Using the Bioconductor ensembldb package
2023-08-28
Last updated: 2023-08-28
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version d9e2337. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: r_packages_4.3.0/
Untracked files:
Untracked: analysis/cell_ranger.Rmd
Untracked: analysis/complex_heatmap.Rmd
Untracked: analysis/sleuth.Rmd
Untracked: analysis/tss_xgboost.Rmd
Untracked: code/multiz100way/
Untracked: data/HG00702_SH089_CHSTrio.chr1.vcf.gz
Untracked: data/HG00702_SH089_CHSTrio.chr1.vcf.gz.tbi
Untracked: data/ncrna_NONCODE[v3.0].fasta.tar.gz
Untracked: data/ncrna_noncode_v3.fa
Untracked: data/netmhciipan.out.gz
Untracked: data/test
Untracked: export/davetang039sblog.WordPress.2023-06-30.xml
Untracked: export/output/
Untracked: women.json
Unstaged changes:
Modified: analysis/graph.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/ensembldb.Rmd) and HTML
(docs/ensembldb.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | d9e2337 | Dave Tang | 2023-08-28 | Genome to transcript |
| html | eac5905 | Dave Tang | 2023-02-13 | Build site. |
| Rmd | 28276f9 | Dave Tang | 2023-02-13 | Using ensembldb |
The ensembldb package can be used to retrieve genomic
and protein annotations and to map between protein, transcript, and
genome coordinates. This mapping relies on annotations of proteins
(their sequences) to their encoding transcripts which are stored in
EnsDb databases.
All functions, except
proteinToGenomeandtranscriptToGenomereturnIRangeswith negative coordinates if the mapping failed (e.g. because the identifier is unknown to the database, or if, for mappings to and from protein coordinates, the input coordinates are not within the coding region of a transcript).proteinToGenomeandtranscriptToGenomereturn emptyGRangesif mappings fail.
Installation
To begin, install the ensembldb and AnnotationHub packages.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
deps <- c("ensembldb", "AnnotationHub", "Gviz")
sapply(deps, function(x){
if (!require(x, quietly = TRUE, character.only = TRUE))
BiocManager::install(x)
})$ensembldb
NULL
$AnnotationHub
NULL
$Gviz
NULLlibrary(ensembldb)
library(AnnotationHub)
library(Gviz)AnnotationHub
The AnnotationHub server provides easy R / Bioconductor access to large collections of publicly available whole genome resources, e.g,. ENSEMBL genome fasta or gtf files, UCSC chain resources, ENCODE data tracks at UCSC, etc.
Create an AnnotationHub object.
ah <- AnnotationHub(ask = FALSE)
ahAnnotationHub with 70130 records
# snapshotDate(): 2023-04-24
# $dataprovider: Ensembl, BroadInstitute, UCSC, ftp://ftp.ncbi.nlm.nih.gov/g...
# $species: Homo sapiens, Mus musculus, Drosophila melanogaster, Bos taurus,...
# $rdataclass: GRanges, TwoBitFile, BigWigFile, EnsDb, Rle, OrgDb, ChainFile...
# additional mcols(): taxonomyid, genome, description,
# coordinate_1_based, maintainer, rdatadateadded, preparerclass, tags,
# rdatapath, sourceurl, sourcetype
# retrieve records with, e.g., 'object[["AH5012"]]'
title
AH5012 | Chromosome Band
AH5013 | STS Markers
AH5014 | FISH Clones
AH5015 | Recomb Rate
AH5016 | ENCODE Pilot
... ...
