Getting started with clusterProfiler
2025-03-09
Last updated: 2025-03-09
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version fc1274d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: data/1M_neurons_filtered_gene_bc_matrices_h5.h5
Ignored: data/293t/
Ignored: data/293t_3t3_filtered_gene_bc_matrices.tar.gz
Ignored: data/293t_filtered_gene_bc_matrices.tar.gz
Ignored: data/5k_Human_Donor1_PBMC_3p_gem-x_5k_Human_Donor1_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor2_PBMC_3p_gem-x_5k_Human_Donor2_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor3_PBMC_3p_gem-x_5k_Human_Donor3_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor4_PBMC_3p_gem-x_5k_Human_Donor4_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/97516b79-8d08-46a6-b329-5d0a25b0be98.h5ad
Ignored: data/Parent_SC3v3_Human_Glioblastoma_filtered_feature_bc_matrix.tar.gz
Ignored: data/brain_counts/
Ignored: data/cl.obo
Ignored: data/cl.owl
Ignored: data/jurkat/
Ignored: data/jurkat:293t_50:50_filtered_gene_bc_matrices.tar.gz
Ignored: data/jurkat_293t/
Ignored: data/jurkat_filtered_gene_bc_matrices.tar.gz
Ignored: data/pbmc20k/
Ignored: data/pbmc20k_seurat/
Ignored: data/pbmc3k/
Ignored: data/pbmc4k_filtered_gene_bc_matrices.tar.gz
Ignored: data/pbmc_1k_v3_filtered_feature_bc_matrix.h5
Ignored: data/pbmc_1k_v3_raw_feature_bc_matrix.h5
Ignored: data/refdata-gex-GRCh38-2020-A.tar.gz
Ignored: data/seurat_1m_neuron.rds
Ignored: data/t_3k_filtered_gene_bc_matrices.tar.gz
Ignored: r_packages_4.4.1/
Untracked files:
Untracked: analysis/bioc_scrnaseq.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/cluster_profiler.Rmd) and
HTML (docs/cluster_profiler.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | fc1274d | Dave Tang | 2025-03-09 | Subset GO terms to only biological processes |
| html | f0166b0 | Dave Tang | 2025-03-09 | Build site. |
| Rmd | 6ad2657 | Dave Tang | 2025-03-09 | Checking missing GO terms |
| html | e815348 | Dave Tang | 2025-03-06 | Build site. |
| Rmd | 1fa9528 | Dave Tang | 2025-03-06 | Universal enrichment analyser |
| html | 2c080dc | Dave Tang | 2024-11-01 | Build site. |
| Rmd | 124a8d9 | Dave Tang | 2024-11-01 | Using clusterProfiler |
Getting started
Install.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
install.packages('ggarchery')
install.packages('ggtangle')
BiocManager::install("clusterProfiler")
BiocManager::install("org.Hs.eg.db")
BiocManager::install("GO.db")Load libraries.
suppressPackageStartupMessages(library(tidyverse))
suppressPackageStartupMessages(library(ggarchery))
suppressPackageStartupMessages(library(clusterProfiler))
suppressPackageStartupMessages(library(org.Hs.eg.db))
suppressPackageStartupMessages(library(GO.db))Example data
Use An example differential gene expression results table.
de_res <- read_csv("https://raw.githubusercontent.com/davetang/muse/refs/heads/main/data/13970886_edger_res.csv", show_col_types = FALSE)
head(de_res)# A tibble: 6 × 6
ensembl_gene_id logFC logCPM F PValue adjusted_pvalue
<chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 ENSG00000000003 2.73 4.83 4.28 0.0684 0.109
2 ENSG00000000005 -7.00 0.541 17.6 0.00216 0.0138
3 ENSG00000000419 0.120 5.34 0.114 0.743 0.776
4 ENSG00000000457 -0.708 5.31 3.35 0.0993 0.145
5 ENSG00000000460 -0.897 3.95 2.66 0.136 0.186
6 ENSG00000000938 1.54 5.60 1.86 0.205 0.258 Gene IDs
The {clusterProfiler} package uses the enrichGO()
function for performing a Gene
Ontology over-representation test. The input for gene
is a vector of Entrez Gene IDs.
Our example data uses Ensembl gene IDs, so we need to convert these into Entrez Gene IDs. We can use the {org.Hs.eg.db} package provided by Bioconductor, which provides genome wide annotation for human, primarily based on mapping using Entrez Gene identifiers.
ensembl_to_entrez <- AnnotationDbi::select(
org.Hs.eg.db,
keys = de_res$ensembl_gene_id,
columns = c("ENSEMBL", "ENTREZID"),
keytype = "ENSEMBL"
)'select()' returned 1:many mapping between keys and columnsde_res |>
dplyr::inner_join(ensembl_to_entrez, by = dplyr::join_by(ensembl_gene_id == ENSEMBL)) |>
dplyr::select(ensembl_gene_id, ENTREZID, dplyr::everything()) -> de_res
head(de_res)# A tibble: 6 × 7
ensembl_gene_id ENTREZID logFC logCPM F PValue adjusted_pvalue
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 ENSG00000000003 7105 2.73 4.83 4.28 0.0684 0.109
2 ENSG00000000005 64102 -7.00 0.541 17.6 0.00216 0.0138
3 ENSG00000000419 8813 0.120 5.34 0.114 0.743 0.776
4 ENSG00000000457 57147 -0.708 5.31 3.35 0.0993 0.145
5 ENSG00000000460 55732 -0.897 3.95 2.66 0.136 0.186
6 ENSG00000000938 2268 1.54 5.60 1.86 0.205 0.258 clusterProfiler
The example data contains results of a comparison between cancer samples and normal samples; positive fold change indicates that genes were expressed higher in cancer. There are a lot of genes up-regulated in cancer.
de_res |>
dplyr::filter(logFC > 0, adjusted_pvalue < 0.05) |>
nrow()[1] 1440Instead of examining the list of up-regulated genes individually, which would take a long time, we can see whether these genes have common Gene Ontology terms more often than expected.
