Getting started with Seurat
2024-04-15
Last updated: 2024-04-15
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 30c319d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: r_packages_4.3.0/
Ignored: r_packages_4.3.2/
Ignored: r_packages_4.3.3/
Untracked files:
Untracked: analysis/breast_cancer.Rmd
Untracked: code/multiz100way/
Untracked: data/dataset.h5ad
Untracked: data/lung_bcell.rds
Untracked: data/pbmc3k.csv
Untracked: data/pbmc3k.csv.gz
Untracked: data/pbmc3k/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/seurat.Rmd) and HTML
(docs/seurat.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 30c319d | Dave Tang | 2024-04-15 | Download example AnnData file |
| html | 237607a | Dave Tang | 2024-04-07 | Build site. |
| Rmd | 2ba0e6d | Dave Tang | 2024-04-07 | Fix zip conditional |
| html | 5f6b5a7 | Dave Tang | 2024-04-07 | Build site. |
| Rmd | f5d29f9 | Dave Tang | 2024-04-07 | Export as CSV |
| html | 261b999 | Dave Tang | 2024-03-20 | Build site. |
| Rmd | de75931 | Dave Tang | 2024-03-20 | Filtering on mitochondrial percent |
| html | 03bb7a1 | Dave Tang | 2024-03-20 | Build site. |
| Rmd | 0887184 | Dave Tang | 2024-03-20 | Complete official tutorial |
| html | 23b21ad | Dave Tang | 2024-03-20 | Build site. |
| Rmd | 64ac12f | Dave Tang | 2024-03-20 | Normalisation, variable genes, and scaling |
| html | 3c41e26 | Dave Tang | 2024-03-20 | Build site. |
| Rmd | d5ab5a8 | Dave Tang | 2024-03-20 | Basic filtering |
| html | 5de3e6c | Dave Tang | 2024-03-19 | Build site. |
| Rmd | 0e1dfa9 | Dave Tang | 2024-03-19 | Sparse matrix |
| html | 944e5f2 | Dave Tang | 2024-03-19 | Build site. |
| Rmd | ae0ea42 | Dave Tang | 2024-03-19 | Getting started with Seurat |
This post follows the Peripheral Blood Mononuclear Cells (PBMCs) tutorial for 2,700 single cells. It was written while I was going through the tutorial and contains my notes. The dataset for this tutorial can be downloaded from the 10X Genomics dataset page but it is also hosted on Amazon (see below). The PBMCs, which are primary cells with relatively small amounts of RNA (around 1pg RNA/cell), come from a healthy donor. There were 2,700 cells detected and sequencing was performed on an Illumina NextSeq 500 with around 69,000 reads per cell. To get started install Seurat by using install.packages().
install.packages("Seurat")If you get the warning:
‘SeuratObject’ was built under R 4.3.0 but the current version is 4.3.2; it is recomended that you reinstall ‘SeuratObject’ as the ABI for R may have changed
re-install the SeuratObject package using a repository
that has an updated copy. The same goes for the htmltools
package.
install.packages("SeuratObject", repos = "https://cran.ism.ac.jp/")
install.packages("htmltools", repos = "https://cran.ism.ac.jp/")
packageVersion("SeuratObject")
packageVersion("htmltools")Load Seurat.
library("Seurat")Loading required package: SeuratObjectLoading required package: sp'SeuratObject' was built under R 4.3.0 but the current version is
4.3.3; it is recomended that you reinstall 'SeuratObject' as the ABI
for R may have changed
Attaching package: 'SeuratObject'The following object is masked from 'package:base':
intersectpackageVersion("Seurat")[1] '5.0.3'Data
To follow the tutorial, you need the 10X data.
mkdir -p data/pbmc3k && cd data/pbmc3k
wget -c https://s3-us-west-2.amazonaws.com/10x.files/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gzThe extracted files.
ls -1 data/pbmc3k/filtered_gene_bc_matrices/hg19barcodes.tsv
genes.tsv
matrix.mtxmatrix.mtx is a MatrixMarket
file. It has the following properties:
- Only non-zero entries are stored in the file
- Comments start with a
%, like LaTeX - The first line indicates the total number of rows, columns, and entries
- The following lines after the first provide a row and column number and the value at that coordinate
head data/pbmc3k/filtered_gene_bc_matrices/hg19/matrix.mtx%%MatrixMarket matrix coordinate real general
%
32738 2700 2286884
32709 1 4
32707 1 1
32706 1 10
32704 1 1
32703 1 5
32702 1 6
32700 1 10Seurat object
Load 10x data into a matrix.
pbmc.data <- Read10X(data.dir = "data/pbmc3k/filtered_gene_bc_matrices/hg19/")class(pbmc.data)[1] "dgCMatrix"
attr(,"package")
[1] "Matrix"32,738 genes and 2,700 cells.
dim(pbmc.data)[1] 32738 2700Save as CSV file.
system.time(
write.csv(x = pbmc.data, file = "data/pbmc3k.csv")
) user system elapsed
54.959 0.572 100.727 Gzip.
if [[ ! -f data/pbmc3k.csv.gz ]]; then
gzip data/pbmc3k.csv
fi
ls -lh data/pbmc3k.csv.gz-rw-r--r-- 1 rstudio rstudio 3.4M Apr 7 13:19 data/pbmc3k.csv.gzCheck out the first six genes and cells
pbmc.data[1:6, 1:6]6 x 6 sparse Matrix of class "dgCMatrix"
AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1
MIR1302-10 . . .
FAM138A . . .
OR4F5 . . .
RP11-34P13.7 . . .
RP11-34P13.8 . . .
AL627309.1 . . .
AAACCGTGCTTCCG-1 AAACCGTGTATGCG-1 AAACGCACTGGTAC-1
MIR1302-10 . . .
FAM138A . . .
OR4F5 . . .
RP11-34P13.7 . . .
RP11-34P13.8 . . .
AL627309.1 . . .Summary of total expression per single cell.
summary(colSums(pbmc.data)) Min. 1st Qu. Median Mean 3rd Qu. Max.
548 1758 2197 2367 2763 15844 Check how many genes have at least one transcript in each cell.
The median number of detected genes among the single cells is 817.
at_least_one <- apply(pbmc.data, 2, function(x) sum(x>0))
hist(
at_least_one,
breaks = 100,
main = "Distribution of detected genes",
xlab = "Genes with at least one tag"
)
abline(v = median(at_least_one), col = 2, lty = 3)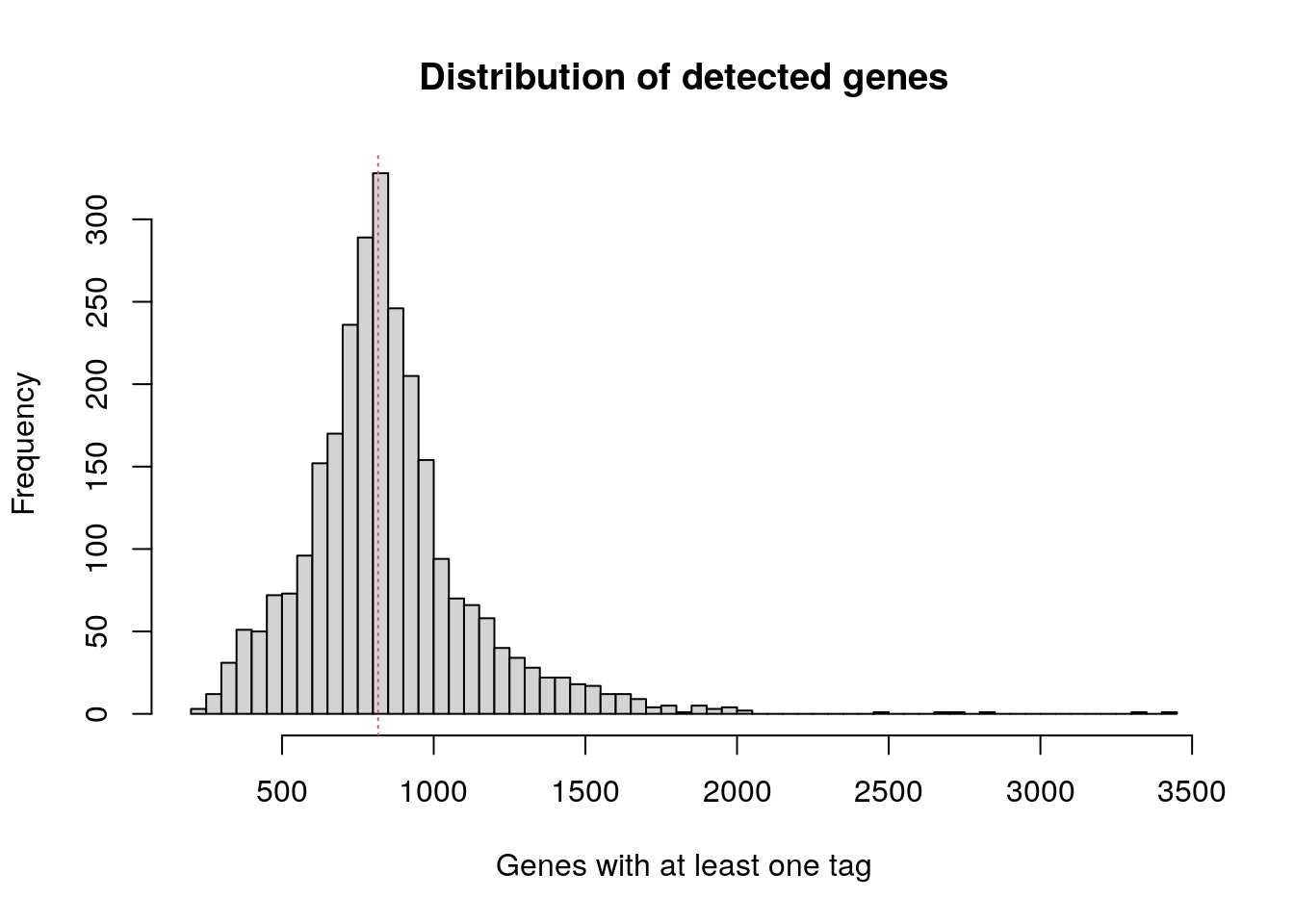
Total expression per cell. The median sum of expression among the single cells is 2,197. This distribution is very similar to the distribution of detected genes shown above.
hist(
colSums(pbmc.data),
breaks = 100,
main = "Expression sum per cell",
xlab = "Sum expression"
)
abline(v = median(colSums(pbmc.data)), col = 2, lty = 3)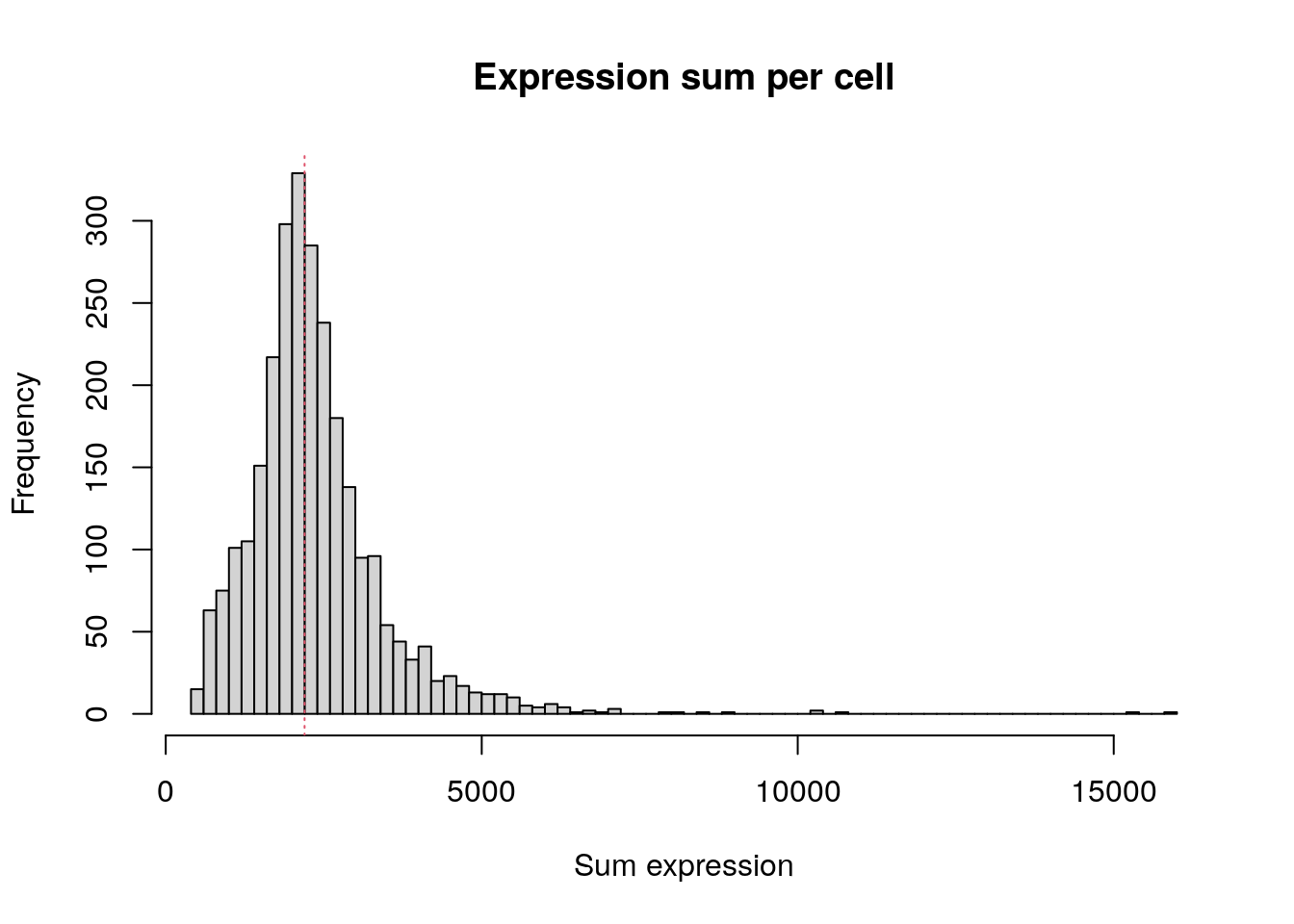
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
We will filter out genes and single cells before we continue with the analysis. The tutorial has arbitrary values of keeping genes expressed in three or more cells and keeping cells with at least 200 detected genes.
