edgeR versus DESeq2
2024-10-23
Last updated: 2024-10-23
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version e0abadf. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/pbmc3k.csv
Ignored: data/pbmc3k.csv.gz
Ignored: data/pbmc3k/
Ignored: r_packages_4.4.0/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/edger_vs_deseq2.Rmd) and
HTML (docs/edger_vs_deseq2.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | e0abadf | Dave Tang | 2024-10-23 | Compare significances |
| html | a658556 | Dave Tang | 2024-10-23 | Build site. |
| Rmd | e471135 | Dave Tang | 2024-10-23 | edgeR versus DESeq2 |
Installation
Install packages using BiocManager::install().
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("edgeR")
BiocManager::install("DESeq2")Count table
https://zenodo.org/records/13970886
my_url <- 'https://zenodo.org/records/13970886/files/rsem.merged.gene_counts.tsv?download=1'
my_file <- 'rsem.merged.gene_counts.tsv'
if(file.exists(my_file) == FALSE){
download.file(url = my_url, destfile = my_file)
}
gene_counts <- read_tsv("rsem.merged.gene_counts.tsv", show_col_types = FALSE)
head(gene_counts)# A tibble: 6 × 10
gene_id `transcript_id(s)` ERR160122 ERR160123 ERR160124 ERR164473 ERR164550
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 ENSG0000… ENST00000373020,E… 2 6 5 374 1637
2 ENSG0000… ENST00000373031,E… 19 40 28 0 1
3 ENSG0000… ENST00000371582,E… 268. 274. 429. 489 637
4 ENSG0000… ENST00000367770,E… 360. 449. 566. 363. 606.
5 ENSG0000… ENST00000286031,E… 156. 185. 265. 85.4 312.
6 ENSG0000… ENST00000374003,E… 24 23 40 1181 423
# ℹ 3 more variables: ERR164551 <dbl>, ERR164552 <dbl>, ERR164554 <dbl>Metadata.
tibble::tribble(
~sample, ~run_id, ~group,
"C2_norm", "ERR160122", "normal",
"C3_norm", "ERR160123", "normal",
"C5_norm", "ERR160124", "normal",
"C1_norm", "ERR164473", "normal",
"C1_cancer", "ERR164550", "cancer",
"C2_cancer", "ERR164551", "cancer",
"C3_cancer", "ERR164552", "cancer",
"C5_cancer", "ERR164554", "cancer"
) -> my_metadata
my_metadata$group <- factor(my_metadata$group, levels = c('normal', 'cancer'))Matrix.
gene_counts |>
dplyr::select(starts_with("ERR")) |>
mutate(across(everything(), as.integer)) |>
as.matrix() -> gene_counts_mat
row.names(gene_counts_mat) <- gene_counts$gene_id
idx <- match(colnames(gene_counts_mat), my_metadata$run_id)
colnames(gene_counts_mat) <- my_metadata$sample[idx]
tail(gene_counts_mat) C2_norm C3_norm C5_norm C1_norm C1_cancer C2_cancer C3_cancer
ENSG00000293594 0 0 0 0 0 0 0
ENSG00000293595 3 5 3 0 0 0 0
ENSG00000293596 0 0 0 0 0 0 0
ENSG00000293597 1 2 11 1 2 3 1
ENSG00000293599 2 0 1 0 1 2 0
ENSG00000293600 45 59 85 561 789 1099 701
C5_cancer
ENSG00000293594 0
ENSG00000293595 0
ENSG00000293596 0
ENSG00000293597 2
ENSG00000293599 0
ENSG00000293600 845Remove genes that are lowly expressed.