First we’ll get the top 500 most significantly up-regulated genes.
de_res |>
dplyr::filter(logFC > 0, adjusted_pvalue < 0.05) |>
dplyr::slice_min(order_by = adjusted_pvalue, n = 500) |>
dplyr::filter(!is.na(ENTREZID)) |>
dplyr::pull(ENTREZID) -> sig_up_genesCreate the background, i.e., the universe, so we can test for over-representation.
de_res |>
dplyr::filter(!is.na(ENTREZID)) |>
dplyr::pull(ENTREZID) -> the_universeGene Ontology terms are grouped into three ontologies:
- Molecular Function (MF)
- Biological Process (BP), and
- Cellular Component (CC)
Here, we’ll perform an over-representation analysis using biological processes.
ego_bp <- enrichGO(
gene = sig_up_genes,
universe = the_universe,
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
minGSSize = 10,
maxGSSize = 500,
readable = TRUE
)
head(ego_bp) ID
GO:0016064 GO:0016064
GO:0019724 GO:0019724
GO:0002460 GO:0002460
GO:0002449 GO:0002449
GO:0002443 GO:0002443
GO:0006959 GO:0006959
Description
GO:0016064 immunoglobulin mediated immune response
GO:0019724 B cell mediated immunity
GO:0002460 adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains
GO:0002449 lymphocyte mediated immunity
GO:0002443 leukocyte mediated immunity
GO:0006959 humoral immune response
GeneRatio BgRatio RichFactor FoldEnrichment zScore pvalue
GO:0016064 30/385 196/17518 0.15306122 6.964484 12.587673 4.569415e-17
GO:0019724 30/385 199/17518 0.15075377 6.859492 12.461452 7.046005e-17
GO:0002460 37/385 382/17518 0.09685864 4.407194 10.092931 3.590714e-14
GO:0002449 35/385 368/17518 0.09510870 4.327569 9.670801 3.134308e-13
GO:0002443 38/385 466/17518 0.08154506 3.710406 8.889621 3.440135e-12
GO:0006959 30/385 299/17518 0.10033445 4.565348 9.321312 4.278292e-12
p.adjust qvalue
GO:0016064 1.319717e-13 1.180762e-13
GO:0019724 1.319717e-13 1.180762e-13
GO:0002460 4.483604e-11 4.011520e-11
GO:0002449 2.935280e-10 2.626220e-10
GO:0002443 2.577349e-09 2.305977e-09
GO:0006959 2.671080e-09 2.389839e-09
geneID
GO:0016064 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/IGHV3-48/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/FOXP3/IGKC/IGHV3-15/TREM2/CD28
GO:0019724 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/IGHV3-48/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/FOXP3/IGKC/IGHV3-15/TREM2/CD28
GO:0002460 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IL4I1/IRF4/JAK3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/CCL19/IGHV3-48/CXCL13/LILRB4/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/FOXP3/IGKC/IGHV3-15/PLA2G4A/TREM2/CD28
GO:0002449 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IL4I1/IGHV3-23/IGHV2-5/CD27/IGHV3-30/SLAMF7/IGHV3-48/LILRB4/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/CD2/FOXP3/LGALS9/IGKC/IGHV3-15/TREM2/CD28
GO:0002443 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IL4I1/JAK3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/SLAMF7/IGHV3-48/LILRB4/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/CCL3/IGHV1-69D/CD84/CD2/FOXP3/LGALS9/IGKC/IGHV3-15/TREM2/CD28
GO:0006959 WFDC2/IGHG4/IGHG2/IGHG3/IGKV3-20/CXCL14/POU2AF1/MMP7/CCL22/BPIFA1/CXCL9/CCL19/CXCL13/CXCL1/LTF/CXCL10/MS4A1/IGHM/IGHG1/POU2F2/CCL7/TNFRSF21/ADM/IGHA2/C4A/CCL3/CXCL8/CCL8/TREM2/CD28
Count
GO:0016064 30
GO:0019724 30
GO:0002460 37
GO:0002449 35
GO:0002443 38
GO:0006959 30Bar plot showing each enriched GO term coloured by the adjusted p-value.
barplot(ego_bp, showCategory=10)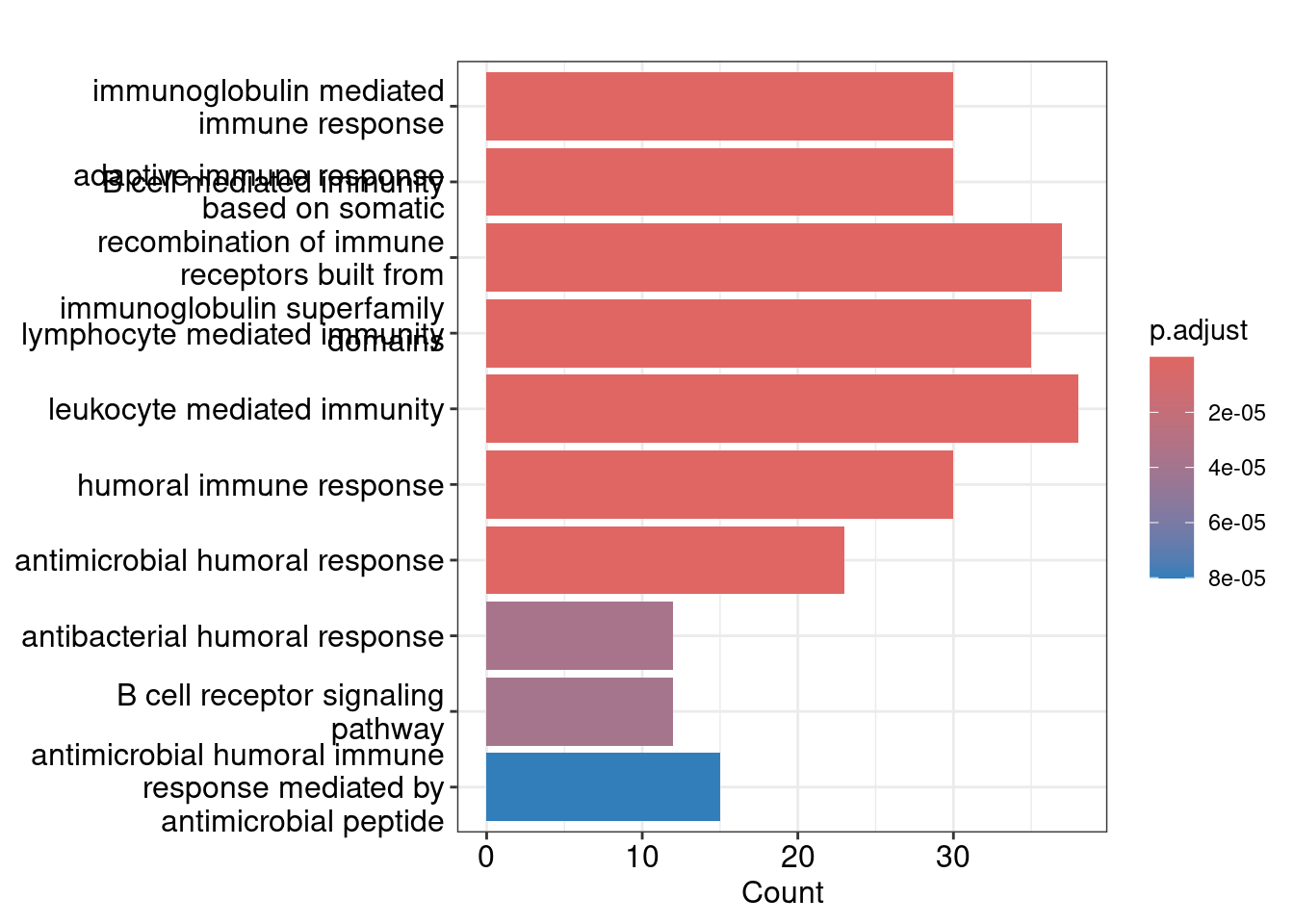
Dot plot showing each enriched GO term with associated statistics.
