppAnnoy_0.0.21
[141] httr_1.4.6 AnnotationDbi_1.60.2
[143] ComplexHeatmap_2.14.0 Rdpack_2.4
[145] colorspace_2.1-0 XML_3.99-0.14
[147] fs_1.6.3 tensor_1.5
[149] reticulate_1.30 splines_4.2.2
[151] uwot_0.1.16 spatstat.utils_3.0-3
[153] scater_1.26.1 sp_2.0-0
[155] plotly_4.10.2 xtable_1.8-4
[157] jsonlite_1.8.7 nloptr_2.0.3
[159] R6_2.5.1 pillar_1.9.0
[161] htmltools_0.5.5 mime_0.12
[163] glue_1.6.2 fastmap_1.1.1
[165] minqa_1.2.5 BiocParallel_1.32.6
[167] BiocNeighbors_1.16.0 codetools_0.2-19
[169] mvtnorm_1.2-2 utf8_1.2.3
[171] lattice_0.21-8 bslib_0.5.0
[173] spatstat.sparse_3.0-2 tibble_3.2.1
[175] pbkrtest_0.5.2 numDeriv_2016.8-1.1
[177] ggbeeswarm_0.7.2 leiden_0.4.3
[179] gtools_3.9.4 survival_3.5-5
[181] limma_3.54.2 glmmTMB_1.1.8
[183] rmarkdown_2.23 munsell_0.5.0
[185] GetoptLong_1.0.5 GenomeInfoDbData_1.2.9
[187] iterators_1.0.14 variancePartition_1.28.9
[189] reshape2_1.4.4 gtable_0.3.3
[191] rbibutils_2.2.13