Manually check the number of genes detected in three or more cells; a lot of genes are not detected in 3 or more cells.
tmp <- apply(pbmc.data, 1, function(x) sum(x>0))
table(tmp>=3)
FALSE TRUE
19024 13714 All cells have at least 200 detected genes
keep <- tmp>=3
tmp <- pbmc.data[keep,]
at_least_one <- apply(tmp, 2, function(x) sum(x>0))
summary(at_least_one) Min. 1st Qu. Median Mean 3rd Qu. Max.
212.0 690.0 816.0 845.5 952.0 3400.0 dim(tmp)[1] 13714 2700See ?SeuratObject for more information on the class.
pbmc <- CreateSeuratObject(
counts = pbmc.data,
min.cells = 3,
min.features = 200,
project = "pbmc3k"
)Warning: Feature names cannot have underscores ('_'), replacing with dashes
('-')class(pbmc)[1] "Seurat"
attr(,"package")
[1] "SeuratObject"Same numbers as above
pbmcAn object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
1 layer present: countsSlots in Seurat object.
SeuratObject: Data Structures for Single Cell Data
Defines S4 classes for single-cell genomic data and associated information, such as dimensionality reduction embeddings, nearest-neighbor graphs, and spatially-resolved coordinates. Provides data access methods and R-native hooks to ensure the Seurat object is familiar to other R users
Read more about the S4 class in the Advanced R book.
slotNames(pbmc) [1] "assays" "meta.data" "active.assay" "active.ident" "graphs"
[6] "neighbors" "reductions" "images" "project.name" "misc"
[11] "version" "commands" "tools" Basic filtering
The tutorial states that “The number of genes and UMIs (nGene and
nUMI) are automatically calculated for every object by Seurat.” The nUMI
is calculated as num.mol <- colSums(object.raw.data),
i.e. each transcript is a unique molecule. The number of genes is simply
the tally of genes with at least 1 transcript;
num.genes <- colSums(object.raw.data > is.expr) where
is.expr is zero.
A common quality control metric is the percentage of transcripts from the mitochondrial genome. According to the paper Classification of low quality cells from single-cell RNA-seq data the reason this is a quality control metric is because if a single cell is lysed, cytoplasmic RNA will be lost apart from the RNA that is enclosed in the mitochondria, which will be retained and sequenced.
Human mitochondria genes conveniently start with MT.
mito.genes <- grep(pattern = "^MT-", x = rownames(x = pbmc@assays$RNA), value = TRUE)
length(mito.genes)[1] 13percent.mito <- Matrix::colSums(pbmc[['RNA']]$counts[mito.genes, ]) / Matrix::colSums(pbmc[['RNA']]$counts) * 100
head(percent.mito)AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
3.0177759 3.7935958 0.8897363 1.7430845
AAACCGTGTATGCG-1 AAACGCACTGGTAC-1
1.2244898 1.6643551 Check out the meta data.
head(pbmc@meta.data) orig.ident nCount_RNA nFeature_RNA
AAACATACAACCAC-1 pbmc3k 2419 779
AAACATTGAGCTAC-1 pbmc3k 4903 1352
AAACATTGATCAGC-1 pbmc3k 3147 1129
AAACCGTGCTTCCG-1 pbmc3k 2639 960
AAACCGTGTATGCG-1 pbmc3k 980 521
AAACGCACTGGTAC-1 pbmc3k 2163 781Add mitochondrial percent to the meta using
AddMetaData.
pbmc <- AddMetaData(
object = pbmc,
metadata = percent.mito,
col.name = "percent.mito"
)
head(pbmc@meta.data) orig.ident nCount_RNA nFeature_RNA percent.mito
AAACATACAACCAC-1 pbmc3k 2419 779 3.0177759
AAACATTGAGCTAC-1 pbmc3k 4903 1352 3.7935958
AAACATTGATCAGC-1 pbmc3k 3147 1129 0.8897363
AAACCGTGCTTCCG-1 pbmc3k 2639 960 1.7430845
AAACCGTGTATGCG-1 pbmc3k 980 521 1.2244898
AAACGCACTGGTAC-1 pbmc3k 2163 781 1.6643551Use PercentageFeatureSet to calculate the percentage of
a set of features, which saves us from some typing. The [[
operator can add columns to object metadata, which is a great place to
stash QC stats.
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
head(pbmc@meta.data[, c('percent.mito', 'percent.mt')]) percent.mito percent.mt
AAACATACAACCAC-1 3.0177759 3.0177759
AAACATTGAGCTAC-1 3.7935958 3.7935958
AAACATTGATCAGC-1 0.8897363 0.8897363
AAACCGTGCTTCCG-1 1.7430845 1.7430845
AAACCGTGTATGCG-1 1.2244898 1.2244898
AAACGCACTGGTAC-1 1.6643551 1.6643551Instead of setting a hard filtering threshold, one can use the 3 * Median Absolute Deviation to determine threshold limits for filtering out cells.
median(pbmc@meta.data$percent.mt) - 3 * mad(pbmc@meta.data$percent.mt)[1] -0.3751742median(pbmc@meta.data$percent.mt) + 3 * mad(pbmc@meta.data$percent.mt)[1] 4.436775Plot number of genes, UMIs, and % mitochondria
Visualize QC metrics as a violin plot
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)Warning: Default search for "data" layer in "RNA" assay yielded no results;
utilizing "counts" layer instead.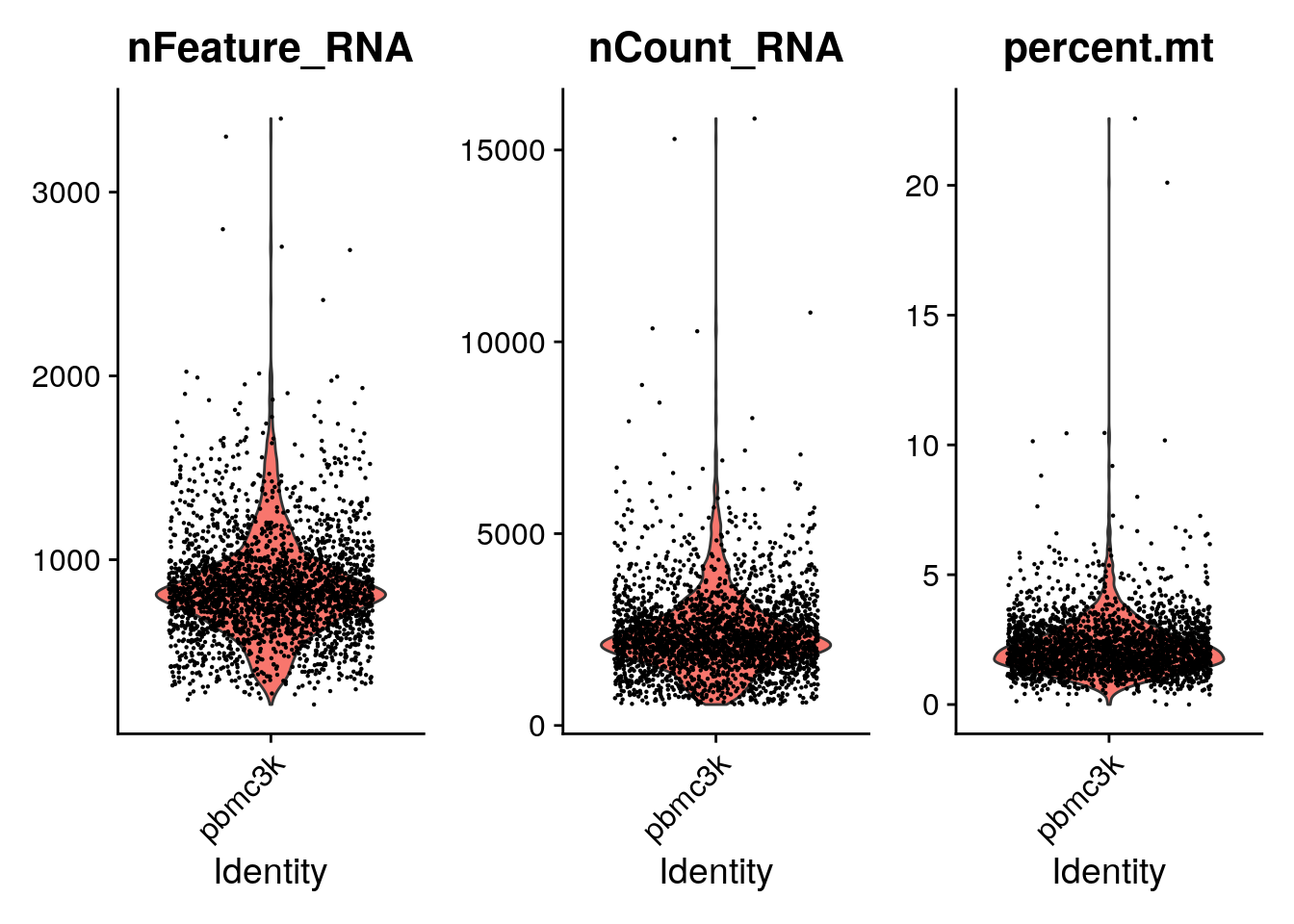
A couple of cells have high mitochondrial percentage which may indicate lost of cytoplasmic RNA.
The GenePlot() function can be used to visualise gene-gene relationships as well as any columns in the seurat object. Below we use the plotting function to spot cells that have a high percentage of mitochondrial RNA and to plot the relationship between the number of unique molecules and the number of genes captured.
FeatureScatter is typically used to visualize feature-feature relationships, but can be used for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2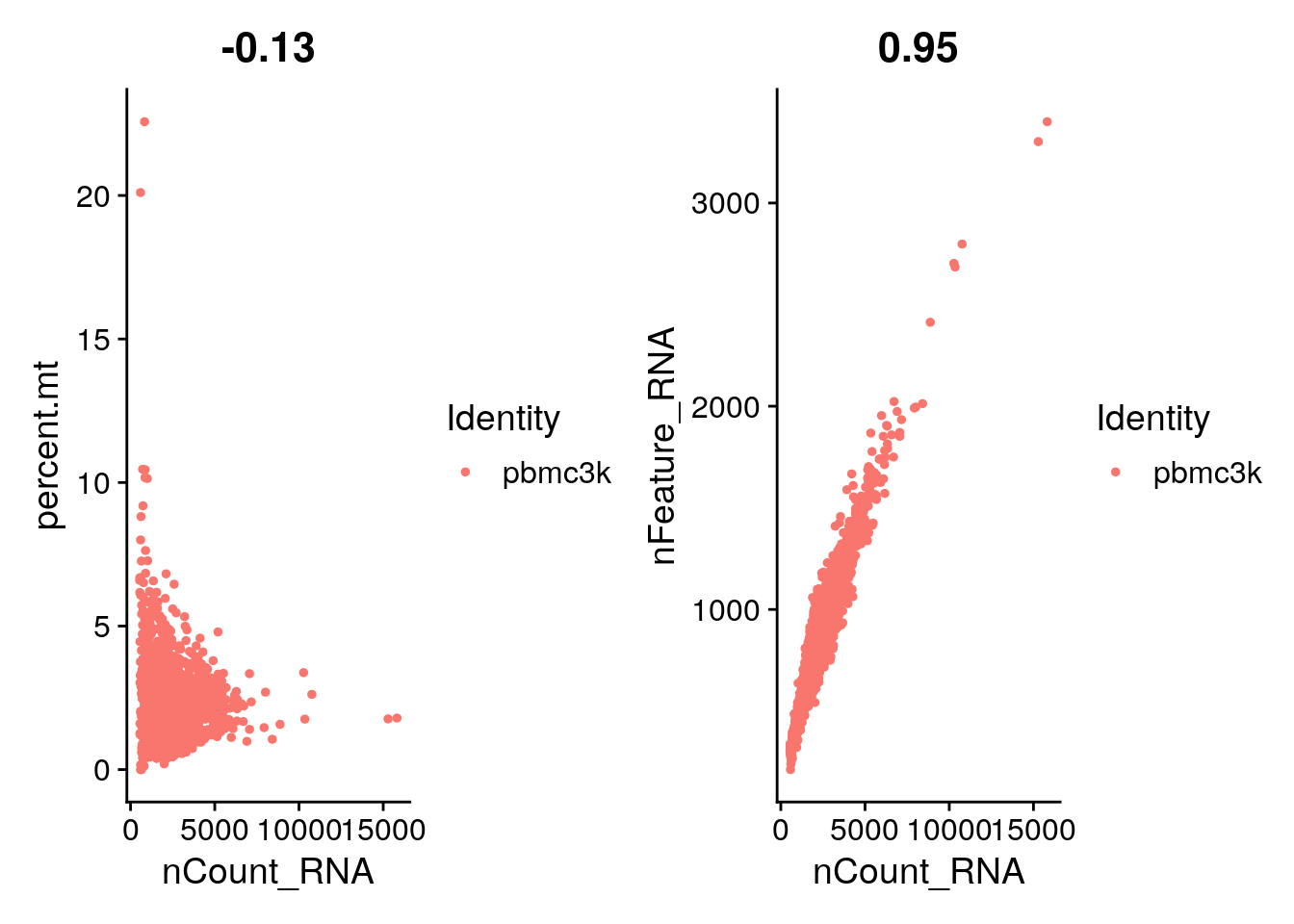
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
Manual check; I already know all cells have >200 genes.