dotplot(ego_bp, showCategory=10)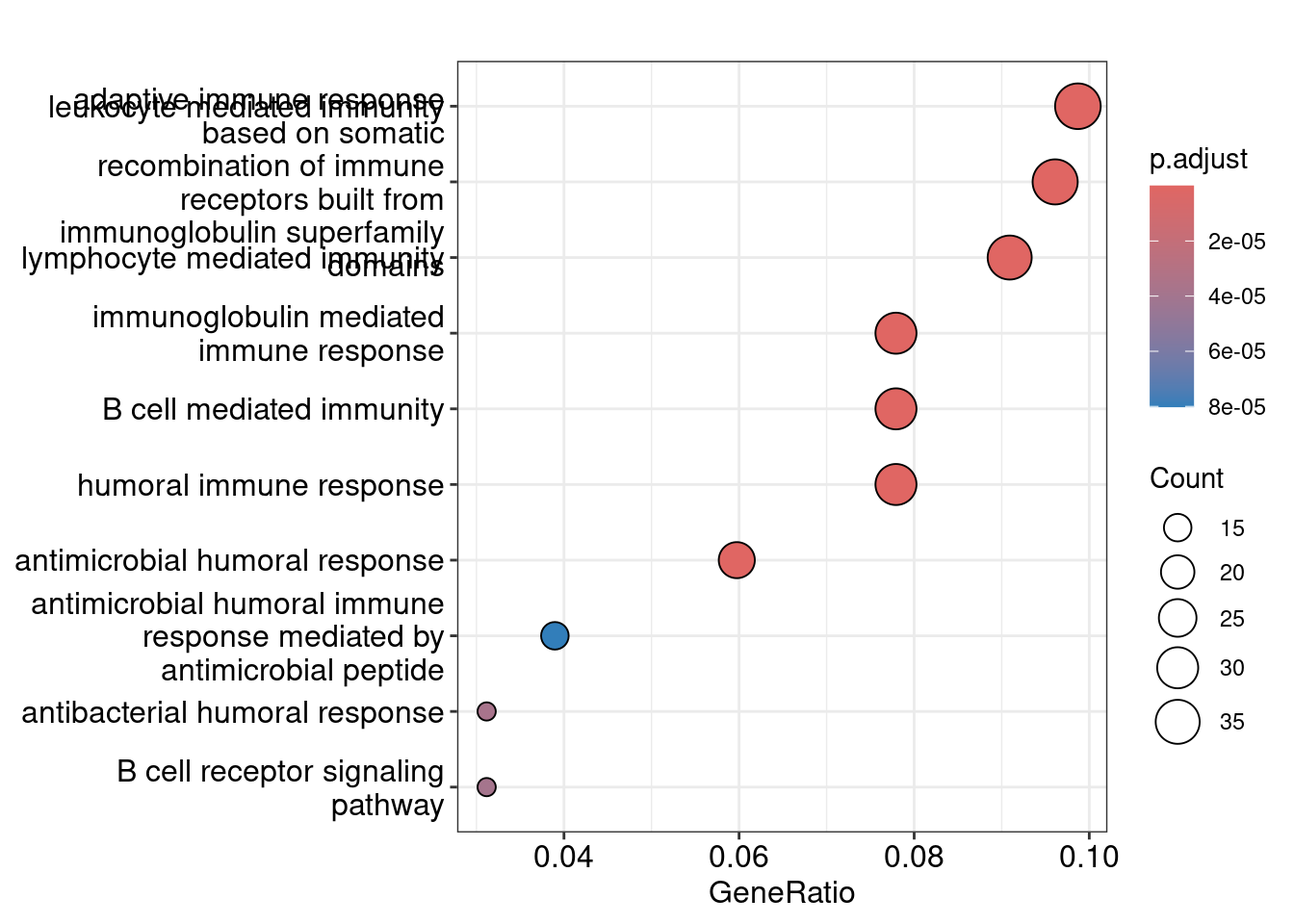
Heat plot showing the enriched GO terms on the y-axis and the genes on the x-axis. Genes with the associated GO term are highlighted.
heatplot(ego_bp, showCategory=10)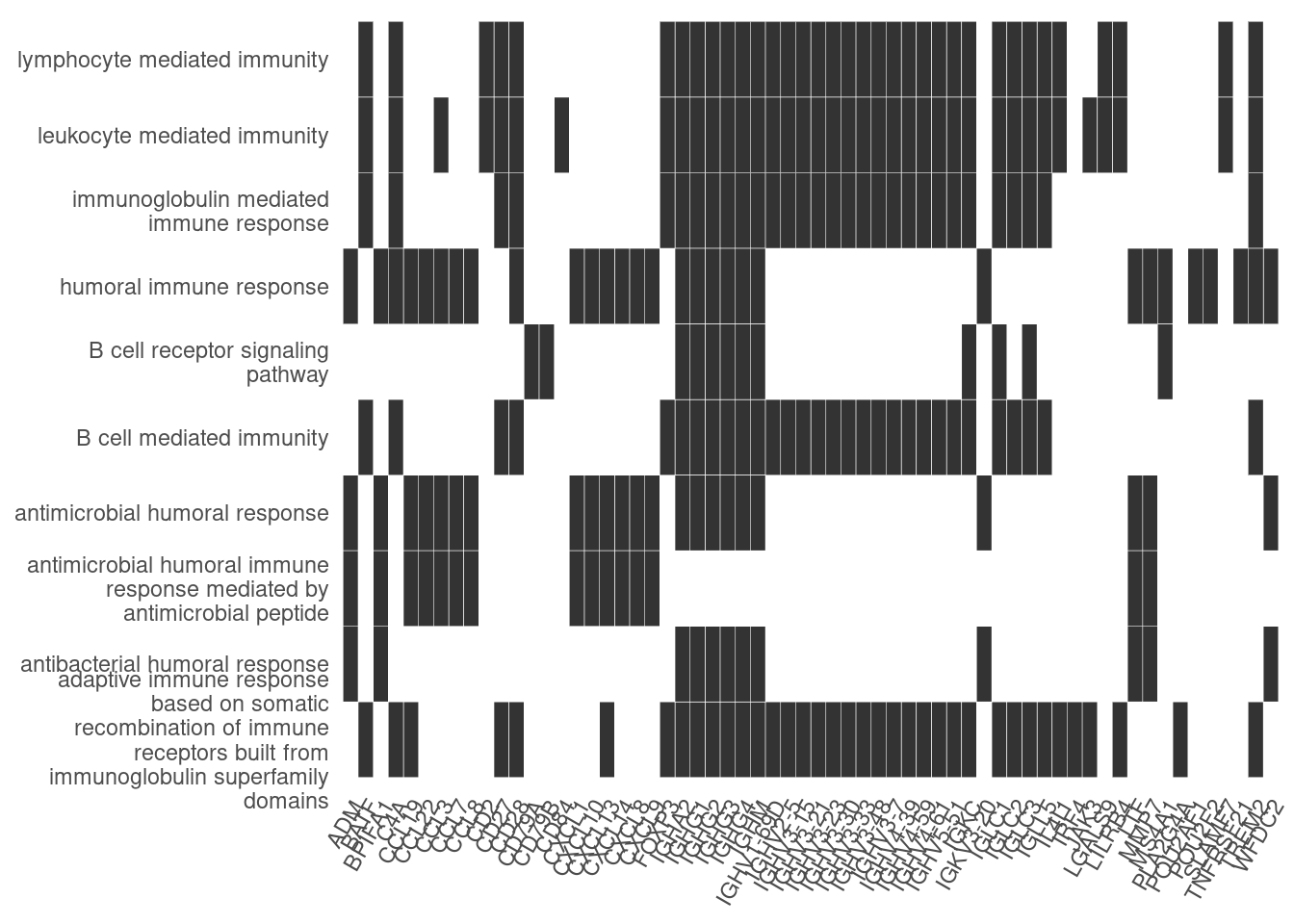
goplot shows the gene ontology graph with the enriched
GO terms highlighted.