AH113536 | org.Alternaria_alternata.eg.sqlite
AH113537 | org.Alternaria_tenuis.eg.sqlite
AH113538 | org.Torula_alternata.eg.sqlite
AH113539 | org.Psilocybe_cubensis.eg.sqlite
AH113540 | org.Stropharia_cubensis.eg.sqlite Query.
ensdb_homo <- query(ah, c("EnsDb", "Homo sapiens"))Latest available GENCODE version, which is quite old.
latest <- nrow(mcols(ensdb_homo))
edb <- ensdb_homo[[latest]]loading from cacheedbEnsDb for Ensembl:
|Backend: SQLite
|Db type: EnsDb
|Type of Gene ID: Ensembl Gene ID
|Supporting package: ensembldb
|Db created by: ensembldb package from Bioconductor
|script_version: 0.3.10
|Creation time: Thu Feb 16 12:36:05 2023
|ensembl_version: 109
|ensembl_host: localhost
|Organism: Homo sapiens
|taxonomy_id: 9606
|genome_build: GRCh38
|DBSCHEMAVERSION: 2.2
|common_name: human
|species: homo_sapiens
| No. of genes: 70623.
| No. of transcripts: 276218.
|Protein data available.Mapping genome coordinates to transcript coordinates
The
genomeToTranscriptfunction maps genomic coordinates to coordinates within the transcript(s) encoded at the specified coordinates. The function takes a GRanges as input and returns an IRangesList of length equal to the length of the input object. Each IRanges in the IRangesList provides the coordinates within the respective transcript.
The genomic region 17:7687460-7687515 contains the start of the TP53 gene (ENST00000269305.9) with some coordinates beyond the start.
gnm <- GRanges("17:7687460-7687515")Visualise using Gviz.
options(ucscChromosomeNames = FALSE)
gat <- GenomeAxisTrack(range = gnm)
gnm_gns <- getGeneRegionTrackForGviz(edb, filter = GRangesFilter(gnm))