table(pbmc@meta.data$percent.mito < 5 & pbmc@meta.data$nFeature_RNA<2500)
FALSE TRUE
62 2638 pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmcAn object of class Seurat
13714 features across 2638 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
1 layer present: countsNormalisation
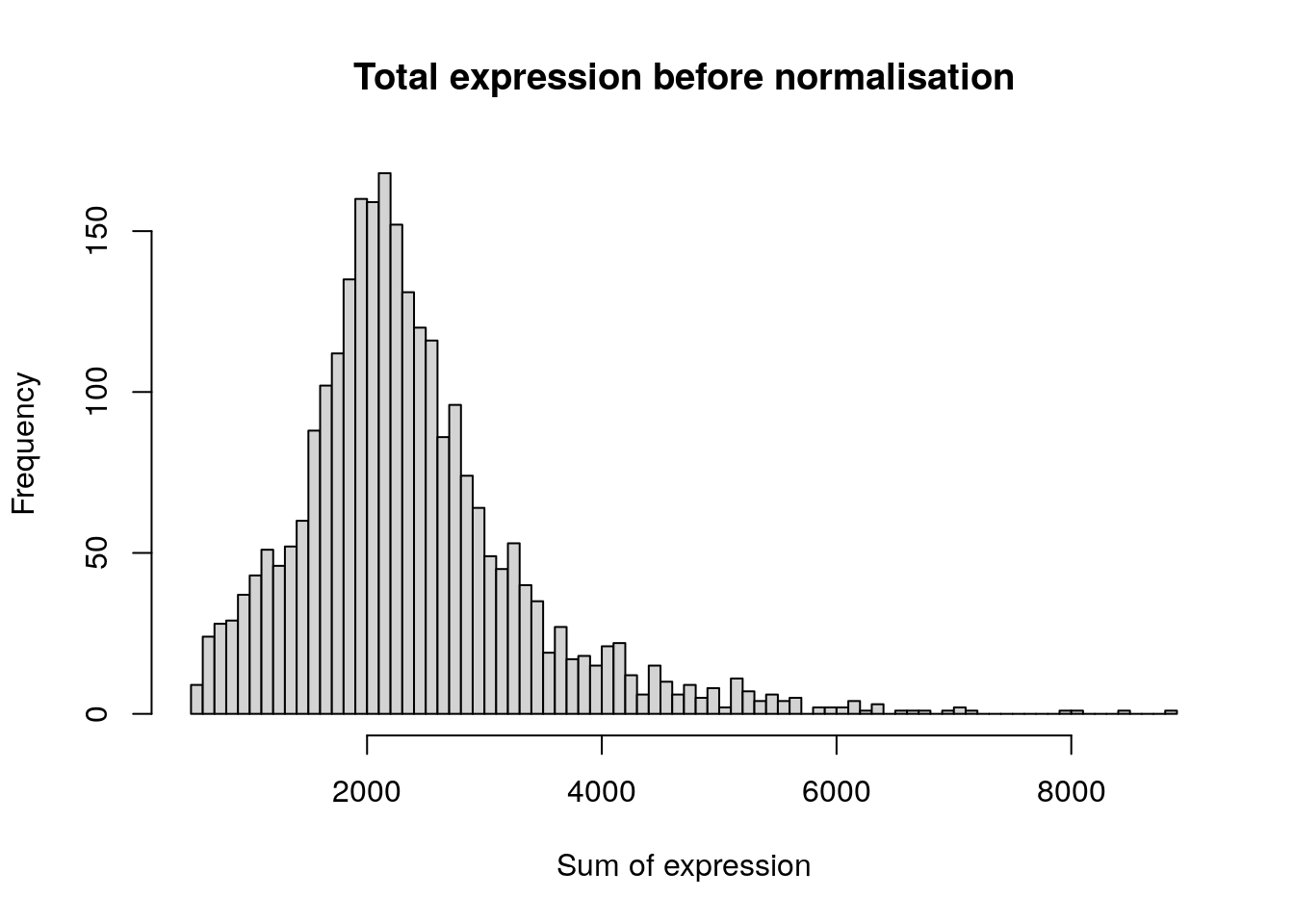
The next step is to normalise the data, so that each cell can be compared against each other. At the time of writing, the only normalisation method implemented in Seurat is by log normalisation. Gene expression measurements for each cell are normalised by its total expression, scaled by 10,000, and log-transformed.
hist(
colSums(pbmc[['RNA']]$counts),
breaks = 100,
main = "Total expression before normalisation",
xlab = "Sum of expression"
)
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
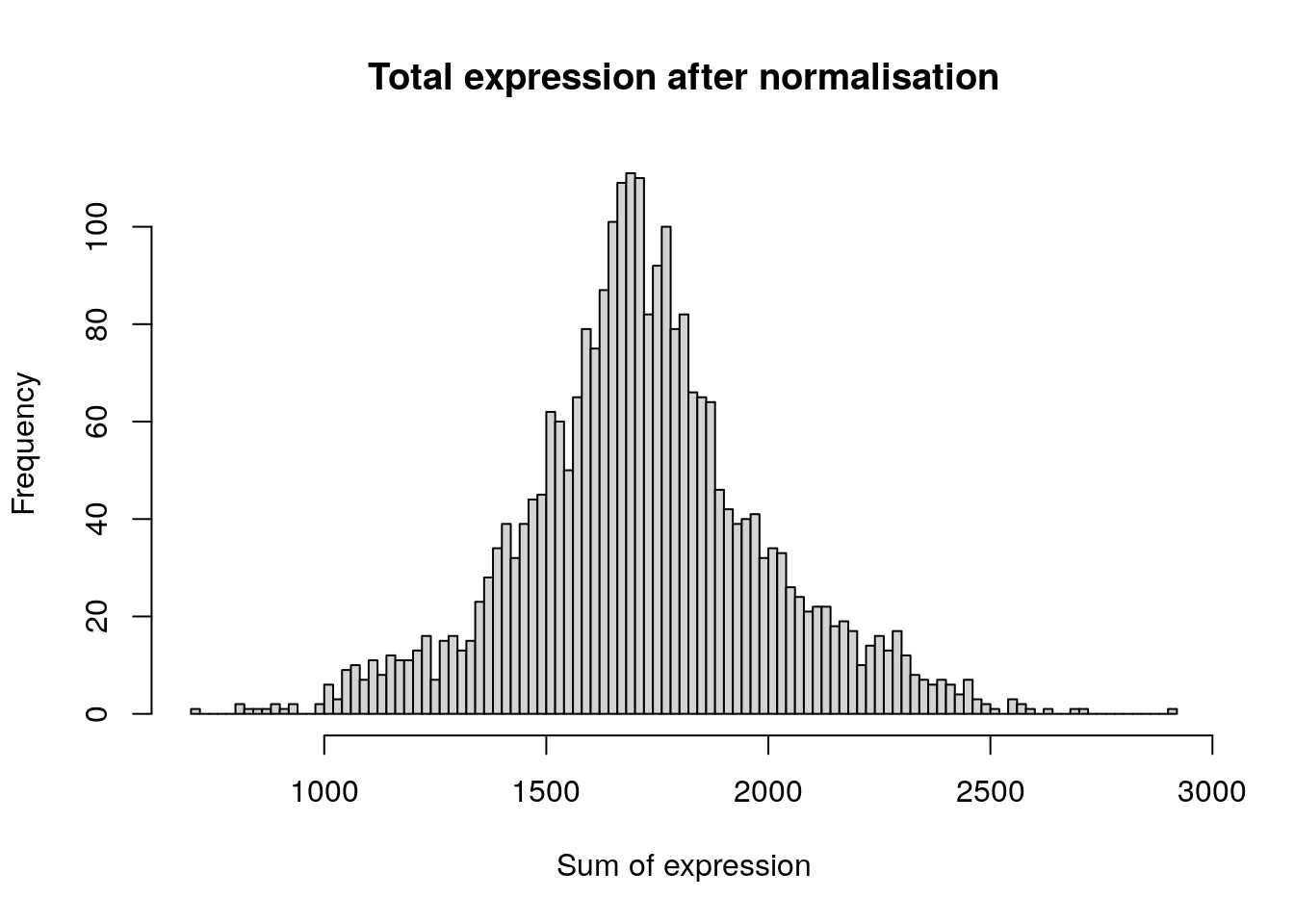
After removing unwanted cells from the dataset, the next step is to normalise the data. By default, we employ a global-scaling normalization method “LogNormalize” that normalises the feature expression measurements for each cell by the total expression, multiplies this by a scale factor (10,000 by default), and log-transforms the result. In Seurat v5, Normalized values are stored in pbmc[[“RNA”]]$data.
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)Normalizing layer: countsFor clarity, in this previous line of code (and in future commands), we provide the default values for certain parameters in the function call. However, this isn’t required and the same behavior can be achieved with:
pbmc <- NormalizeData(pbmc)Normalizing layer: countsWhile this method of normalization is standard and widely used in scRNA-seq analysis, global-scaling relies on an assumption that each cell originally contains the same number of RNA molecules. We and others have developed alternative workflows for the single cell preprocessing that do not make these assumptions. For users who are interested, please check out our SCTransform() normalization workflow. The method is described in ourpaper, with a separate vignette using Seurat here. The use of SCTransform replaces the need to run NormalizeData, FindVariableFeatures, or ScaleData (described below.)
hist(
colSums(pbmc[['RNA']]$data),
breaks = 100,
main = "Total expression after normalisation",
xlab = "Sum of expression"
)
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
Identification of highly variable features (feature selection)
Once the data is normalised, the next step is to find genes are vary
between single cells; genes that are constant among all cells have no
distinguishing power. The FindVariableFeatures() function
calculates the average expression and dispersion for each gene, places
these genes into bins, and then calculates a z-score for dispersion
within each bin. I interpret that as take each gene, get the average
expression and variance of the gene across the 2,638 cells, categorise
genes into bins (default is 20) based on their expression and variance,
and finally normalise the variance in each bin. This was the same
approach in Macosko et al.
and new methods for detecting genes with variable expression patterns
will be implemented in Seurat soon (according to the tutorial). The
parameters used below are typical settings for UMI data that is
normalised to a total of 10,000 molecules and will identify around 2,000
variable genes. The tutorial recommends that users should explore the
parameters themselves since each dataset is different.
We next calculate a subset of features that exhibit high cell-to-cell variation in the dataset (i.e, they are highly expressed in some cells, and lowly expressed in others). We and others have found that focusing on these genes in downstream analysis helps to highlight biological signal in single-cell datasets.
Our procedure in Seurat is described in detail here, and improves on previous versions by directly modeling the mean-variance relationship inherent in single-cell data, and is implemented in the FindVariableFeatures() function. By default, we return 2,000 features per dataset. These will be used in downstream analysis, like PCA.