goplot(ego_bp)Warning: ggrepel: 1 unlabeled data points (too many overlaps). Consider
increasing max.overlaps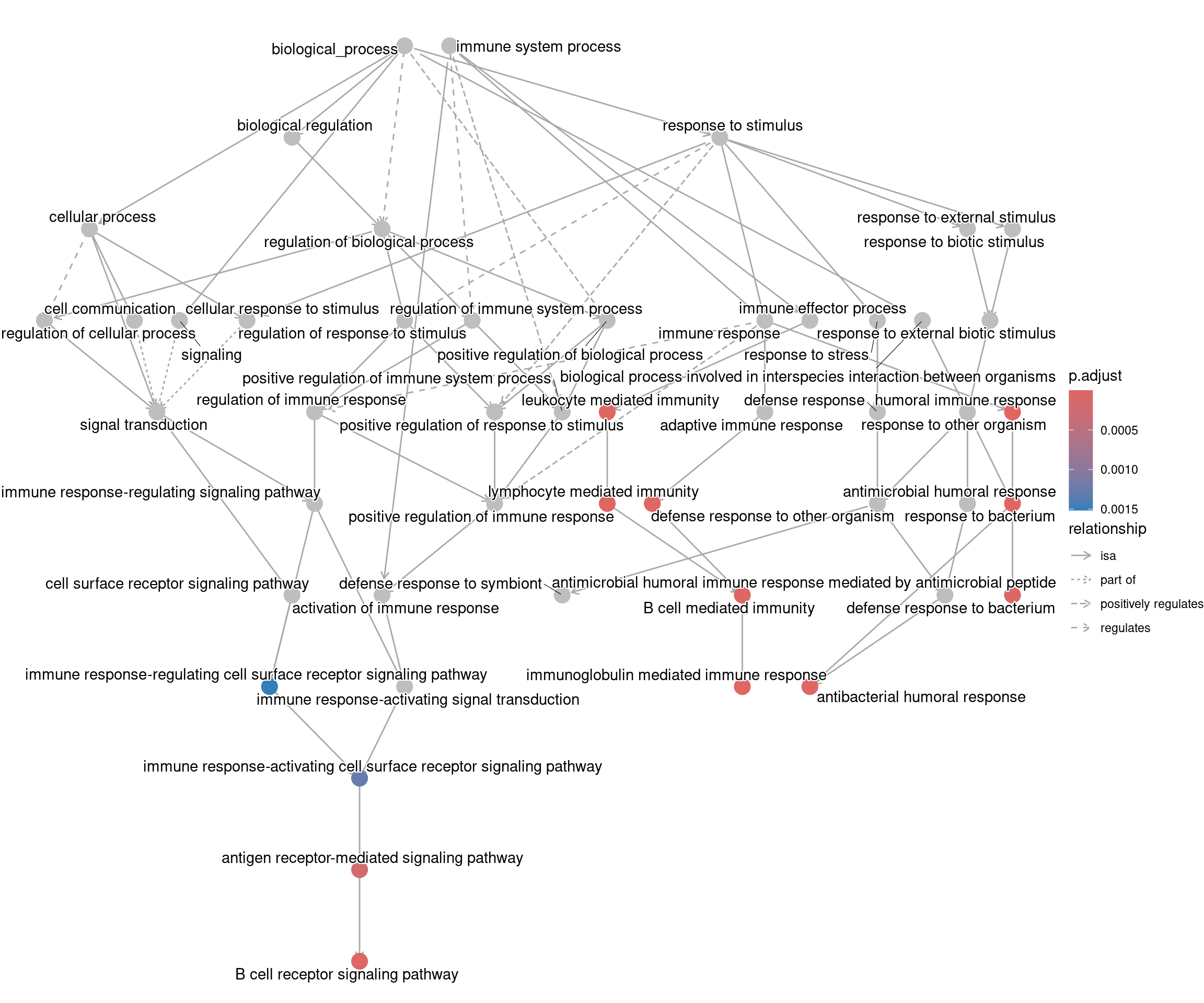
Another nice feature of {clusterProfiler} is that you can plot multiple gene lists together. We can create a list of down-regulated genes.
de_res |>
dplyr::filter(logFC < 0, adjusted_pvalue < 0.05) |>
dplyr::slice_min(order_by = adjusted_pvalue, n = 500) |>
dplyr::filter(!is.na(ENTREZID)) |>
dplyr::pull(ENTREZID) -> sig_down_genesPerform GO enrichment on two gene lists.