gtx <- GeneRegionTrack(gnm_gns, name = "tx", geneSymbol = TRUE,
showId = TRUE)
ht <- HighlightTrack(trackList = list(gat, gtx), range = gnm)
plotTracks(list(ht))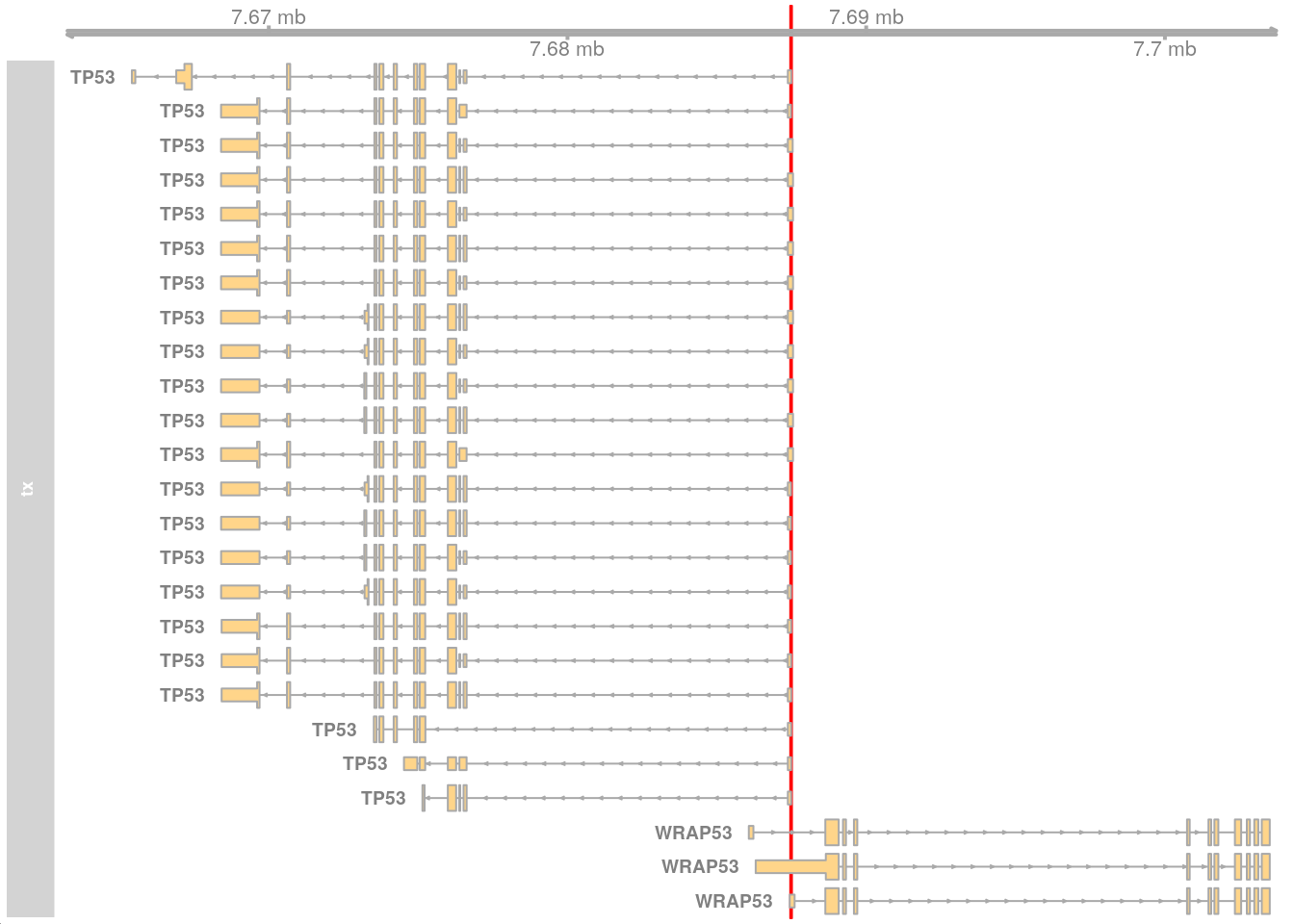
This works but the Ensembl ID does not match the GENCODE ID (ENST00000269305.9).
gnm_tx <- genomeToTranscript(gnm, edb)
gnm_txIRangesList object of length 1:
[[1]]
IRanges object with 12 ranges and 7 metadata columns:
start end width | tx_id
<integer> <integer> <integer> | <character>
ENST00000316024 1160 1215 56 | ENST00000316024
ENST00000457584 21 76 56 | ENST00000457584
ENST00000620739 24 79 56 | ENST00000620739
LRG_321t1-1 36 91 56 | LRG_321t1-1
LRG_321t1-2 36 91 56 | LRG_321t1-2
... ... ... ... . ...
LRG_321t3-1 36 91 56 | LRG_321t3-1
LRG_321t3-2 36 91 56 | LRG_321t3-2
LRG_321t4-1 36 91 56 | LRG_321t4-1
LRG_321t4-2 36 91 56 | LRG_321t4-2
LRG_321t8 36 91 56 | LRG_321t8
exon_id exon_rank seq_start seq_end seq_name
<character> <integer> <integer> <integer> <character>
ENST00000316024 ENSE00001897389 1 7687460 7687515 17
ENST00000457584 ENSE00001710635 1 7687460 7687515 17
ENST00000620739 ENSE00001146308 1 7687460 7687515 17
LRG_321t1-1 LRG_321t1e1 1 7687460 7687515 17
LRG_321t1-2 LRG_321t1e1 1 7687460 7687515 17
... ... ... ... ... ...
LRG_321t3-1 LRG_321t1e1 1 7687460 7687515 17
LRG_321t3-2 LRG_321t1e1 1 7687460 7687515 17
LRG_321t4-1 LRG_321t1e1 1 7687460 7687515 17
LRG_321t4-2 LRG_321t1e1 1 7687460 7687515 17
LRG_321t8 LRG_321t1e1 1 7687460 7687515 17
seq_strand
<character>
ENST00000316024 *
ENST00000457584 *
ENST00000620739 *
LRG_321t1-1 *
LRG_321t1-2 *
... ...