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)Finding variable features for layer countslength(VariableFeatures(pbmc))[1] 2000Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(pbmc), 10)
top10 [1] "PPBP" "LYZ" "S100A9" "IGLL5" "GNLY" "FTL" "PF4" "FTH1"
[9] "GNG11" "S100A8"Plot variable features with and without labels
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)When using repel, set xnudge and ynudge to 0 for optimal resultsplot1 + plot2Warning in scale_x_log10(): log-10 transformation introduced infinite values.
log-10 transformation introduced infinite values.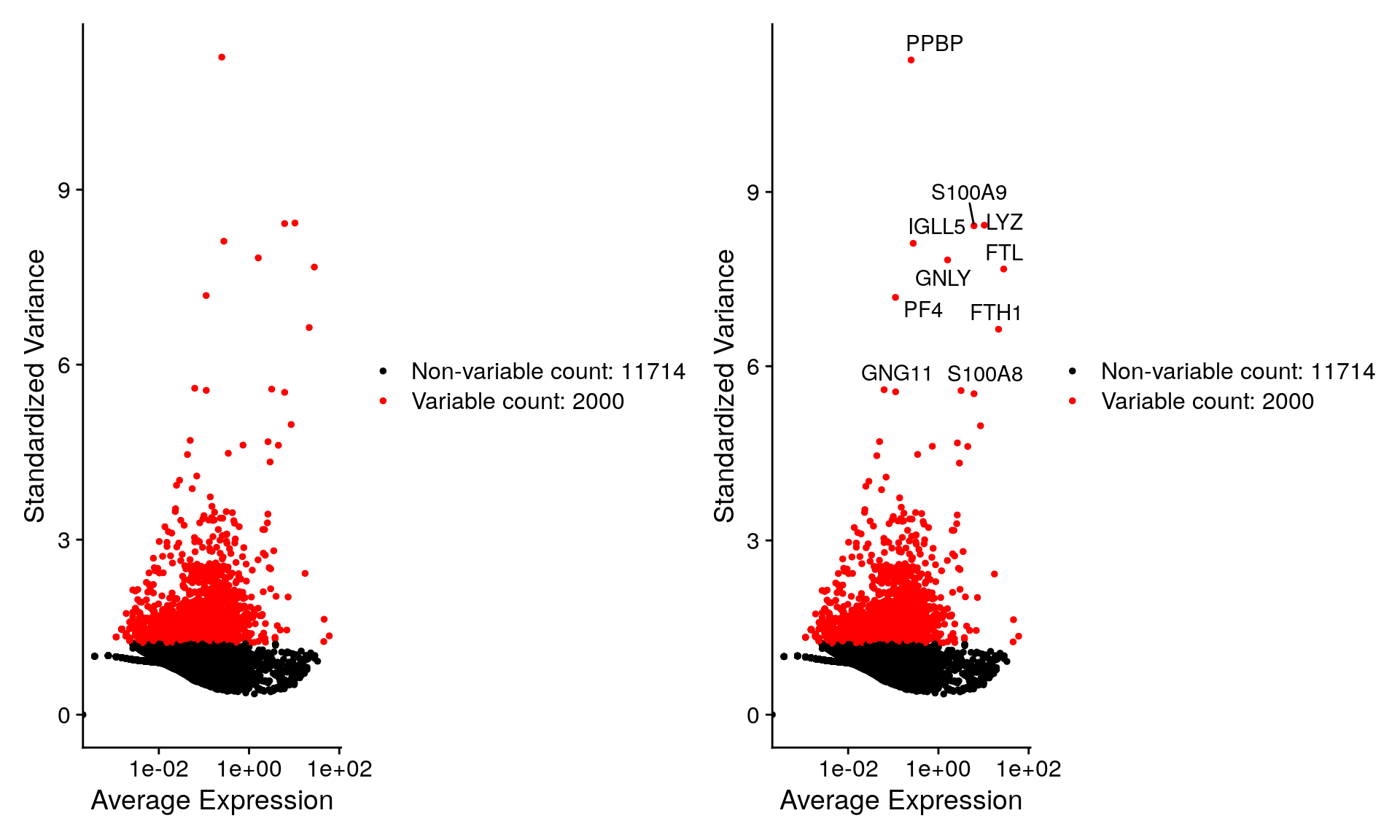
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
Scaling the data
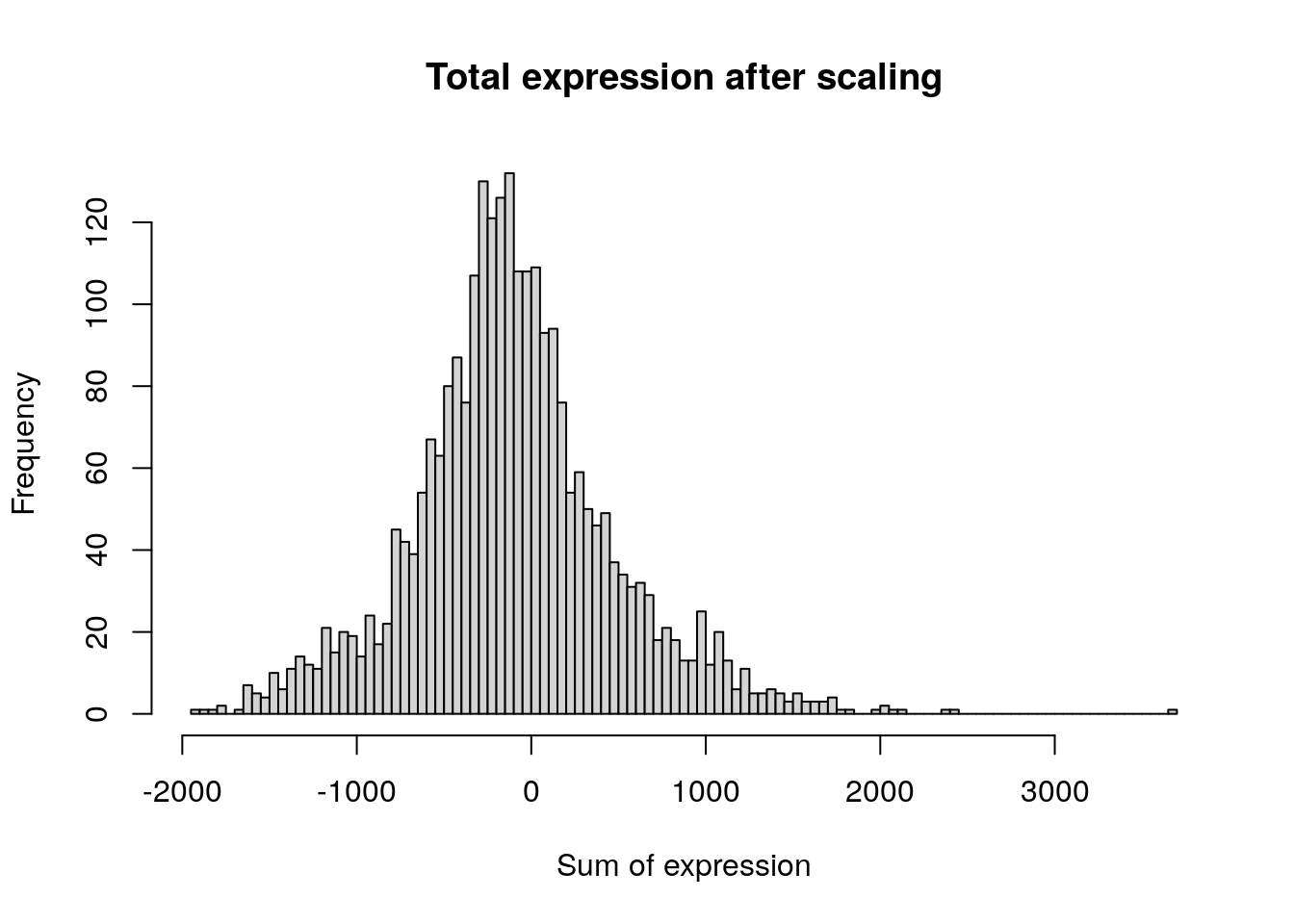
Next, we apply a linear transformation (“scaling”) that is a standard
pre-processing step prior to dimensional reduction techniques like PCA.
The ScaleData() function:
- Shifts the expression of each gene, so that the mean expression across cells is 0
- Scales the expression of each gene, so that the variance across
cells is 1
- This step gives equal weight in downstream analyses, so that highly-expressed genes do not dominate
- The results of this are stored in pbmc[[“RNA”]]$scale.data
- By default, only variable features are scaled.
- You can specify the features argument to scale additional features
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)Centering and scaling data matrixdim(pbmc[["RNA"]]$scale.data)[1] 13714 2638hist(
colSums(pbmc[['RNA']]$scale.data),
breaks = 100,
main = "Total expression after scaling",
xlab = "Sum of expression"
)
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
How can I remove unwanted sources of variation?
In Seurat, we also use the ScaleData() function to
remove unwanted sources of variation from a single-cell dataset. For
example, we could “regress out” heterogeneity associated with (for
example) cell cycle stage, or mitochondrial contamination i.e.:
pbmc <- ScaleData(pbmc, features = all.genes, vars.to.regress = "percent.mt")However, particularly for advanced users who would like to use this
functionality, we strongly recommend the use of our new normalization
workflow, SCTransform(). The method is described in this
paper, with a separate vignette
using Seurat. As with ScaleData(), the function
SCTransform() also includes a vars.to.regress
parameter.
Perform linear dimensional reduction
Next we perform PCA on the scaled data. By default, only the
previously determined variable features are used as input, but can be
defined using features argument if you wish to choose a different subset
(if you do want to use a custom subset of features, make sure you pass
these to ScaleData first).
For the first principal components, Seurat outputs a list of genes with the most positive and negative loadings, representing modules of genes that exhibit either correlation (or anti-correlation) across single-cells in the dataset.
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL, FTH1, LYZ, FCN1, S100A9, TYMP
FCER1G, CFD, LGALS1, S100A8, CTSS, LGALS2, SERPINA1, IFITM3, SPI1, CFP
PSAP, IFI30, SAT1, COTL1, S100A11, NPC2, GRN, LGALS3, GSTP1, PYCARD
Negative: MALAT1, LTB, IL32, IL7R, CD2, B2M, ACAP1, CD27, STK17A, CTSW
CD247, GIMAP5, AQP3, CCL5, SELL, TRAF3IP3, GZMA, MAL, CST7, ITM2A
MYC, GIMAP7, HOPX, BEX2, LDLRAP1, GZMK, ETS1, ZAP70, TNFAIP8, RIC3
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1, HLA-DRA, LINC00926, CD79B, HLA-DRB1, CD74
HLA-DMA, HLA-DPB1, HLA-DQA2, CD37, HLA-DRB5, HLA-DMB, HLA-DPA1, FCRLA, HVCN1, LTB
BLNK, P2RX5, IGLL5, IRF8, SWAP70, ARHGAP24, FCGR2B, SMIM14, PPP1R14A, C16orf74
Negative: NKG7, PRF1, CST7, GZMB, GZMA, FGFBP2, CTSW, GNLY, B2M, SPON2
CCL4, GZMH, FCGR3A, CCL5, CD247, XCL2, CLIC3, AKR1C3, SRGN, HOPX
TTC38, APMAP, CTSC, S100A4, IGFBP7, ANXA1, ID2, IL32, XCL1, RHOC
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1, HLA-DPA1, CD74, MS4A1, HLA-DRB1, HLA-DRA
HLA-DRB5, HLA-DQA2, TCL1A, LINC00926, HLA-DMB, HLA-DMA, CD37, HVCN1, FCRLA, IRF8
PLAC8, BLNK, MALAT1, SMIM14, PLD4, LAT2, IGLL5, P2RX5, SWAP70, FCGR2B
Negative: PPBP, PF4, SDPR, SPARC, GNG11, NRGN, GP9, RGS18, TUBB1, CLU
HIST1H2AC, AP001189.4, ITGA2B, CD9, TMEM40, PTCRA, CA2, ACRBP, MMD, TREML1
NGFRAP1, F13A1, SEPT5, RUFY1, TSC22D1, MPP1, CMTM5, RP11-367G6.3, MYL9, GP1BA
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1, CD74, HLA-DPB1, HIST1H2AC, PF4, TCL1A
SDPR, HLA-DPA1, HLA-DRB1, HLA-DQA2, HLA-DRA, PPBP, LINC00926, GNG11, HLA-DRB5, SPARC
GP9, AP001189.4, CA2, PTCRA, CD9, NRGN, RGS18, GZMB, CLU, TUBB1
Negative: VIM, IL7R, S100A6, IL32, S100A8, S100A4, GIMAP7, S100A10, S100A9, MAL
AQP3, CD2, CD14, FYB, LGALS2, GIMAP4, ANXA1, CD27, FCN1, RBP7
LYZ, S100A11, GIMAP5, MS4A6A, S100A12, FOLR3, TRABD2A, AIF1, IL8, IFI6
PC_ 5
Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY, CCL4, CST7, PRF1, GZMA, SPON2
GZMH, S100A9, LGALS2, CCL3, CTSW, XCL2, CD14, CLIC3, S100A12, CCL5
RBP7, MS4A6A, GSTP1, FOLR3, IGFBP7, TYROBP, TTC38, AKR1C3, XCL1, HOPX
Negative: LTB, IL7R, CKB, VIM, MS4A7, AQP3, CYTIP, RP11-290F20.3, SIGLEC10, HMOX1
PTGES3, LILRB2, MAL, CD27, HN1, CD2, GDI2, ANXA5, CORO1B, TUBA1B
FAM110A, ATP1A1, TRADD, PPA1, CCDC109B, ABRACL, CTD-2006K23.1, WARS, VMO1, FYB Seurat provides several useful ways of visualizing both cells and
features that define the PCA, including VizDimReduction(),
DimPlot(), and DimHeatmap().