my_gene_list <- list(
up_gene = sig_up_genes,
down_gene = sig_down_genes
)
ego_bp_both <- compareCluster(
geneCluster = my_gene_list,
fun = "enrichGO",
universe = the_universe,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "BP",
pvalueCutoff = 0.01,
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
minGSSize = 10,
maxGSSize = 500,
readable = TRUE
)
head(as.data.frame(ego_bp_both)) Cluster ID
1 up_gene GO:0016064
2 up_gene GO:0019724
3 up_gene GO:0002460
4 up_gene GO:0002449
5 up_gene GO:0002443
6 up_gene GO:0006959
Description
1 immunoglobulin mediated immune response
2 B cell mediated immunity
3 adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains
4 lymphocyte mediated immunity
5 leukocyte mediated immunity
6 humoral immune response
GeneRatio BgRatio RichFactor FoldEnrichment zScore pvalue
1 30/385 196/17518 0.15306122 6.964484 12.587673 4.569415e-17
2 30/385 199/17518 0.15075377 6.859492 12.461452 7.046005e-17
3 37/385 382/17518 0.09685864 4.407194 10.092931 3.590714e-14
4 35/385 368/17518 0.09510870 4.327569 9.670801 3.134308e-13
5 38/385 466/17518 0.08154506 3.710406 8.889621 3.440135e-12
6 30/385 299/17518 0.10033445 4.565348 9.321312 4.278292e-12
p.adjust qvalue
1 1.319717e-13 1.180762e-13
2 1.319717e-13 1.180762e-13
3 4.483604e-11 4.011520e-11
4 2.935280e-10 2.626220e-10
5 2.577349e-09 2.305977e-09
6 2.671080e-09 2.389839e-09
geneID
1 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/IGHV3-48/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/FOXP3/IGKC/IGHV3-15/TREM2/CD28
2 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/IGHV3-48/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/FOXP3/IGKC/IGHV3-15/TREM2/CD28
3 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IL4I1/IRF4/JAK3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/CCL19/IGHV3-48/CXCL13/LILRB4/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/FOXP3/IGKC/IGHV3-15/PLA2G4A/TREM2/CD28
4 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IL4I1/IGHV3-23/IGHV2-5/CD27/IGHV3-30/SLAMF7/IGHV3-48/LILRB4/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/IGHV1-69D/CD2/FOXP3/LGALS9/IGKC/IGHV3-15/TREM2/CD28
5 IGLC1/IGHG4/IGHG2/IGHG3/IGHV5-51/IGLL5/IGHV4-39/IGLC3/IL4I1/JAK3/IGHV3-23/IGHV2-5/CD27/IGHV3-30/SLAMF7/IGHV3-48/LILRB4/BATF/IGHM/IGHG1/IGLC2/IGHV4-61/IGHA2/IGHV3-7/IGHV3-21/IGHV3-33/IGHV4-59/C4A/CCL3/IGHV1-69D/CD84/CD2/FOXP3/LGALS9/IGKC/IGHV3-15/TREM2/CD28
6 WFDC2/IGHG4/IGHG2/IGHG3/IGKV3-20/CXCL14/POU2AF1/MMP7/CCL22/BPIFA1/CXCL9/CCL19/CXCL13/CXCL1/LTF/CXCL10/MS4A1/IGHM/IGHG1/POU2F2/CCL7/TNFRSF21/ADM/IGHA2/C4A/CCL3/CXCL8/CCL8/TREM2/CD28
Count
1 30
2 30
3 37
4 35
5 38
6 30Dot plot with enriched GO terms by gene list.
dotplot(ego_bp_both, showCategory = 15)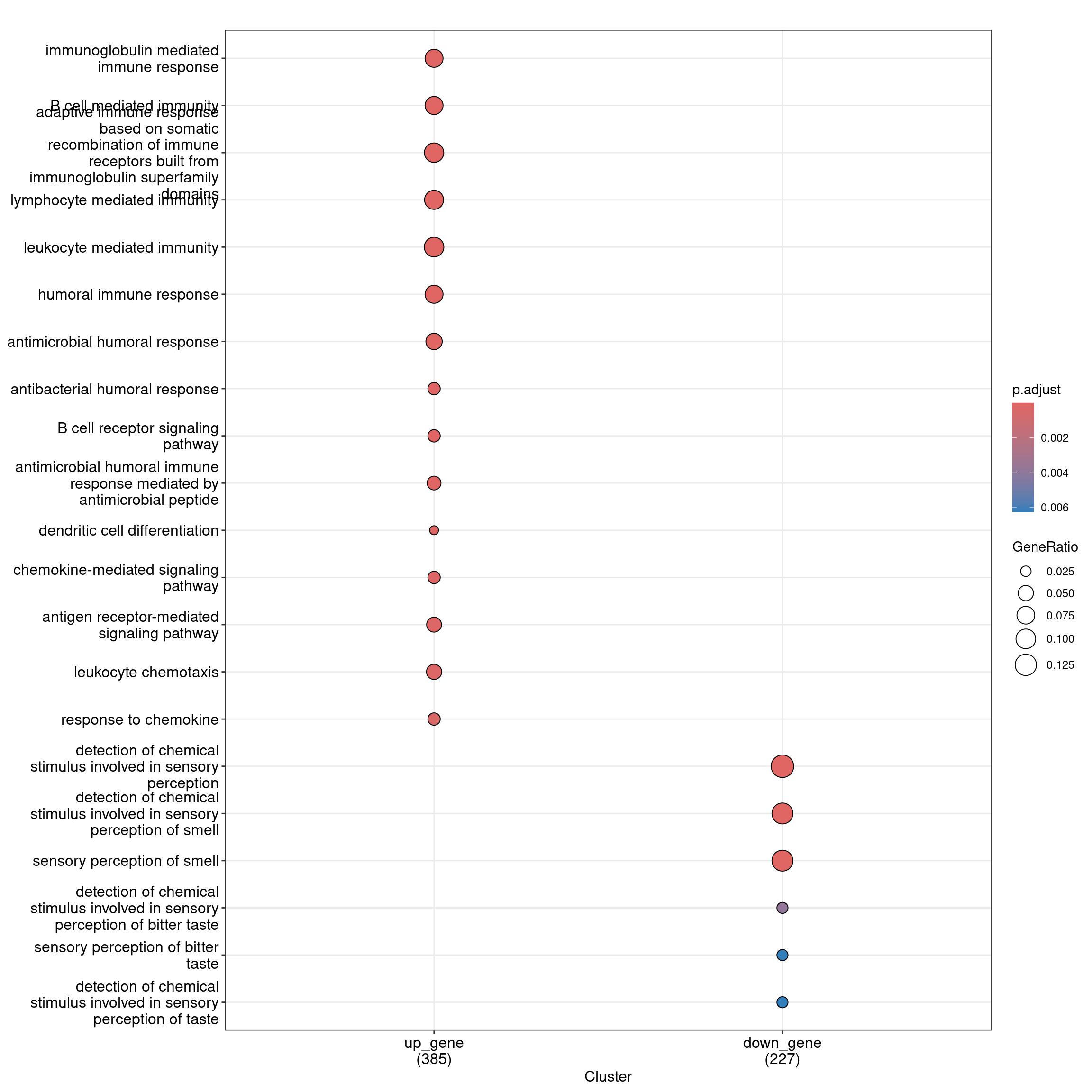
enricher
enricher() is a universal enrichment analyzer.
- gene - a vector of gene id
- universe - background genes. If missing, the all genes listed in the database (eg TERM2GENE table) will be used as background.
- minGSSize - minimal size of genes annotated for testing
- maxGSSize - maximal size of genes annotated for testing
- TERM2GENE - user input annotation of TERM TO GENE mapping, a data.frame of 2 column with term and gene. Only used when gson is NULL.
- TERM2NAME - user input of TERM TO NAME mapping, a data.frame of 2 column with term and name. Only used when gson is NULL.
TERM2NAME is needed to map the GOID to its term (description).
An easy way of obtaining this mapping is by extracting the information from the Bioconductor GO annotation database package ({GO.db}).