LRG_321t3-1 *
LRG_321t3-2 *
LRG_321t4-1 *
LRG_321t4-2 *
LRG_321t8 *Mapping protein coordinates to the genome coordinates
The
proteinToGenomefunction allows to map coordinates within the amino acid sequence of a protein to the corresponding DNA sequence on the genome. A protein identifier and the coordinates of the sequence within its amino acid sequence are required and have to be passed as anIRangesobject to the function. The protein identifier can either be passed as names of this object, or provided in a metadata column (mcols).
The example below (from the vignette) maps positions 5 to 9 within the amino acid sequence of the protein ENSP00000385415.
GAGE10_prt <- IRanges(start = 5, end = 9, names = "ENSP00000385415")
GAGE10_gnm <- proteinToGenome(GAGE10_prt, edb)Fetching CDS for 1 proteins ... 1 found
Checking CDS and protein sequence lengths ... 1/1 OKGAGE10_gnm$ENSP00000385415
GRanges object with 1 range and 7 metadata columns:
seqnames ranges strand | protein_id tx_id
<Rle> <IRanges> <Rle> | <character> <character>
[1] X 49304872-49304886 + | ENSP00000385415 ENST00000407599
exon_id exon_rank cds_ok protein_start protein_end
<character> <integer> <logical> <integer> <integer>
[1] ENSE00001692657 2 TRUE 5 9
-------
seqinfo: 1 sequence from GRCh38 genomeThe result is returned in a list, with one element for each range in
the input IRanges.
Below is an example with two proteins.
two_prt <- IRanges(
start = c(6, 15),
end = c(6, 15),
names = c("ENSP00000366863", "ENSP00000358262")
)
two_prt_to_gnm <- proteinToGenome(two_prt, edb)Fetching CDS for 2 proteins ... 2 found
Checking CDS and protein sequence lengths ... 2/2 OKtwo_prt_to_gnm$ENSP00000366863
GRanges object with 1 range and 7 metadata columns:
seqnames ranges strand | protein_id tx_id
<Rle> <IRanges> <Rle> | <character> <character>
[1] 13 75481750-75481752 - | ENSP00000366863 ENST00000377636
exon_id exon_rank cds_ok protein_start protein_end
<character> <integer> <logical> <integer> <integer>
[1] ENSE00003893703 1 TRUE 6 6
-------
seqinfo: 2 sequences from GRCh38 genome
$ENSP00000358262
GRanges object with 1 range and 7 metadata columns:
seqnames ranges strand | protein_id tx_id
<Rle> <IRanges> <Rle> | <character> <character>
[1] 1 147242746-147242748 + | ENSP00000358262 ENST00000369258
exon_id exon_rank cds_ok protein_start protein_end
<character> <integer> <logical> <integer> <integer>
[1] ENSE00003728289 1 TRUE 15 15
-------
seqinfo: 2 sequences from GRCh38 genomeWe use sapply() to convert the results into a data
frame.