Examine and visualize PCA results a few different ways
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL
Negative: MALAT1, LTB, IL32, IL7R, CD2
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
Negative: NKG7, PRF1, CST7, GZMB, GZMA
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1
Negative: PPBP, PF4, SDPR, SPARC, GNG11
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1
Negative: VIM, IL7R, S100A6, IL32, S100A8
PC_ 5
Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY
Negative: LTB, IL7R, CKB, VIM, MS4A7 VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")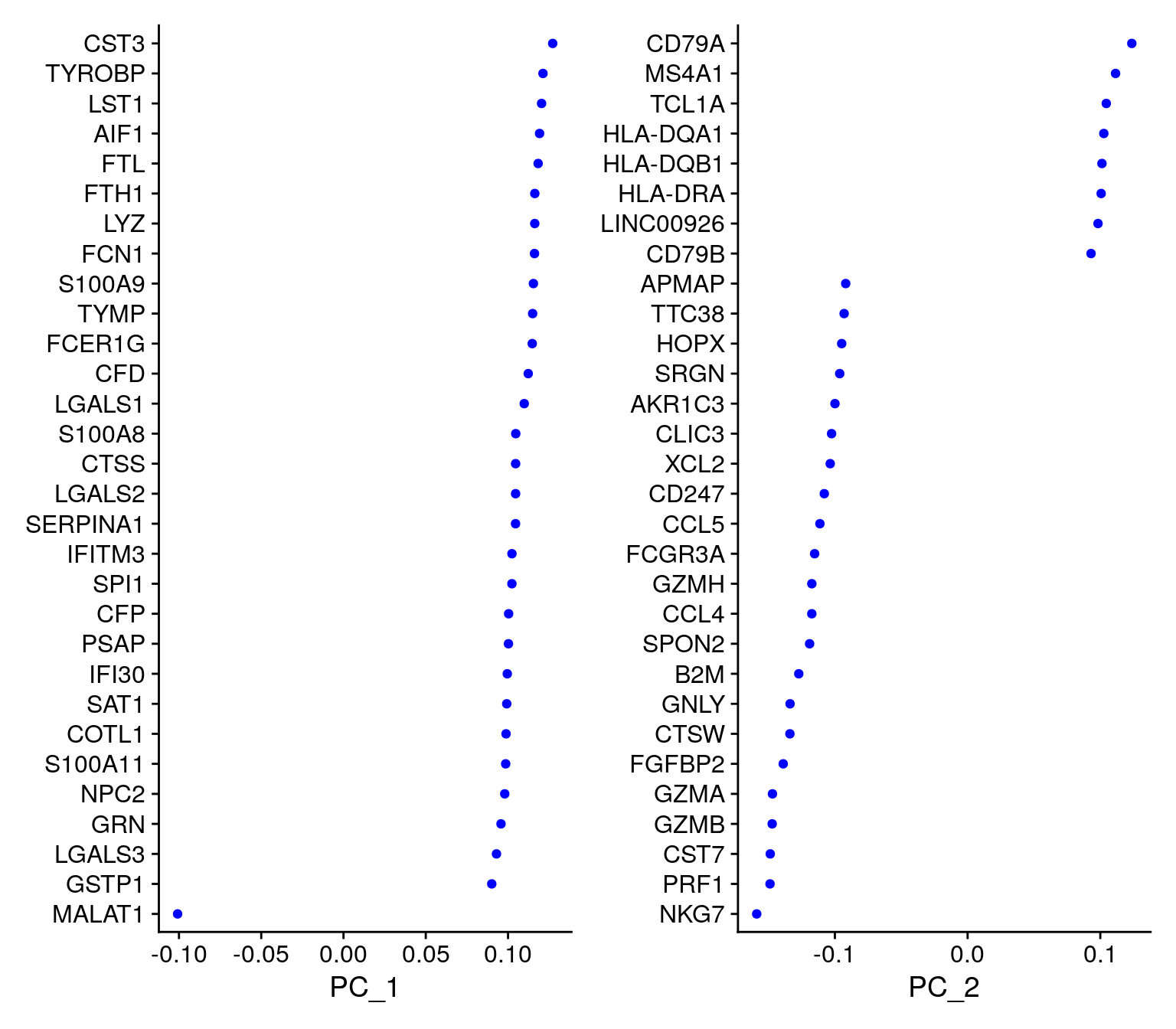
DimPlot(pbmc, reduction = "pca") + NoLegend()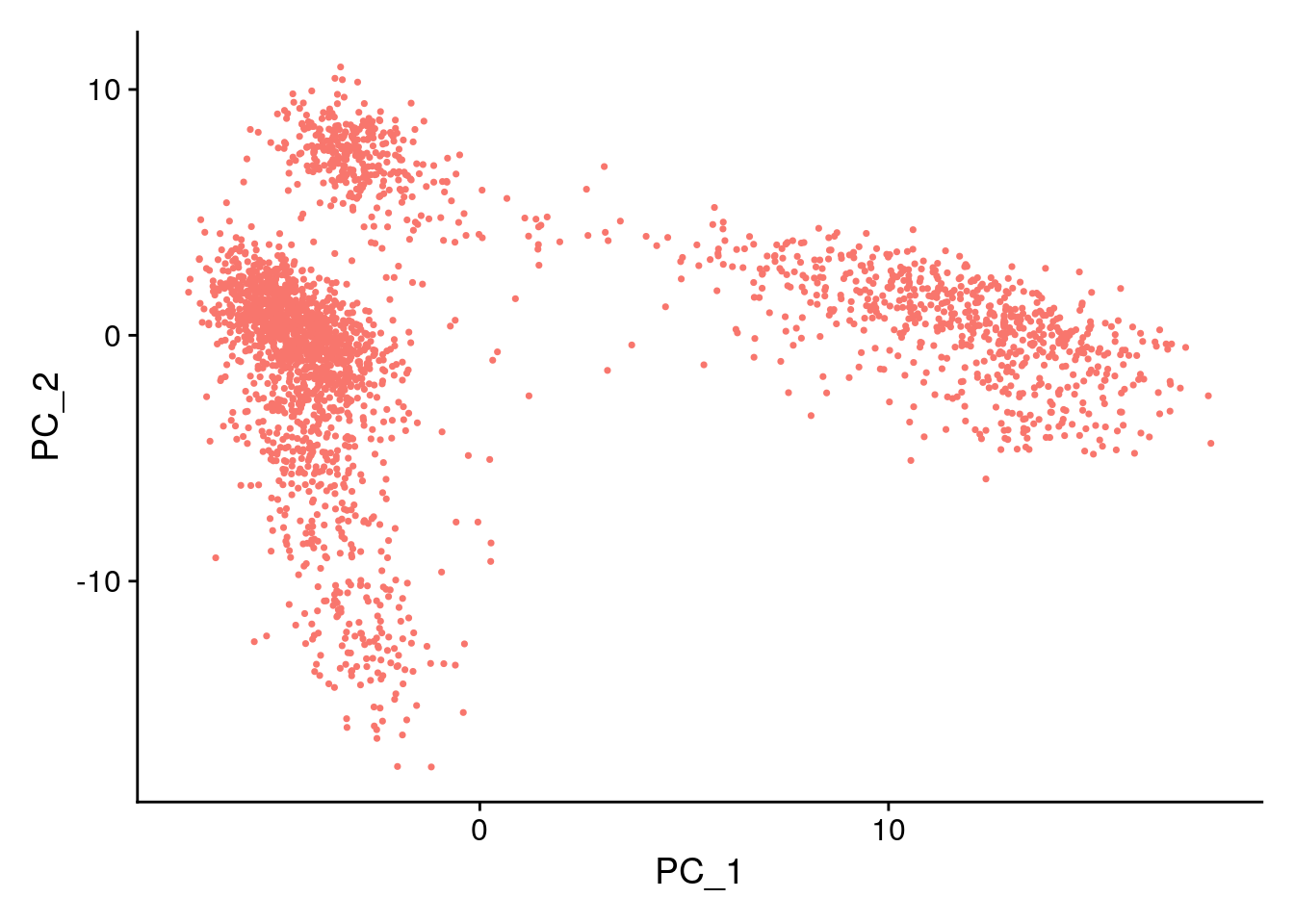
In particular DimHeatmap() allows for easy exploration
of the primary sources of heterogeneity in a dataset, and can be useful
when trying to decide which PCs to include for further downstream
analyses. Both cells and features are ordered according to their PCA
scores. Setting cells to a number plots the ‘extreme’ cells on both ends
of the spectrum, which dramatically speeds plotting for large datasets.
Though clearly a supervised analysis, we find this to be a valuable tool
for exploring correlated feature sets.
DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)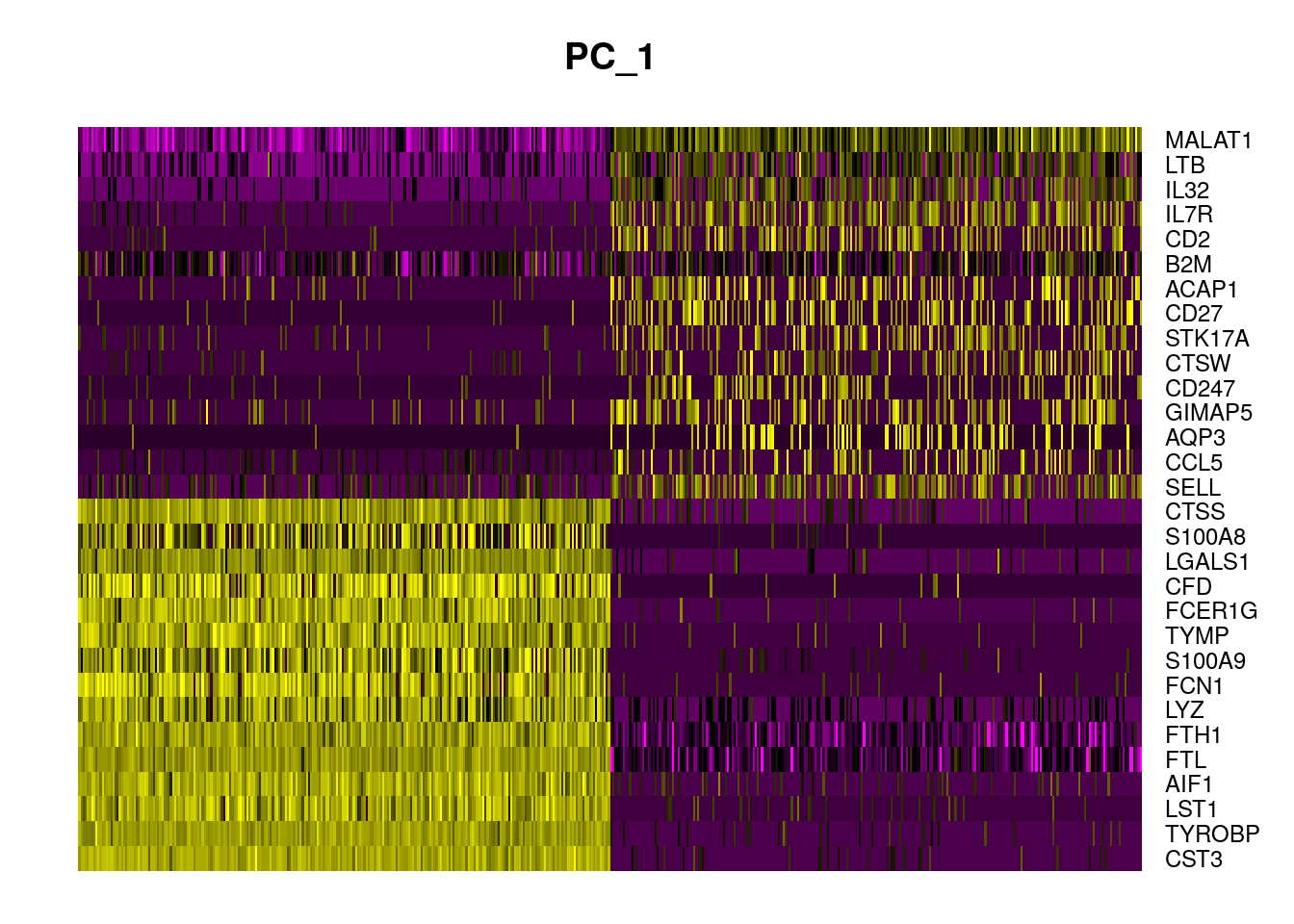
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)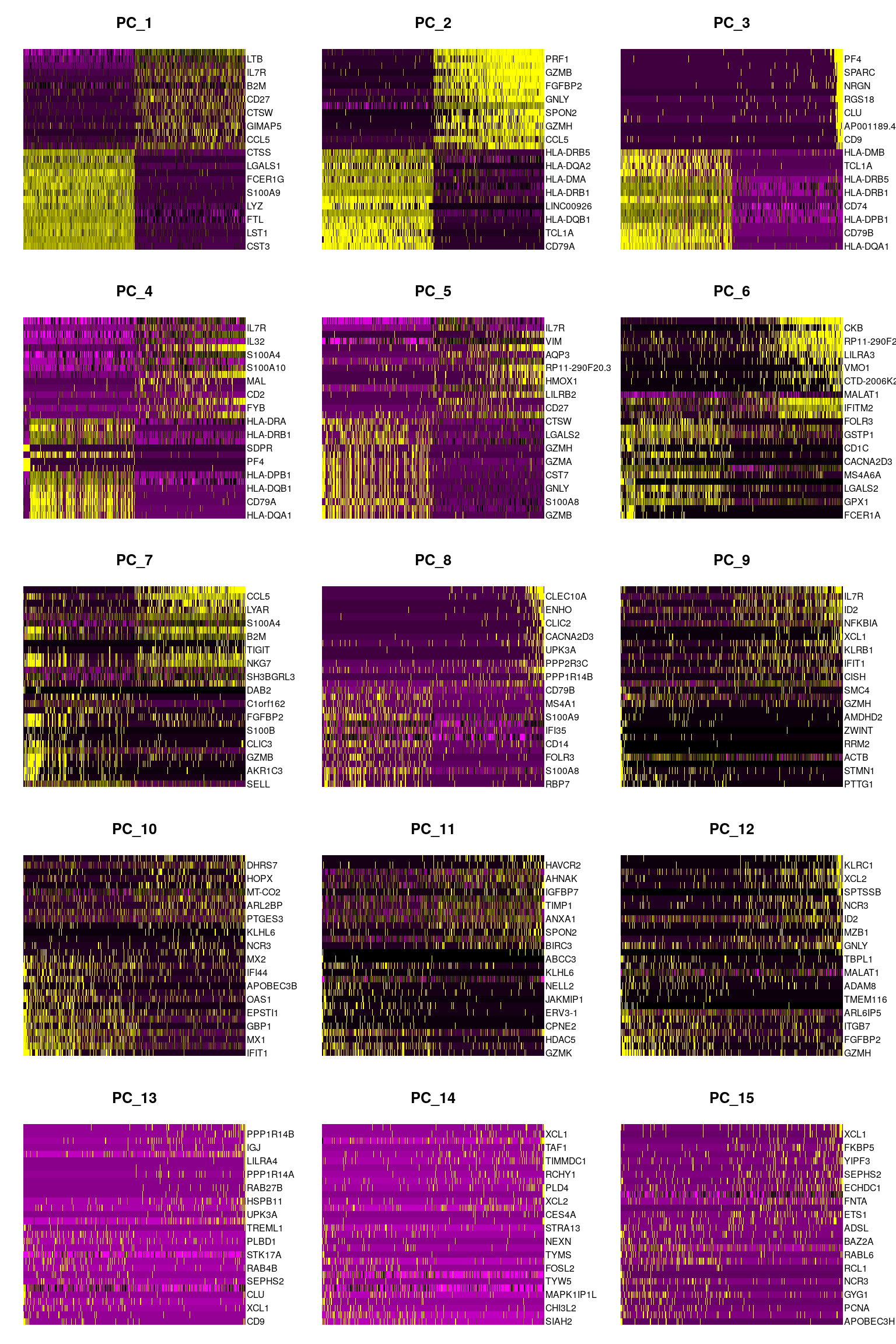
Determine the dimensionality of the dataset
To overcome the extensive technical noise in any single feature for scRNA-seq data, Seurat clusters cells based on their PCA scores, with each PC essentially representing a ‘metafeature’ that combines information across a correlated feature set. The top principal components therefore represent a robust compression of the dataset. However, how many components should we choose to include? 10? 20? 100?