keep <- rowSums(cpm(gene_counts_mat) > 0.5) >= 2
gene_counts_mat <- gene_counts_mat[keep, ]
tail(gene_counts_mat) C2_norm C3_norm C5_norm C1_norm C1_cancer C2_cancer C3_cancer
ENSG00000293576 0 7 12 0 0 0 0
ENSG00000293586 157 157 193 21 40 15 0
ENSG00000293587 3 3 5 0 2 1 0
ENSG00000293588 4 5 6 1 2 5 2
ENSG00000293595 3 5 3 0 0 0 0
ENSG00000293600 45 59 85 561 789 1099 701
C5_cancer
ENSG00000293576 0
ENSG00000293586 10
ENSG00000293587 3
ENSG00000293588 3
ENSG00000293595 0
ENSG00000293600 845edgeR workflow
y <- DGEList(
counts = gene_counts_mat,
group = my_metadata$group
)
y <- normLibSizes(y)
design <- model.matrix(~y$samples$group)
y <- estimateDisp(y, design, robust=TRUE)
fit <- glmQLFit(y, design, robust=TRUE)
res <- glmQLFTest(fit)
topTags(res, adjust.method = "BH")Coefficient: y$samples$groupcancer
logFC logCPM F PValue FDR
ENSG00000289381 -7.412756 2.341373 127.60623 2.767866e-08 0.001035846
ENSG00000151834 -8.027915 3.644102 64.37842 1.049799e-06 0.003612063
ENSG00000250696 -8.602174 3.809297 63.66033 1.249401e-06 0.003612063
ENSG00000229894 -9.123230 4.910552 60.16361 1.751012e-06 0.003612063
ENSG00000100985 5.735426 5.769059 99.70828 2.012691e-06 0.003612063
ENSG00000167910 -8.022082 3.453041 56.63286 2.199494e-06 0.003612063
ENSG00000196778 -8.815389 4.237774 56.19864 2.296578e-06 0.003612063
ENSG00000166091 -7.247200 2.881457 54.63510 2.658697e-06 0.003612063
ENSG00000240890 -9.077199 4.702122 53.69560 3.074356e-06 0.003612063
ENSG00000224781 -7.116543 2.556254 51.43346 3.645089e-06 0.003612063DESeq2 workflow
lung_cancer <- DESeqDataSetFromMatrix(
countData = gene_counts_mat,
colData = my_metadata,
design = ~ group
)
lung_cancer <- DESeq(lung_cancer)estimating size factorsestimating dispersionsgene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatesfitting model and testinglung_cancer_res <- results(lung_cancer, pAdjustMethod = "BH")
lung_cancer_res[order(lung_cancer_res$padj), ] |> head(10)log2 fold change (MLE): group cancer vs normal
Wald test p-value: group cancer vs normal
DataFrame with 10 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000211893 13264.627 6.54532 0.564034 11.60448 3.91049e-31
ENSG00000100985 627.926 5.35553 0.494870 10.82209 2.70522e-27
ENSG00000211892 4174.271 5.09062 0.482865 10.54252 5.50060e-26
ENSG00000169385 214.909 -4.60663 0.486305 -9.47271 2.72667e-21
ENSG00000172288 760.700 -27.98296 2.956493 -9.46492 2.93793e-21
ENSG00000236424 2182.510 -29.42478 3.099437 -9.49359 2.23219e-21
ENSG00000211897 6680.207 5.40162 0.574687 9.39924 5.49588e-21
ENSG00000211966 262.489 5.05816 0.539184 9.38115 6.52570e-21
ENSG00000290677 155.595 -4.71772 0.505092 -9.34033 9.60378e-21
ENSG00000182415 515.257 -27.44538 2.958942 -9.27541 1.76945e-20
padj
<numeric>
ENSG00000211893 1.43867e-26
ENSG00000100985 4.97625e-23
ENSG00000211892 6.74557e-22
ENSG00000169385 1.80144e-17
ENSG00000172288 1.80144e-17
ENSG00000236424 1.80144e-17
ENSG00000211897 2.88848e-17
ENSG00000211966 3.00101e-17
ENSG00000290677 3.92581e-17
ENSG00000182415 5.91801e-17Compare differentially expressed genes
my_thres <- 0.01
topTags(res, n = Inf, adjust.method = "BH") |>
as.data.frame() |>
dplyr::filter(FDR < my_thres) |>
row.names() -> edger_degs
lung_cancer_res |>
as.data.frame() |>
dplyr::filter(padj < my_thres) |>
row.names() -> deseq2_degs
jaccard_index <- function(set1, set2) {
length(intersect(set1, set2)) / length(union(set1, set2))
}
jaccard_index(edger_degs, deseq2_degs)[1] 0.2256522DESeq2 returns a lot more differentially expressed genes (DEGs) than edgeR.