# extract a named vector of all terms
goterms <- AnnotationDbi::Term(GOTERM)
#convert into a data frame
term2name <- data.frame(
"term"=names(goterms),
"name"=goterms
)
dim(term2name)[1] 40940 2head(term2name) term name
GO:0000001 GO:0000001 mitochondrion inheritance
GO:0000002 GO:0000002 mitochondrial genome maintenance
GO:0000006 GO:0000006 high-affinity zinc transmembrane transporter activity
GO:0000007 GO:0000007 low-affinity zinc ion transmembrane transporter activity
GO:0000009 GO:0000009 alpha-1,6-mannosyltransferase activity
GO:0000010 GO:0000010 heptaprenyl diphosphate synthase activityGet the ontologies and store in term2name.
ontologies <- AnnotationDbi::select(x = GO.db, keys = names(goterms), columns = c("GOID", "ONTOLOGY"))'select()' returned 1:1 mapping between keys and columnslength(unique(term2name$term))[1] 40940length(unique(ontologies$GOID))[1] 40940stopifnot(all(term2name$term == ontologies$GOID))
term2name$ontology <- ontologies$ONTOLOGY
head(term2name) term name
GO:0000001 GO:0000001 mitochondrion inheritance
GO:0000002 GO:0000002 mitochondrial genome maintenance
GO:0000006 GO:0000006 high-affinity zinc transmembrane transporter activity
GO:0000007 GO:0000007 low-affinity zinc ion transmembrane transporter activity
GO:0000009 GO:0000009 alpha-1,6-mannosyltransferase activity
GO:0000010 GO:0000010 heptaprenyl diphosphate synthase activity
ontology
GO:0000001 BP
GO:0000002 BP
GO:0000006 MF
GO:0000007 MF
GO:0000009 MF
GO:0000010 MFany(is.na(term2name$term))[1] FALSEterm2name can then be used when calling
enrichr(), by specifying TERM2NAME=term2name.
Be sure, though, to check that term2name contains all GOIDs
present in your TERM2GENE mapping.
Demo input.
demo <- readr::read_csv("data/nfurzeri_gene_id_to_go_id.csv.gz", show_col_types = FALSE)
head(demo)# A tibble: 6 × 2
ensembl_gene_id go_id
<chr> <chr>
1 ENSNFUG00015000040 <NA>
2 ENSNFUG00015000041 GO:0007156
3 ENSNFUG00015000041 GO:0005886
4 ENSNFUG00015000041 GO:0005737
5 ENSNFUG00015000041 GO:0050808
6 ENSNFUG00015000041 GO:0007411Check if we have all the GO IDs.
goids <- demo$go_id
goids <- goids[!is.na(goids)]
table(unique(goids) %in% term2name$term)
FALSE TRUE
16 6531 Which GO IDs are missing?
missing_goids <- setdiff(unique(goids), term2name$term)
missing_goids [1] "GO:0008272" "GO:0102769" "GO:0006211" "GO:0004024" "GO:0090179"
[6] "GO:0004310" "GO:0035308" "GO:0042543" "GO:0052794" "GO:0052795"
[11] "GO:0052796" "GO:0034998" "GO:0102148" "GO:0005355" "GO:0003867"
[16] "GO:0060775"They are missing because they have become obsolete.
missing_goids %in% keys(GOOBSOLETE) [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[16] TRUEPrepare data frame of 2 column with term and gene.
demo |>
dplyr::filter(!is.na(go_id)) |>
dplyr::rename(term = go_id, gene = ensembl_gene_id) |>
dplyr::select(term, gene) |>
dplyr::arrange(term) -> term2gene
lookup <- AnnotationDbi::select(x = GO.db, keys = term2gene$term, columns = c("GOID", "ONTOLOGY"))'select()' returned many:1 mapping between keys and columnsstopifnot(all(lookup$GOID == term2gene$term))
term2gene$ontology <- lookup$ONTOLOGY
head(term2gene)# A tibble: 6 × 3
term gene ontology
<chr> <chr> <chr>
1 GO:0000002 ENSNFUG00015006992 BP
2 GO:0000002 ENSNFUG00015020070 BP
3 GO:0000002 ENSNFUG00015013825 BP
4 GO:0000002 ENSNFUG00015019642 BP
5 GO:0000009 ENSNFUG00015008531 MF
6 GO:0000012 ENSNFUG00015012664 BP Test with random genes.
set.seed(1984)
my_genes <- sample(x = unique(term2gene$gene), size = 100, replace = FALSE)
res <- enricher(
gene = my_genes,
TERM2GENE = dplyr::filter(term2gene, ontology == "BP"),
TERM2NAME = dplyr::filter(term2name, ontology == "BP")
)
res#
# over-representation test
#
#...@organism UNKNOWN
#...@ontology UNKNOWN
#...@gene chr [1:100] "ENSNFUG00015003803" "ENSNFUG00015000174" "ENSNFUG00015015379" ...
#...pvalues adjusted by 'BH' with cutoff <0.05
#...0 enriched terms found
#...Citation
S Xu, E Hu, Y Cai, Z Xie, X Luo, L Zhan, W Tang, Q Wang, B Liu, R Wang, W Xie, T Wu, L Xie, G Yu. Using clusterProfiler to characterize multiomics data. Nature Protocols. 2024, 19(11):3292-3320 Test with genes associated with same term.