In Macosko et al, we implemented a resampling test inspired by the JackStraw procedure. While still available in Seurat (see previous vignette), this is a slow and computationally expensive procedure, and we is no longer routinely used in single cell analysis.
An alternative heuristic method generates an ‘Elbow plot’: a ranking of principle components based on the percentage of variance explained by each one (ElbowPlot() function). In this example, we can observe an ‘elbow’ around PC9-10, suggesting that the majority of true signal is captured in the first 10 PCs.
ElbowPlot(pbmc)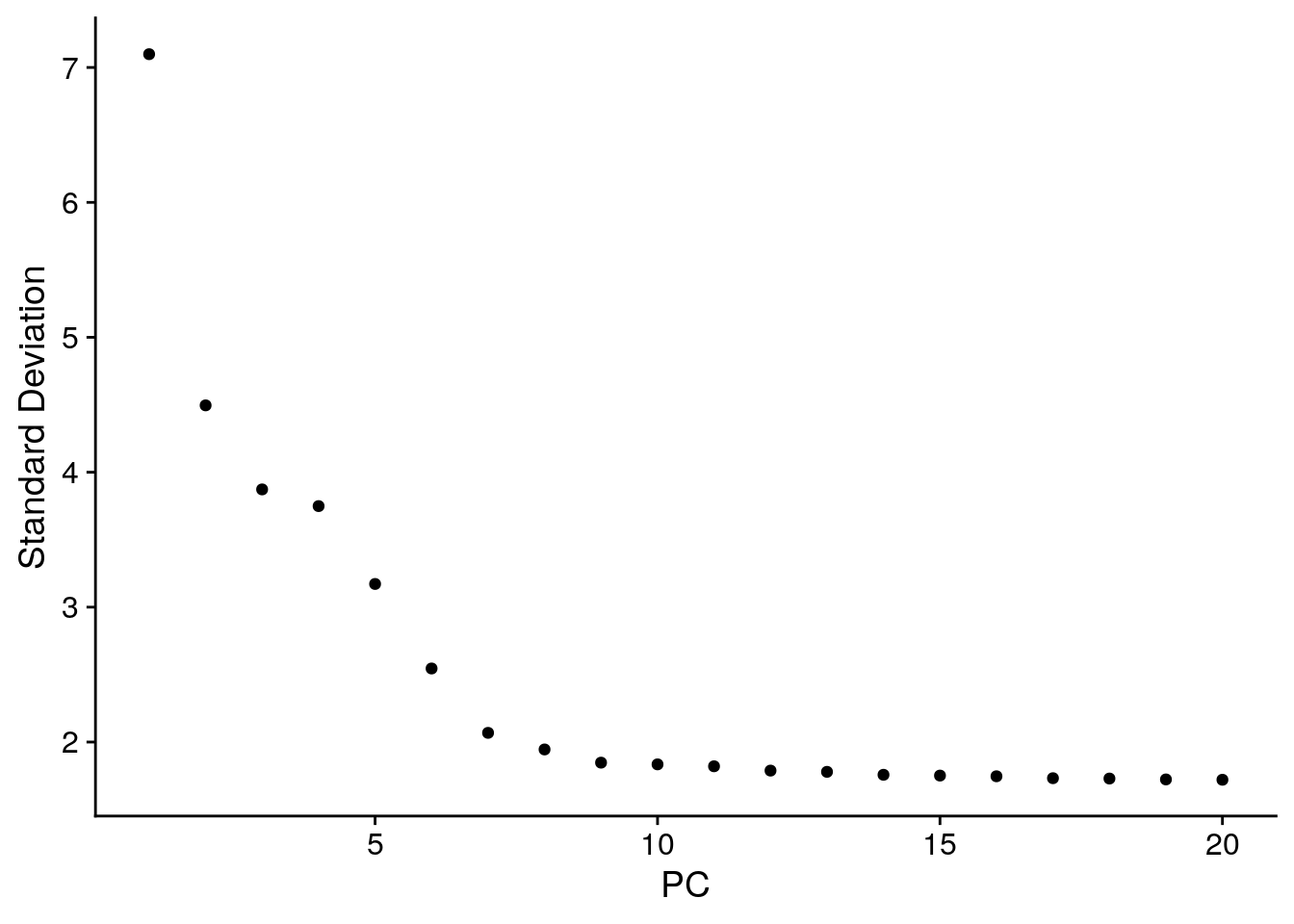
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
Identifying the true dimensionality of a dataset – can be challenging/uncertain for the user. We therefore suggest these multiple approaches for users. The first is more supervised, exploring PCs to determine relevant sources of heterogeneity, and could be used in conjunction with GSEA for example. The second (ElbowPlot) The third is a heuristic that is commonly used, and can be calculated instantly. In this example, we might have been justified in choosing anything between PC 7-12 as a cutoff.
We chose 10 here, but encourage users to consider the following:
- Dendritic cell and NK aficionados may recognize that genes strongly associated with PCs 12 and 13 define rare immune subsets (i.e. MZB1 is a marker for plasmacytoid DCs). However, these groups are so rare, they are difficult to distinguish from background noise for a dataset of this size without prior knowledge.
- We encourage users to repeat downstream analyses with a different number of PCs (10, 15, or even 50!). As you will observe, the results often do not differ dramatically.
- We advise users to err on the higher side when choosing this parameter. For example, performing downstream analyses with only 5 PCs does significantly and adversely affect results.
Cluster the cells
Seurat applies a graph-based clustering approach, building upon initial strategies in (Macosko et al). Importantly, the distance metric which drives the clustering analysis (based on previously identified PCs) remains the same. However, our approach to partitioning the cellular distance matrix into clusters has dramatically improved. Our approach was heavily inspired by recent manuscripts which applied graph-based clustering approaches to scRNA-seq data [SNN-Cliq, Xu and Su, Bioinformatics, 2015] and CyTOF data [PhenoGraph, Levine et al., Cell, 2015]. Briefly, these methods embed cells in a graph structure - for example a K-nearest neighbor (KNN) graph, with edges drawn between cells with similar feature expression patterns, and then attempt to partition this graph into highly interconnected ‘quasi-cliques’ or ‘communities’.
As in PhenoGraph, we first construct a KNN graph based on the euclidean distance in PCA space, and refine the edge weights between any two cells based on the shared overlap in their local neighborhoods (Jaccard similarity). This step is performed using the FindNeighbors() function, and takes as input the previously defined dimensionality of the dataset (first 10 PCs).
To cluster the cells, we next apply modularity optimization techniques such as the Louvain algorithm (default) or SLM [SLM, Blondel et al., Journal of Statistical Mechanics], to iteratively group cells together, with the goal of optimizing the standard modularity function. The FindClusters() function implements this procedure, and contains a resolution parameter that sets the ‘granularity’ of the downstream clustering, with increased values leading to a greater number of clusters. We find that setting this parameter between 0.4-1.2 typically returns good results for single-cell datasets of around 3K cells. Optimal resolution often increases for larger datasets. The clusters can be found using the Idents() function.
pbmc <- FindNeighbors(pbmc, dims = 1:10)Computing nearest neighbor graphComputing SNNpbmc <- FindClusters(pbmc, resolution = 0.5)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2638
Number of edges: 95965
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8723
Number of communities: 9
Elapsed time: 0 secondsLook at cluster IDs of the first 5 cells
head(Idents(pbmc), 5)AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
2 3 2 1
AAACCGTGTATGCG-1
6
Levels: 0 1 2 3 4 5 6 7 8Non-linear dimensional reduction
Seurat offers several non-linear dimensional reduction techniques, such as tSNE and UMAP, to visualize and explore these datasets. The goal of these algorithms is to learn underlying structure in the dataset, in order to place similar cells together in low-dimensional space. Therefore, cells that are grouped together within graph-based clusters determined above should co-localize on these dimension reduction plots.
While we and others have routinely found 2D visualization techniques like tSNE and UMAP to be valuable tools for exploring datasets, all visualization techniques have limitations, and cannot fully represent the complexity of the underlying data. In particular, these methods aim to preserve local distances in the dataset (i.e. ensuring that cells with very similar gene expression profiles co-localize), but often do not preserve more global relationships. We encourage users to leverage techniques like UMAP for visualization, but to avoid drawing biological conclusions solely on the basis of visualization techniques.
pbmc <- RunUMAP(pbmc, dims = 1:10)Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per session04:11:10 UMAP embedding parameters a = 0.9922 b = 1.11204:11:10 Read 2638 rows and found 10 numeric columns04:11:10 Using Annoy for neighbor search, n_neighbors = 3004:11:10 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
04:11:11 Writing NN index file to temp file /tmp/RtmpLVAiWh/file8234212b9c4b
04:11:11 Searching Annoy index using 1 thread, search_k = 3000
04:11:11 Annoy recall = 100%
04:11:11 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30
04:11:12 Initializing from normalized Laplacian + noise (using RSpectra)
04:11:12 Commencing optimization for 500 epochs, with 105124 positive edges
04:11:14 Optimization finishedNote that you can set label = TRUE or use the
LabelClusters function to help label individual clusters
DimPlot(pbmc, reduction = "umap")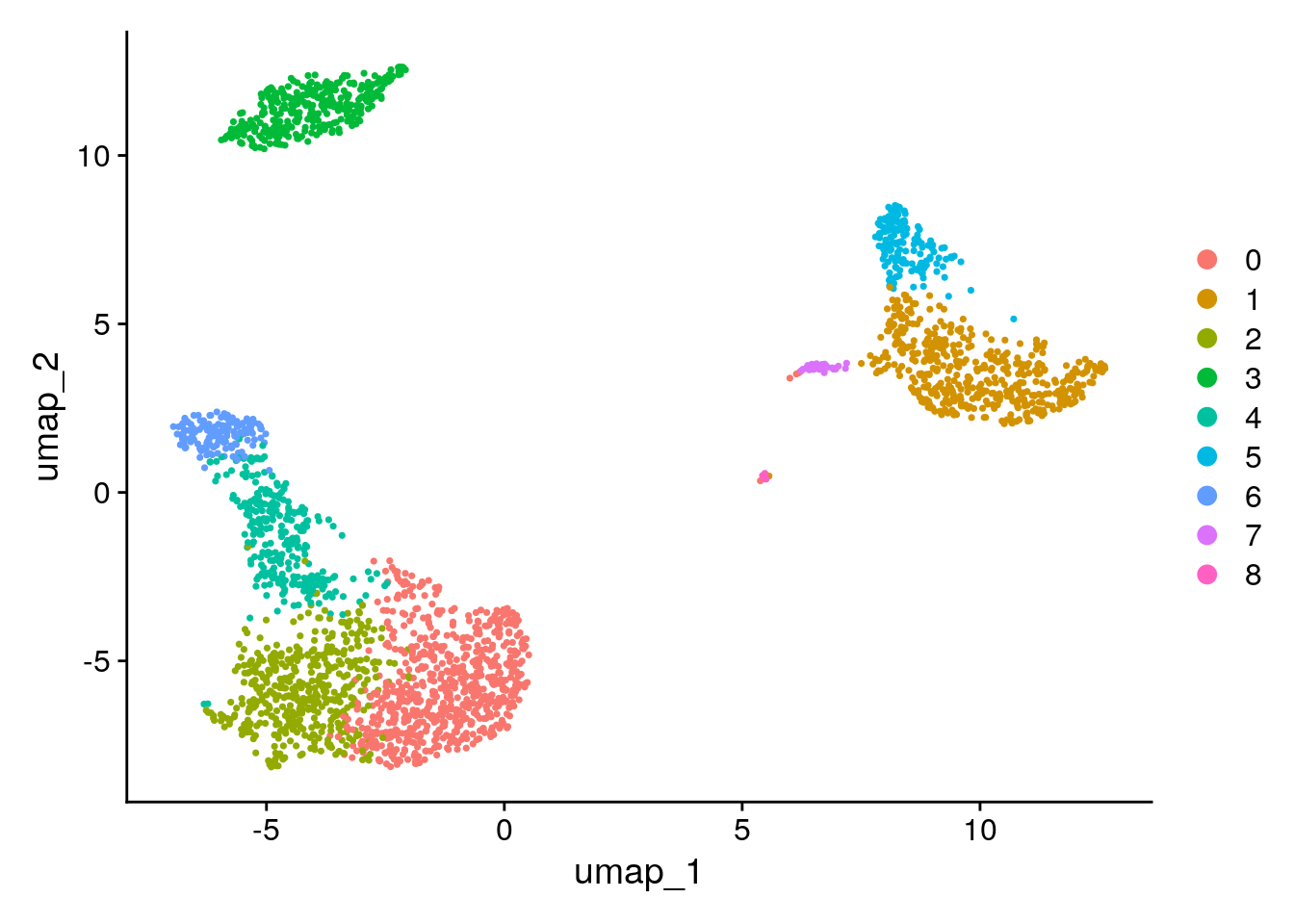
| Version | Author | Date |
|---|---|---|
| 5f6b5a7 | Dave Tang | 2024-04-07 |
Finding differentially expressed features
Seurat can help you find markers that define clusters via differential expression (DE). By default, it identifies positive and negative markers of a single cluster (specified in ident.1), compared to all other cells. FindAllMarkers() automates this process for all clusters, but you can also test groups of clusters vs. each other, or against all cells.