length(edger_degs)[1] 3918length(deseq2_degs)[1] 17363Compare top subset.
compare_degs <- function(my_topn){
topTags(res, n = Inf, adjust.method = "BH") |>
as.data.frame() |>
dplyr::filter(FDR < my_thres) |>
dplyr::slice_min(order_by = FDR, n = my_topn) |>
row.names() -> edger_degs_topn
lung_cancer_res |>
as.data.frame() |>
dplyr::filter(padj < my_thres) |>
dplyr::slice_min(order_by = padj, n = my_topn) |>
row.names() -> deseq2_degs_topn
jaccard_index(edger_degs_topn, deseq2_degs_topn)
}
compare_degs(500)[1] 0.245122Jaccard indexes.
ns <- seq(100, 3500, 100)
jis <- sapply(ns, compare_degs)Plot.
data.frame(n = ns, index = jis) |>
ggplot(aes(n, index)) +
geom_point() +
theme_minimal() +
labs(x = 'Subset size', y = 'Jaccard Index')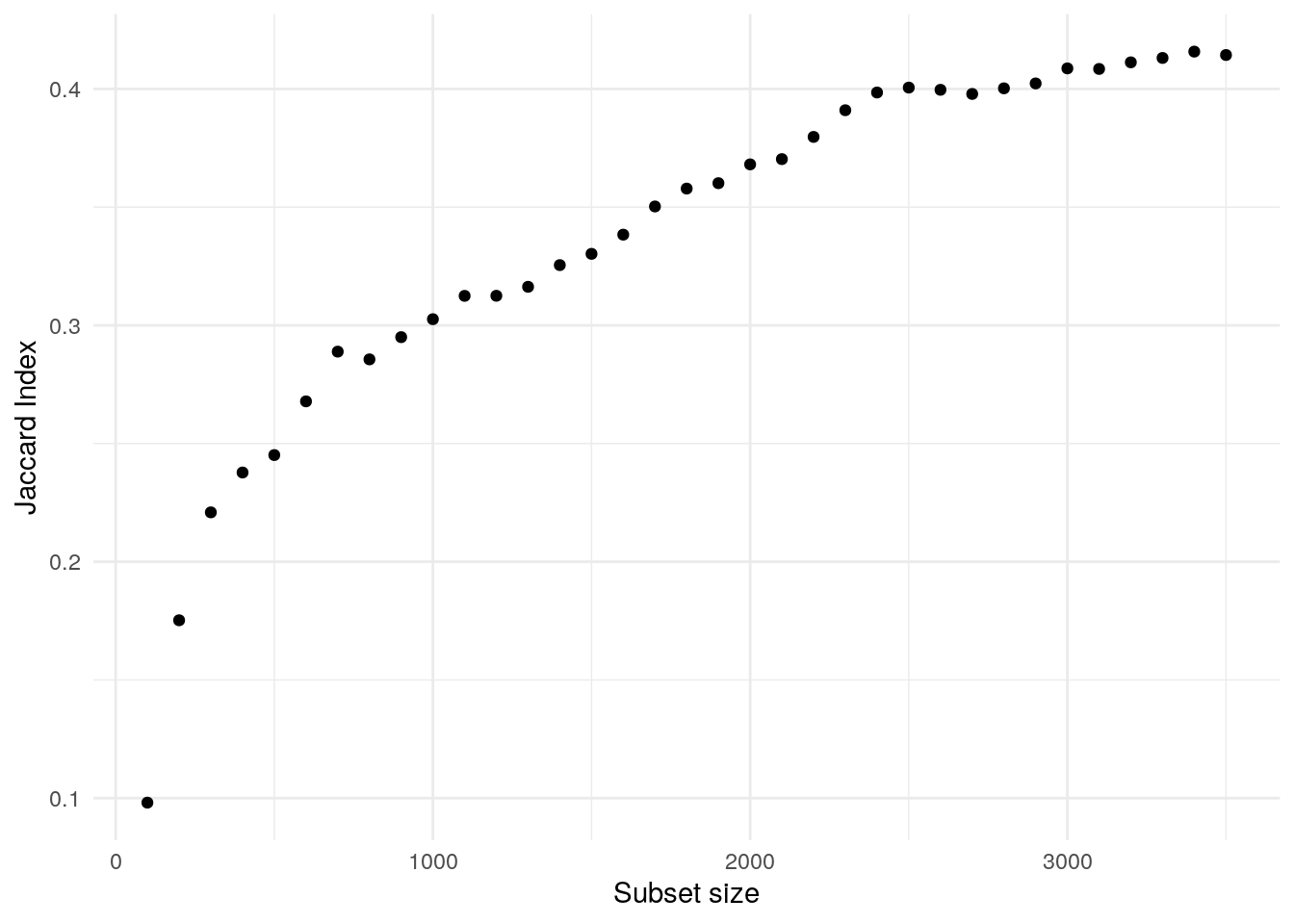
| Version | Author | Date |
|---|---|---|
| a658556 | Dave Tang | 2024-10-23 |
Compare significances
topTags(res, n = Inf, adjust.method = "BH") |>
as.data.frame() -> edger_signif
lung_cancer_res |>
as.data.frame() -> deseq2_signif
idx <- match(row.names(deseq2_signif), row.names(edger_signif))
my_signif <- cbind(edger_signif, deseq2_signif[idx, ])
my_signif |>
dplyr::filter(FDR < my_thres) |>
ggplot(aes(PValue, pvalue)) +
geom_point() +
theme_minimal() +
labs(x = "edgeR p-value", y = "DESeq2 p-value")Warning: Removed 41 rows containing missing values or values outside the scale range
(`geom_point()`).
Small subset of highly significant genes using edgeR but not significant using DESeq2.
my_signif |>
dplyr::filter(FDR < my_thres) |>
dplyr::filter(padj > 0.95) |>
row.names() -> discordant