term2gene |>
dplyr::group_by(term) |>
dplyr::summarise(n = n()) |>
dplyr::arrange(-n) -> dev_null
my_term <- "GO:0006397"
dplyr::filter(term2name, term == my_term) term name ontology
GO:0006397 GO:0006397 mRNA processing BPterm2gene |>
dplyr::filter(term == my_term) |>
dplyr::pull(gene) |>
head(75) -> enriched_genes
set.seed(1984)
my_genes <- sample(x = unique(term2gene$gene), size = 25, replace = FALSE)
my_genes <- union(enriched_genes, my_genes)
res <- enricher(
gene = my_genes,
TERM2GENE = dplyr::filter(term2gene, ontology == "BP"),
TERM2NAME = dplyr::filter(term2name, ontology == "BP")
)
res |>
as.data.frame() ID Description
GO:0006397 GO:0006397 mRNA processing
GO:0008380 GO:0008380 RNA splicing
GO:0000398 GO:0000398 mRNA splicing, via spliceosome
GO:0000381 GO:0000381 regulation of alternative mRNA splicing, via spliceosome
GO:0000387 GO:0000387 spliceosomal snRNP assembly
GO:0043484 GO:0043484 regulation of RNA splicing
GO:0080090 GO:0080090 regulation of primary metabolic process
GO:0030968 GO:0030968 endoplasmic reticulum unfolded protein response
GeneRatio BgRatio RichFactor FoldEnrichment zScore
GO:0006397 75/92 125/12886 0.6000000 84.03913 79.109456
GO:0008380 43/92 89/12886 0.4831461 67.67196 53.520121
GO:0000398 21/92 71/12886 0.2957746 41.42774 28.965584
GO:0000381 7/92 20/12886 0.3500000 49.02283 18.225267
GO:0000387 3/92 12/12886 0.2500000 35.01630 9.996638
GO:0043484 3/92 14/12886 0.2142857 30.01398 9.210457
GO:0080090 2/92 12/12886 0.1666667 23.34420 6.566466
GO:0030968 2/92 14/12886 0.1428571 20.00932 6.034488
pvalue p.adjust qvalue
GO:0006397 5.586297e-146 4.133859e-144 3.586990e-144
GO:0008380 1.687611e-72 6.244159e-71 5.418118e-71
GO:0000398 3.041395e-29 7.502109e-28 6.509653e-28
GO:0000381 5.389679e-11 9.970906e-10 8.651853e-10
GO:0000387 7.395374e-05 1.094515e-03 9.497217e-04
GO:0043484 1.210986e-04 1.493549e-03 1.295967e-03
GO:0080090 3.176485e-03 3.357999e-02 2.913769e-02
GO:0030968 4.339229e-03 4.013787e-02 3.482802e-02
geneID
GO:0006397 ENSNFUG00015000047/ENSNFUG00015000176/ENSNFUG00015000433/ENSNFUG00015000717/ENSNFUG00015000995/ENSNFUG00015002878/ENSNFUG00015003278/ENSNFUG00015003412/ENSNFUG00015003645/ENSNFUG00015006374/ENSNFUG00015008466/ENSNFUG00015010592/ENSNFUG00015011476/ENSNFUG00015011629/ENSNFUG00015012572/ENSNFUG00015015893/ENSNFUG00015018395/ENSNFUG00015021027/ENSNFUG00015023111/ENSNFUG00015024532/ENSNFUG00015000276/ENSNFUG00015000347/ENSNFUG00015000470/ENSNFUG00015000633/ENSNFUG00015000679/ENSNFUG00015001197/ENSNFUG00015001330/ENSNFUG00015002687/ENSNFUG00015003553/ENSNFUG00015003613/ENSNFUG00015004037/ENSNFUG00015004391/ENSNFUG00015008722/ENSNFUG00015009124/ENSNFUG00015010820/ENSNFUG00015011580/ENSNFUG00015013273/ENSNFUG00015014123/ENSNFUG00015014399/ENSNFUG00015014448/ENSNFUG00015019426/ENSNFUG00015020527/ENSNFUG00015020545/ENSNFUG00015021436/ENSNFUG00015004299/ENSNFUG00015006219/ENSNFUG00015006538/ENSNFUG00015007623/ENSNFUG00015008859/ENSNFUG00015008871/ENSNFUG00015008954/ENSNFUG00015013147/ENSNFUG00015014506/ENSNFUG00015014956/ENSNFUG00015015816/ENSNFUG00015017783/ENSNFUG00015019674/ENSNFUG00015019903/ENSNFUG00015020693/ENSNFUG00015021220/ENSNFUG00015022748/ENSNFUG00015023101/ENSNFUG00015023143/ENSNFUG00015004762/ENSNFUG00015005084/ENSNFUG00015005095/ENSNFUG00015006742/ENSNFUG00015008749/ENSNFUG00015009426/ENSNFUG00015013451/ENSNFUG00015014267/ENSNFUG00015014760/ENSNFUG00015014763/ENSNFUG00015018080/ENSNFUG00015018929
GO:0008380 ENSNFUG00015000047/ENSNFUG00015000433/ENSNFUG00015002878/ENSNFUG00015003412/ENSNFUG00015008466/ENSNFUG00015010592/ENSNFUG00015011629/ENSNFUG00015012572/ENSNFUG00015015893/ENSNFUG00015018395/ENSNFUG00015021027/ENSNFUG00015023111/ENSNFUG00015000276/ENSNFUG00015000347/ENSNFUG00015000470/ENSNFUG00015001197/ENSNFUG00015002687/ENSNFUG00015004391/ENSNFUG00015009124/ENSNFUG00015010820/ENSNFUG00015011580/ENSNFUG00015013273/ENSNFUG00015014123/ENSNFUG00015014448/ENSNFUG00015019426/ENSNFUG00015020527/ENSNFUG00015008859/ENSNFUG00015008871/ENSNFUG00015013147/ENSNFUG00015014506/ENSNFUG00015019674/ENSNFUG00015019903/ENSNFUG00015020693/ENSNFUG00015021220/ENSNFUG00015022748/ENSNFUG00015023101/ENSNFUG00015005084/ENSNFUG00015008749/ENSNFUG00015014267/ENSNFUG00015014760/ENSNFUG00015014763/ENSNFUG00015018080/ENSNFUG00015018929
GO:0000398 ENSNFUG00015000433/ENSNFUG00015002878/ENSNFUG00015003412/ENSNFUG00015010592/ENSNFUG00015011629/ENSNFUG00015012572/ENSNFUG00015015893/ENSNFUG00015023111/ENSNFUG00015001197/ENSNFUG00015002687/ENSNFUG00015014123/ENSNFUG00015008871/ENSNFUG00015014506/ENSNFUG00015019903/ENSNFUG00015020693/ENSNFUG00015022748/ENSNFUG00015005084/ENSNFUG00015008749/ENSNFUG00015014267/ENSNFUG00015014760/ENSNFUG00015014763
GO:0000381 ENSNFUG00015024532/ENSNFUG00015000470/ENSNFUG00015011580/ENSNFUG00015014123/ENSNFUG00015014448/ENSNFUG00015008859/ENSNFUG00015013147
GO:0000387 ENSNFUG00015010592/ENSNFUG00015018395/ENSNFUG00015019674
GO:0043484 ENSNFUG00015000995/ENSNFUG00015014448/ENSNFUG00015018080
GO:0080090 ENSNFUG00015011476/ENSNFUG00015021436
GO:0030968 ENSNFUG00015011476/ENSNFUG00015021436
Count
GO:0006397 75
GO:0008380 43
GO:0000398 21
GO:0000381 7
GO:0000387 3
GO:0043484 3
GO:0080090 2
GO:0030968 2Check some of the results.
go_term <- "GO:0007156"
dplyr::filter(term2name, term == go_term) term
GO:0007156 GO:0007156
name
GO:0007156 homophilic cell adhesion via plasma membrane adhesion molecules
ontology
GO:0007156 BPdplyr::filter(term2gene, gene %in% my_genes, term == go_term) |>
nrow()[1] 1dplyr::filter(term2gene, term == go_term) |>
nrow()[1] 130Dot plot.