In Seurat v5, we use the presto package (as described here and available for installation here), to dramatically improve the speed of DE analysis, particularly for large datasets. For users who are not using presto, you can examine the documentation for this function (?FindMarkers) to explore the min.pct and logfc.threshold parameters, which can be increased in order to increase the speed of DE testing.
For a (much!) faster implementation of the Wilcoxon Rank Sum Test, (default method for FindMarkers) please install the {presto} package. After installation of {presto}, Seurat will automatically use the more efficient implementation (no further action necessary).
install.packages("remotes")
remotes::install_github('immunogenomics/presto')Load {presto}.
library('presto')Loading required package: RcppLoading required package: data.tablepackageVersion('presto')[1] '1.0.0'Find all markers of cluster 2.
cluster2.markers <- FindMarkers(pbmc, ident.1 = 2)
head(cluster2.markers, n = 5) p_val avg_log2FC pct.1 pct.2 p_val_adj
IL32 2.593535e-91 1.3221171 0.949 0.466 3.556774e-87
LTB 7.994465e-87 1.3450377 0.981 0.644 1.096361e-82
CD3D 3.922451e-70 1.0562099 0.922 0.433 5.379250e-66
IL7R 1.130870e-66 1.4256944 0.748 0.327 1.550876e-62
LDHB 4.082189e-65 0.9765875 0.953 0.614 5.598314e-61Find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3))
head(cluster5.markers, n = 5) p_val avg_log2FC pct.1 pct.2 p_val_adj
FCGR3A 2.150929e-209 6.832372 0.975 0.039 2.949784e-205
IFITM3 6.103366e-199 6.181000 0.975 0.048 8.370156e-195
CFD 8.891428e-198 6.052575 0.938 0.037 1.219370e-193
CD68 2.374425e-194 5.493138 0.926 0.035 3.256286e-190
RP11-290F20.3 9.308287e-191 6.335402 0.840 0.016 1.276538e-186Find markers for every cluster compared to all remaining cells, report only the positive ones.
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE)
pbmc.markers %>%
dplyr::group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1)# A tibble: 7,046 × 7
# Groups: cluster [9]
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
<dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
1 1.74e-109 1.19 0.897 0.593 2.39e-105 0 LDHB
2 1.17e- 83 2.37 0.435 0.108 1.60e- 79 0 CCR7
3 8.94e- 79 1.09 0.838 0.403 1.23e- 74 0 CD3D
4 3.05e- 53 1.02 0.722 0.399 4.19e- 49 0 CD3E
5 3.28e- 49 2.10 0.333 0.103 4.50e- 45 0 LEF1
6 6.66e- 49 1.25 0.623 0.358 9.13e- 45 0 NOSIP
7 9.31e- 44 2.02 0.328 0.11 1.28e- 39 0 PRKCQ-AS1
8 4.69e- 43 1.53 0.435 0.184 6.43e- 39 0 PIK3IP1
9 1.47e- 39 2.70 0.195 0.04 2.01e- 35 0 FHIT
10 2.44e- 33 1.94 0.262 0.087 3.34e- 29 0 MAL
# ℹ 7,036 more rowsSeurat has several tests for differential expression which can be set with the test.use parameter (see our DE vignette for details). For example, the ROC test returns the ‘classification power’ for any individual marker (ranging from 0 - random, to 1 - perfect).
cluster0.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)We include several tools for visualizing marker expression.
VlnPlot() (shows expression probability distributions
across clusters), and FeaturePlot() (visualizes feature
expression on a tSNE or PCA plot) are our most commonly used
visualizations. We also suggest exploring RidgePlot(),
CellScatter(), and DotPlot() as additional
methods to view your dataset.
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))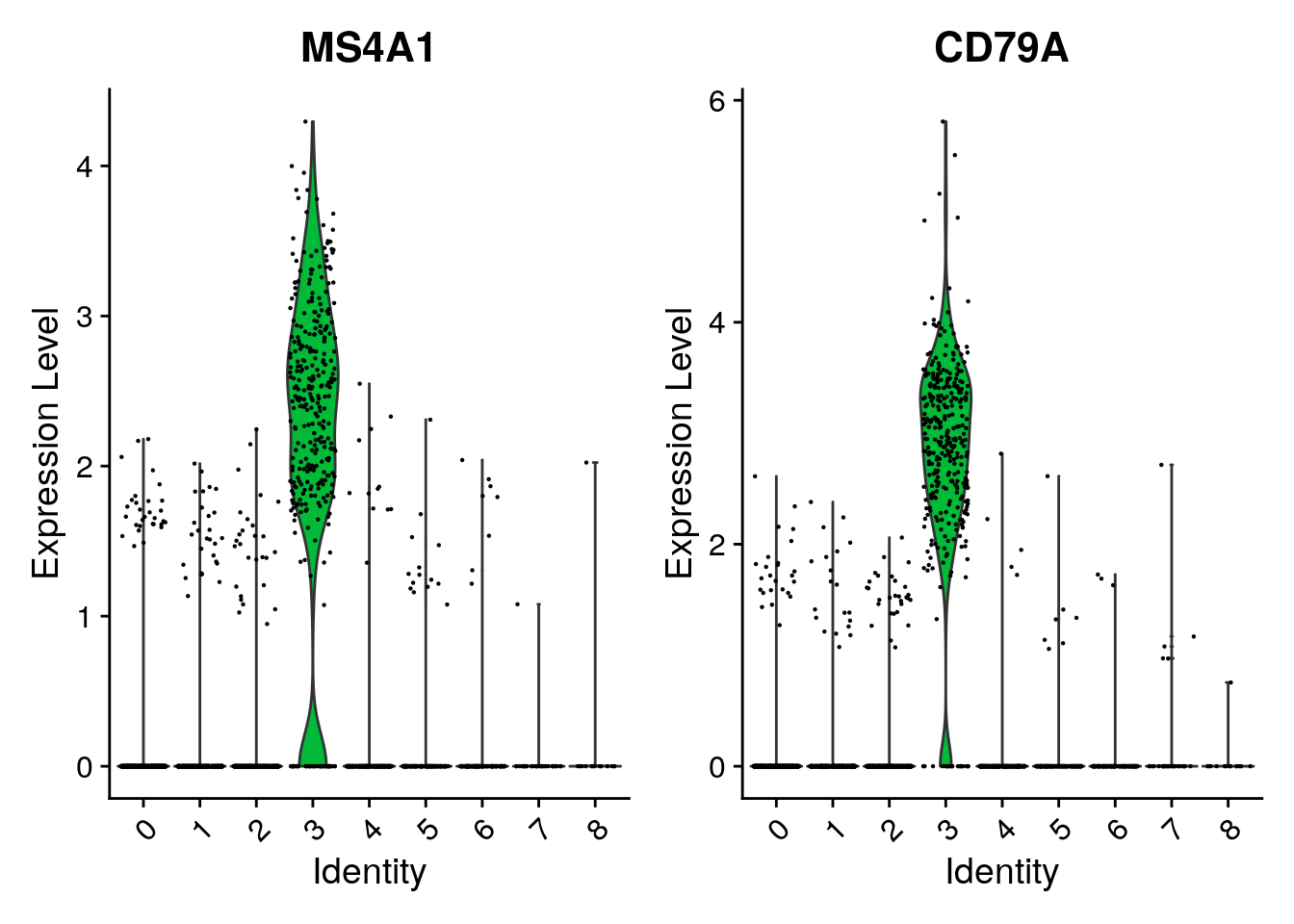
You can plot raw counts as well.