gene_counts_mat[discordant, ] C2_norm C3_norm C5_norm C1_norm C1_cancer C2_cancer C3_cancer
ENSG00000229894 480 620 873 7 1 2 3
ENSG00000237931 76 165 117 2 3 2 0
ENSG00000236761 2682 3073 4410 8 9 2 9
ENSG00000245205 199 195 315 0 2 1 5
ENSG00000250130 30 36 47 2 2 3 1
ENSG00000231292 51 53 66 1 0 3 2
ENSG00000230445 2060 2282 3393 12 8 14 3
ENSG00000235847 81 102 147 0 1 0 0
ENSG00000155875 158 211 295 5 6 6 11
ENSG00000184100 676 765 1046 9 14 13 14
ENSG00000254042 26 21 31 0 2 1 1
ENSG00000215241 174 314 325 13 7 18 20
ENSG00000279058 892 1175 1370 13 26 20 14
ENSG00000290655 210 169 368 0 0 0 0
ENSG00000249077 16 27 19 0 1 1 1
ENSG00000257818 110 140 192 10 4 3 14
ENSG00000257225 236 337 532 11 0 2 17
ENSG00000233771 21 37 71 0 0 0 1
ENSG00000228960 201 485 665 0 0 14 0
ENSG00000164743 290 337 399 21 14 44 31
ENSG00000214184 47 33 56 0 4 4 3
ENSG00000242199 17 18 36 0 0 2 1
ENSG00000167941 11 17 37 1 1 0 3
ENSG00000229492 9 47 76 1 0 1 1
ENSG00000289604 2543 2097 3181 56 158 116 89
C5_cancer
ENSG00000229894 2
ENSG00000237931 1
ENSG00000236761 5
ENSG00000245205 4
ENSG00000250130 4
ENSG00000231292 0
ENSG00000230445 18
ENSG00000235847 3
ENSG00000155875 7
ENSG00000184100 20
ENSG00000254042 0
ENSG00000215241 12
ENSG00000279058 31
ENSG00000290655 2
ENSG00000249077 0
ENSG00000257818 12
ENSG00000257225 10
ENSG00000233771 2
ENSG00000228960 0
ENSG00000164743 26
ENSG00000214184 0
ENSG00000242199 0
ENSG00000167941 0
ENSG00000229492 0
ENSG00000289604 123
sessionInfo()R version 4.4.0 (2024-04-24)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] pheatmap_1.0.12 ggrepel_0.9.5