dotplot(res, showCategory=10) +
ggtitle("Biological Processes")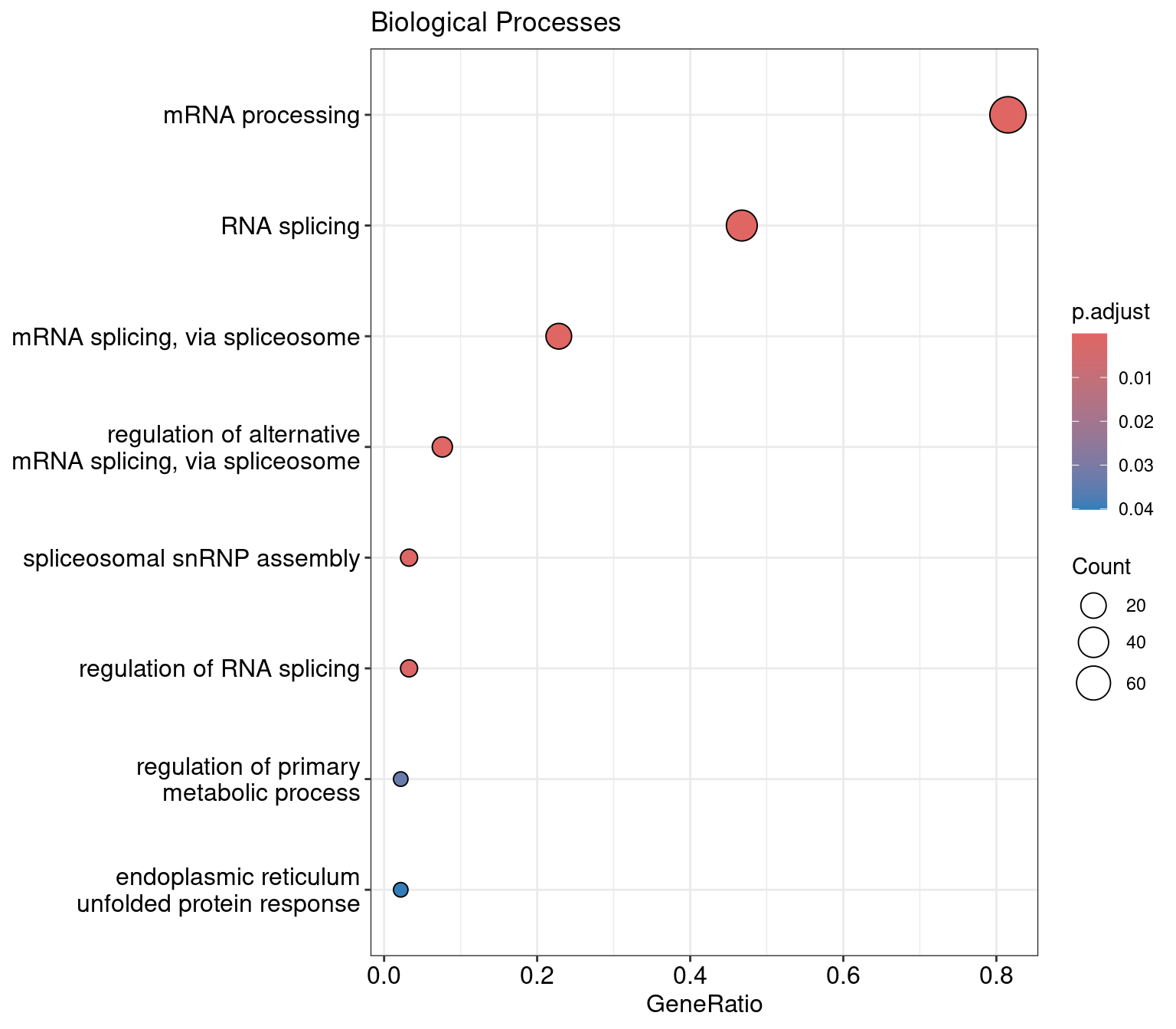
| Version | Author | Date |
|---|---|---|
| e815348 | Dave Tang | 2025-03-06 |
sessionInfo()R version 4.4.1 (2024-06-14)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.5 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] GO.db_3.20.0 org.Hs.eg.db_3.20.0 AnnotationDbi_1.68.0
[4] IRanges_2.40.1 S4Vectors_0.44.0 Biobase_2.66.0
[7] BiocGenerics_0.52.0 clusterProfiler_4.14.6 ggarchery_0.4.3
[10] lubridate_1.9.4 forcats_1.0.0 stringr_1.5.1
[13] dplyr_1.1.4 purrr_1.0.4 readr_2.1.5
[16] tidyr_1.3.1 tibble_3.2.1 ggplot2_3.5.1
[19] tidyverse_2.0.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] DBI_1.2.3 gson_0.1.0 rlang_1.1.5
[4] magrittr_2.0.3 DOSE_4.0.0 git2r_0.35.0
[7] compiler_4.4.1 RSQLite_2.3.9 getPass_0.2-4
[10] png_0.1-8 callr_3.7.6 vctrs_0.6.5
[13] reshape2_1.4.4 pkgconfig_2.0.3 crayon_1.5.3
[16] fastmap_1.2.0 XVector_0.46.0 labeling_0.4.3
[19] utf8_1.2.4 promises_1.3.2 rmarkdown_2.29
[22] tzdb_0.4.0 enrichplot_1.26.6 UCSC.utils_1.2.0
[25] ps_1.9.0 bit_4.5.0.1 xfun_0.51
[28] zlibbioc_1.52.0 cachem_1.1.0 aplot_0.2.5
[31] GenomeInfoDb_1.42.3 jsonlite_1.9.1 blob_1.2.4
[34] later_1.4.1 BiocParallel_1.40.0 parallel_4.4.1
[37] R6_2.6.1 bslib_0.9.0 stringi_1.8.4
[40] RColorBrewer_1.1-3 jquerylib_0.1.4 GOSemSim_2.32.0
[43] Rcpp_1.0.14 knitr_1.49 ggtangle_0.0.6
[46] R.utils_2.13.0 igraph_2.1.4 httpuv_1.6.15
[49] Matrix_1.7-0 splines_4.4.1 timechange_0.3.0
[52] tidyselect_1.2.1 qvalue_2.38.0 rstudioapi_0.17.1
[55] yaml_2.3.10 codetools_0.2-20 curl_6.2.1
[58] processx_3.8.6 lattice_0.22-6 plyr_1.8.9
[61] treeio_1.30.0 withr_3.0.2 KEGGREST_1.46.0
[64] evaluate_1.0.3 gridGraphics_0.5-1 Biostrings_2.74.1
[67] ggtree_3.14.0 pillar_1.10.1 whisker_0.4.1
[70] ggfun_0.1.8 generics_0.1.3 vroom_1.6.5
[73] rprojroot_2.0.4 hms_1.1.3 tidytree_0.4.6
[76] munsell_0.5.1 scales_1.3.0 glue_1.8.0
[79] lazyeval_0.2.2 tools_4.4.1 data.table_1.17.0
[82] fgsea_1.32.2 fs_1.6.5 fastmatch_1.1-6
[85] cowplot_1.1.3 grid_4.4.1 ape_5.8-1
[88] colorspace_2.1-1 nlme_3.1-164 patchwork_1.3.0
[91] GenomeInfoDbData_1.2.13 cli_3.6.4 gtable_0.3.6
[94] R.methodsS3_1.8.2 yulab.utils_0.2.0 sass_0.4.9
[97] digest_0.6.37 ggrepel_0.9.6 ggplotify_0.1.2
[100] farver_2.1.2 memoise_2.0.1 htmltools_0.5.8.1
[103] R.oo_1.27.0 lifecycle_1.0.4 httr_1.4.7
[106] bit64_4.6.0-1