get_pos <- function(x, add_chr = TRUE){
chr <- as.character(seqnames(x))
if(add_chr){
chr <- paste0("chr", chr)
}
list(
chr = chr,
start = start(x),
end = end(x)
)
}
as.data.frame(
t(sapply(two_prt_to_gnm, get_pos))
) chr start end
ENSP00000366863 chr13 75481750 75481752
ENSP00000358262 chr1 147242746 147242748Further reading
sessionInfo()R version 4.3.0 (2023-04-21)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] Gviz_1.44.1 AnnotationHub_3.8.0 BiocFileCache_2.8.0
[4] dbplyr_2.3.2 ensembldb_2.24.0 AnnotationFilter_1.24.0
[7] GenomicFeatures_1.52.2 AnnotationDbi_1.62.2 Biobase_2.60.0
[10] GenomicRanges_1.52.0 GenomeInfoDb_1.36.2 IRanges_2.34.1
[13] S4Vectors_0.38.1 BiocGenerics_0.46.0 BiocManager_1.30.21
[16] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 rstudioapi_0.14
[3] jsonlite_1.8.5 magrittr_2.0.3
[5] rmarkdown_2.22 fs_1.6.2
[7] BiocIO_1.10.0 zlibbioc_1.46.0
[9] vctrs_0.6.2 memoise_2.0.1
[11] Rsamtools_2.16.0 RCurl_1.98-1.12
[13] base64enc_0.1-3 htmltools_0.5.5
[15] S4Arrays_1.0.5 progress_1.2.2
[17] curl_5.0.1 Formula_1.2-5
[19] sass_0.4.6 bslib_0.5.0
[21] htmlwidgets_1.6.2 cachem_1.0.8
[23] GenomicAlignments_1.36.0 whisker_0.4.1
[25] mime_0.12 lifecycle_1.0.3
[27] pkgconfig_2.0.3 Matrix_1.5-4
[29] R6_2.5.1 fastmap_1.1.1
[31] GenomeInfoDbData_1.2.10 MatrixGenerics_1.12.3
[33] shiny_1.7.4 digest_0.6.31
[35] colorspace_2.1-0 ps_1.7.5
[37] rprojroot_2.0.3 Hmisc_5.1-0
[39] RSQLite_2.3.1 filelock_1.0.2
[41] fansi_1.0.4 httr_1.4.6
[43] abind_1.4-5 compiler_4.3.0
[45] withr_2.5.0 bit64_4.0.5
[47] backports_1.4.1 htmlTable_2.4.1
[49] BiocParallel_1.34.2 DBI_1.1.3
[51] highr_0.10 biomaRt_2.56.1
[53] rappdirs_0.3.3 DelayedArray_0.26.7
[55] rjson_0.2.21 tools_4.3.0
[57] foreign_0.8-84 interactiveDisplayBase_1.38.0
[59] httpuv_1.6.11 nnet_7.3-18
[61] glue_1.6.2 restfulr_0.0.15
[63] callr_3.7.3 promises_1.2.0.1
[65] checkmate_2.2.0 getPass_0.2-2
[67] cluster_2.1.4 generics_0.1.3
[69] gtable_0.3.3 BSgenome_1.68.0
[71] data.table_1.14.8 hms_1.1.3
[73] xml2_1.3.4 utf8_1.2.3
[75] XVector_0.40.0 BiocVersion_3.17.1
[77] pillar_1.9.0 stringr_1.5.0
[79] later_1.3.1 dplyr_1.1.2
[81] lattice_0.21-8 deldir_1.0-9
[83] rtracklayer_1.60.1 bit_4.0.5
[85] biovizBase_1.48.0 tidyselect_1.2.0
[87] Biostrings_2.68.1 knitr_1.43
[89] git2r_0.32.0 gridExtra_2.3
[91] ProtGenerics_1.32.0 SummarizedExperiment_1.30.2
[93] xfun_0.39 matrixStats_1.0.0
[95] stringi_1.7.12 lazyeval_0.2.2
[97] yaml_2.3.7 evaluate_0.21
[99] codetools_0.2-19 interp_1.1-4
[101] tibble_3.2.1 cli_3.6.1
[103] rpart_4.1.19 xtable_1.8-4
[105] munsell_0.5.0 processx_3.8.1
[107] jquerylib_0.1.4 dichromat_2.0-0.1
[109] Rcpp_1.0.10 png_0.1-8
[111] XML_3.99-0.14 parallel_4.3.0
[113] ellipsis_0.3.2 ggplot2_3.4.2
[115] blob_1.2.4 prettyunits_1.1.1
[117] jpeg_0.1-10 latticeExtra_0.6-30
[119] bitops_1.0-7 VariantAnnotation_1.46.0
[121] scales_1.2.1 purrr_1.0.1
[123] crayon_1.5.2 rlang_1.1.1
[125] KEGGREST_1.40.0