[3] DESeq2_1.44.0 SummarizedExperiment_1.34.0
[5] Biobase_2.64.0 MatrixGenerics_1.16.0
[7] matrixStats_1.3.0 GenomicRanges_1.56.1
[9] GenomeInfoDb_1.40.1 IRanges_2.38.1
[11] S4Vectors_0.42.1 BiocGenerics_0.50.0
[13] edgeR_4.2.1 limma_3.60.4
[15] lubridate_1.9.3 forcats_1.0.0
[17] stringr_1.5.1 dplyr_1.1.4
[19] purrr_1.0.2 readr_2.1.5
[21] tidyr_1.3.1 tibble_3.2.1
[23] ggplot2_3.5.1 tidyverse_2.0.0
[25] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 farver_2.1.2 fastmap_1.2.0
[4] promises_1.3.0 digest_0.6.37 timechange_0.3.0
[7] lifecycle_1.0.4 statmod_1.5.0 processx_3.8.4
[10] magrittr_2.0.3 compiler_4.4.0 rlang_1.1.4
[13] sass_0.4.9 tools_4.4.0 utf8_1.2.4
[16] yaml_2.3.8 knitr_1.47 labeling_0.4.3
[19] S4Arrays_1.4.1 bit_4.0.5 DelayedArray_0.30.1
[22] RColorBrewer_1.1-3 abind_1.4-5 BiocParallel_1.38.0
[25] withr_3.0.1 grid_4.4.0 fansi_1.0.6
[28] git2r_0.33.0 colorspace_2.1-0 scales_1.3.0
[31] cli_3.6.3 rmarkdown_2.27 crayon_1.5.2
[34] generics_0.1.3 rstudioapi_0.16.0 httr_1.4.7
[37] tzdb_0.4.0 cachem_1.1.0 splines_4.4.0
[40] zlibbioc_1.50.0 parallel_4.4.0 XVector_0.44.0
[43] vctrs_0.6.5 Matrix_1.7-0 jsonlite_1.8.8
[46] callr_3.7.6 hms_1.1.3 bit64_4.0.5
[49] locfit_1.5-9.9 jquerylib_0.1.4 glue_1.7.0
[52] codetools_0.2-20 ps_1.7.6 stringi_1.8.4
[55] gtable_0.3.5 later_1.3.2 UCSC.utils_1.0.0
[58] munsell_0.5.1 pillar_1.9.0 htmltools_0.5.8.1
[61] GenomeInfoDbData_1.2.12 R6_2.5.1 rprojroot_2.0.4
[64] vroom_1.6.5 evaluate_0.24.0 lattice_0.22-6
[67] highr_0.11 httpuv_1.6.15 bslib_0.7.0
[70] Rcpp_1.0.12 SparseArray_1.4.8 whisker_0.4.1
[73] xfun_0.44 fs_1.6.4 getPass_0.2-4
[76] pkgconfig_2.0.3