VlnPlot(pbmc, features = c("NKG7", "PF4"), layer = "counts", log = TRUE)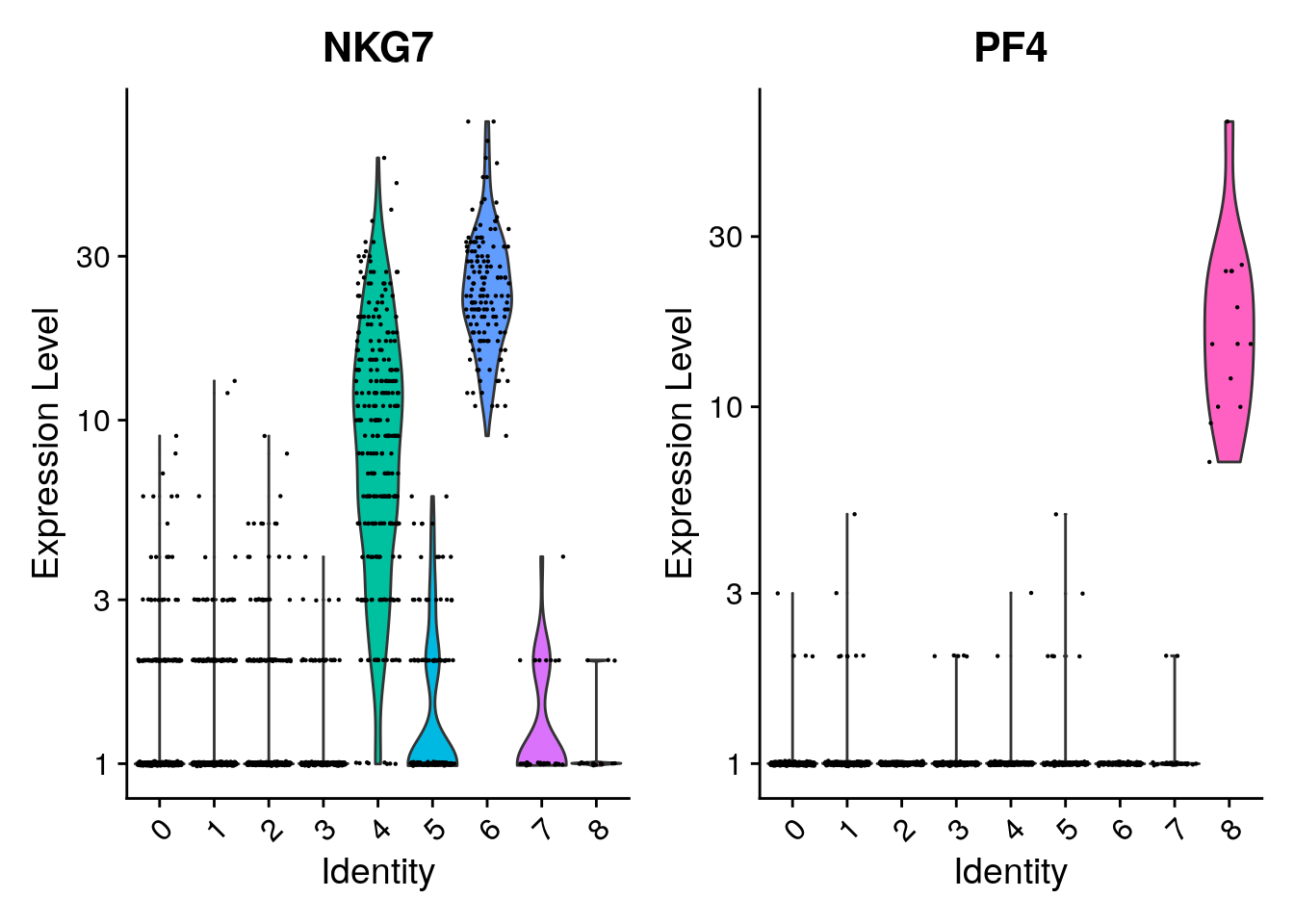
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))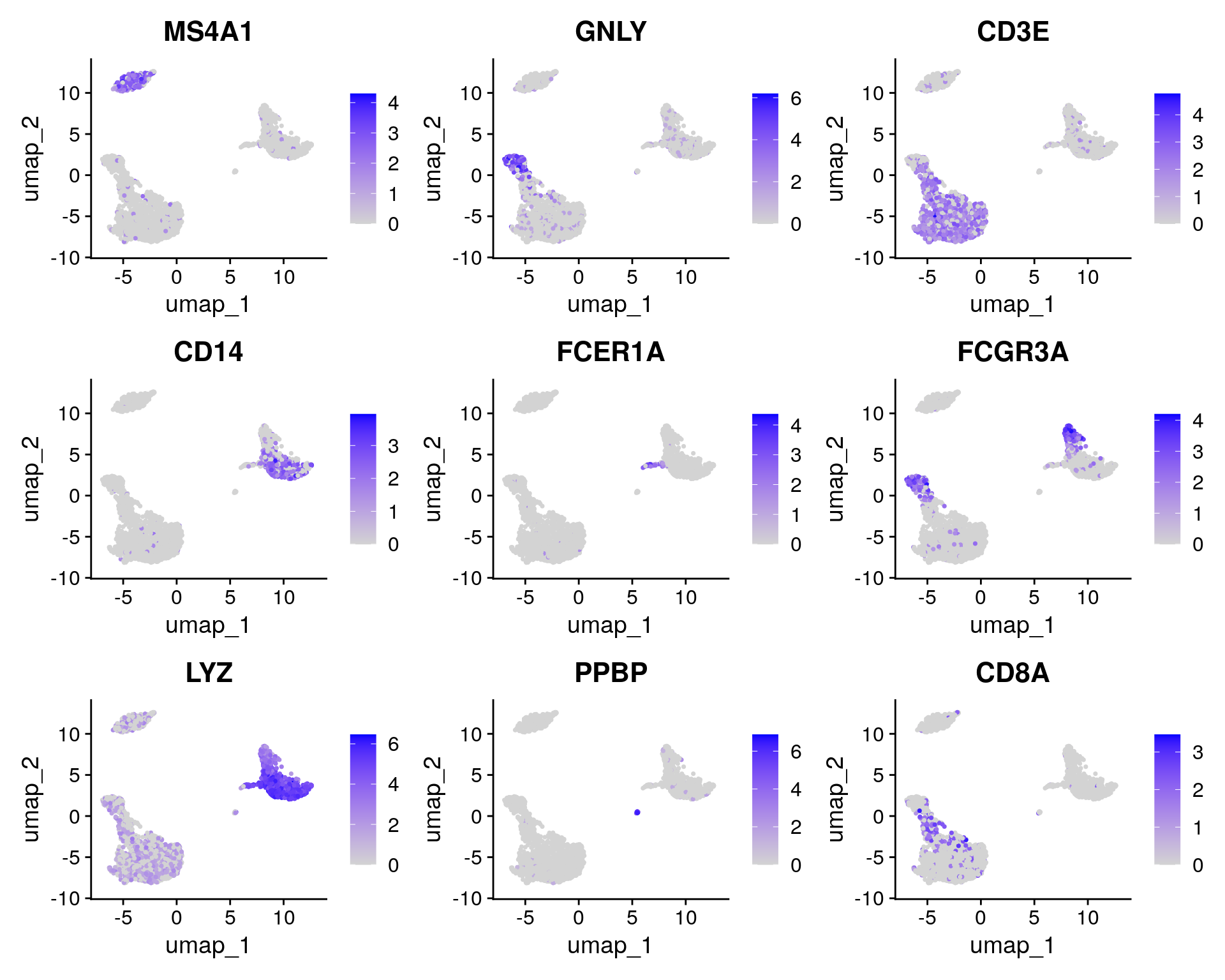
DoHeatmap() generates an expression heatmap for given
cells and features. In this case, we are plotting the top 20 markers (or
all markers if less than 20) for each cluster.
pbmc.markers %>%
dplyr::group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
dplyr::slice_head(n = 10) %>%
dplyr::ungroup() -> top10
DoHeatmap(pbmc, features = top10$gene) + NoLegend()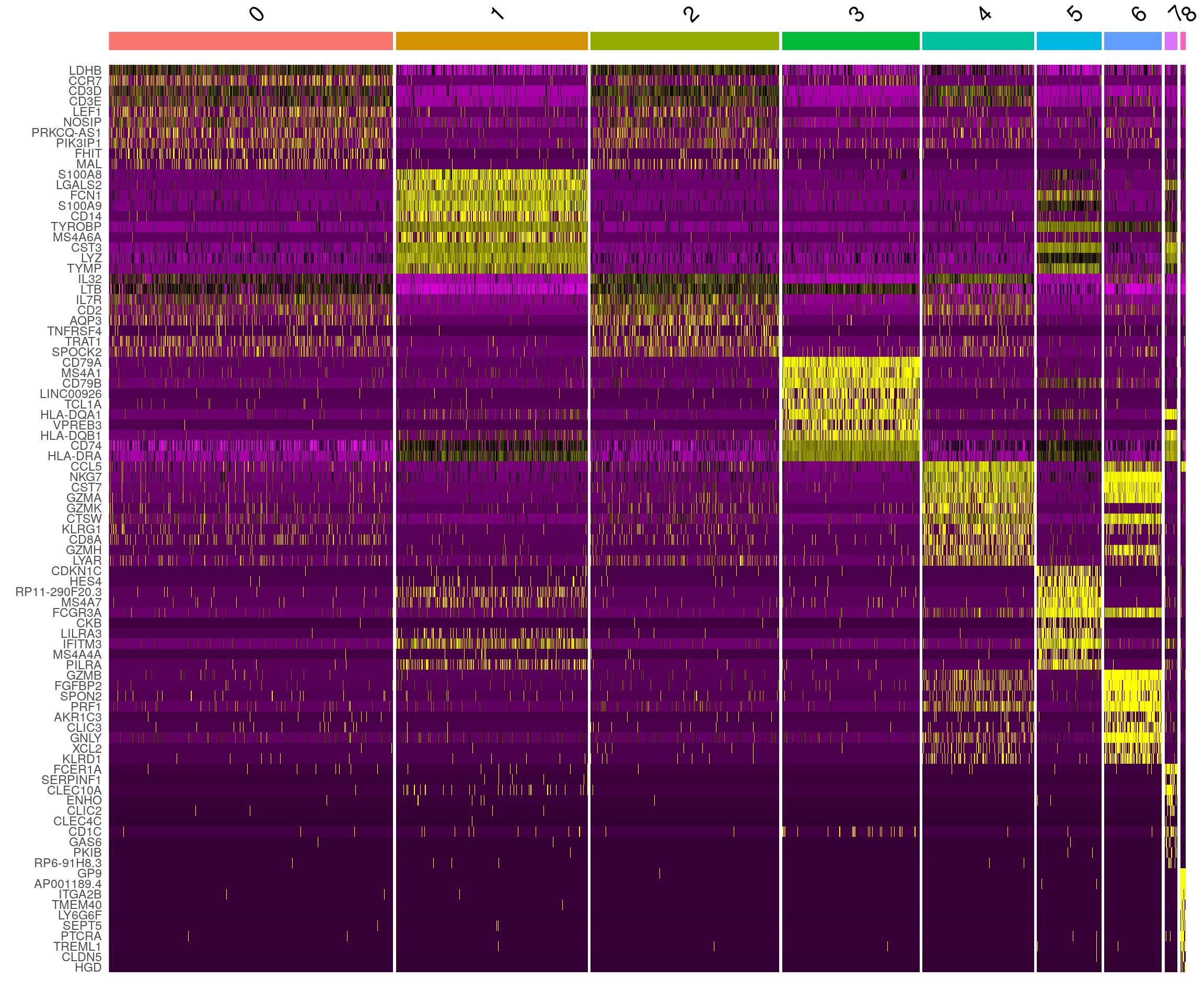
Assigning cell type identity to clusters
Fortunately in the case of this dataset, we can use canonical markers to easily match the unbiased clustering to known cell types:
| Cluster ID | Markers | Cell Type |
|---|---|---|
| 0 | IL7R, CCR7 | Naive CD4+ T |
| 1 | CD14, LYZ | CD14+ Mono |
| 2 | IL7R, S100A4 | Memory CD4+ |
| 3 | MS4A1 | B |
| 4 | CD8A | CD8+ T |
| 5 | FCGR3A, MS4A7 | FCGR3A+ Mono |
| 6 | GNLY, NKG7 | NK |
| 7 | FCER1A, CST3 | DC |
| 8 | PPBP | Platelet |
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()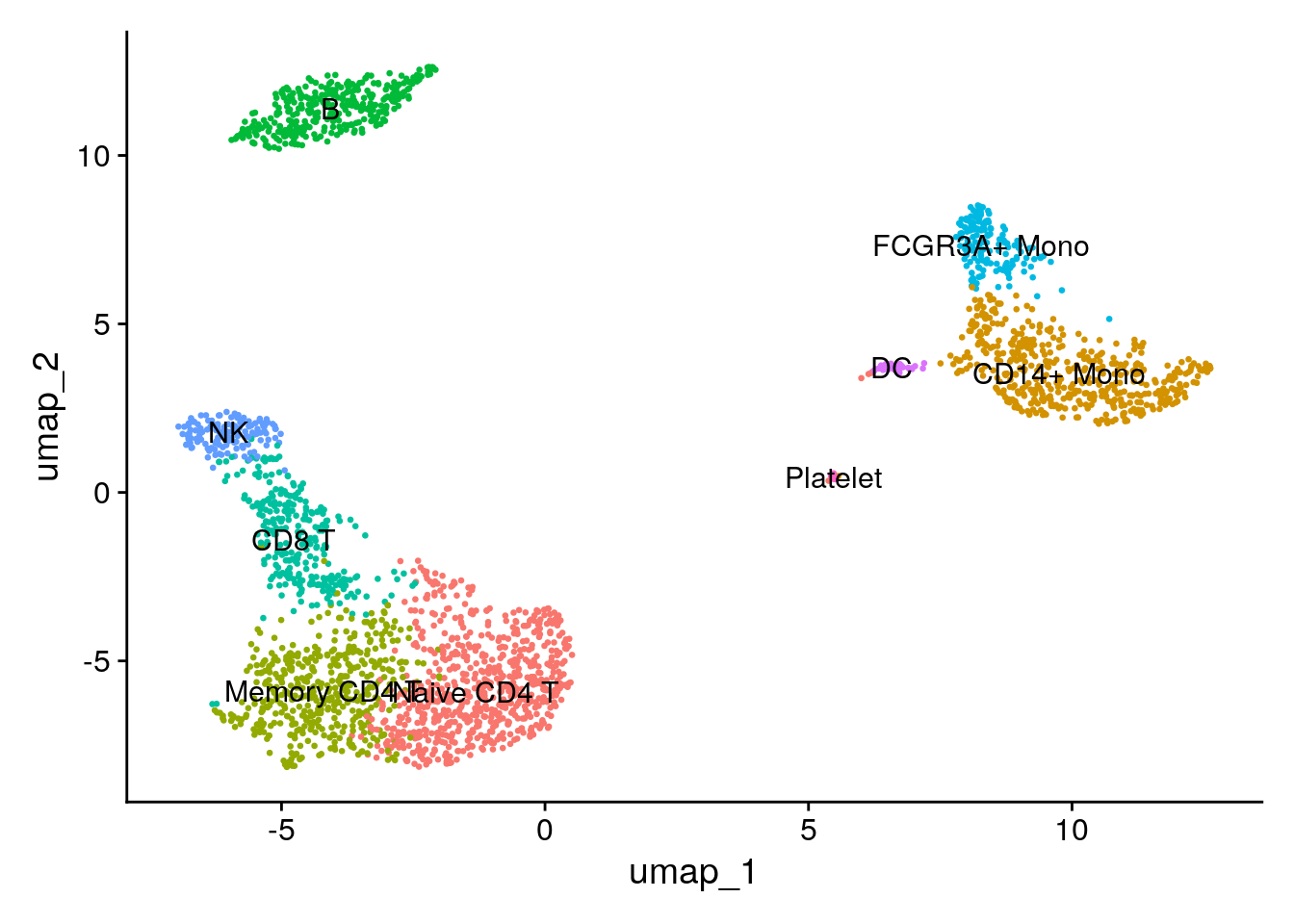
library(ggplot2)
DimPlot(pbmc, reduction = "umap", label = TRUE, label.size = 4.5) + xlab("UMAP 1") +
ylab("UMAP 2") +
theme(axis.title = element_text(size = 18), legend.text = element_text(size = 18)) +
guides(colour = guide_legend(override.aes = list(size = 10)))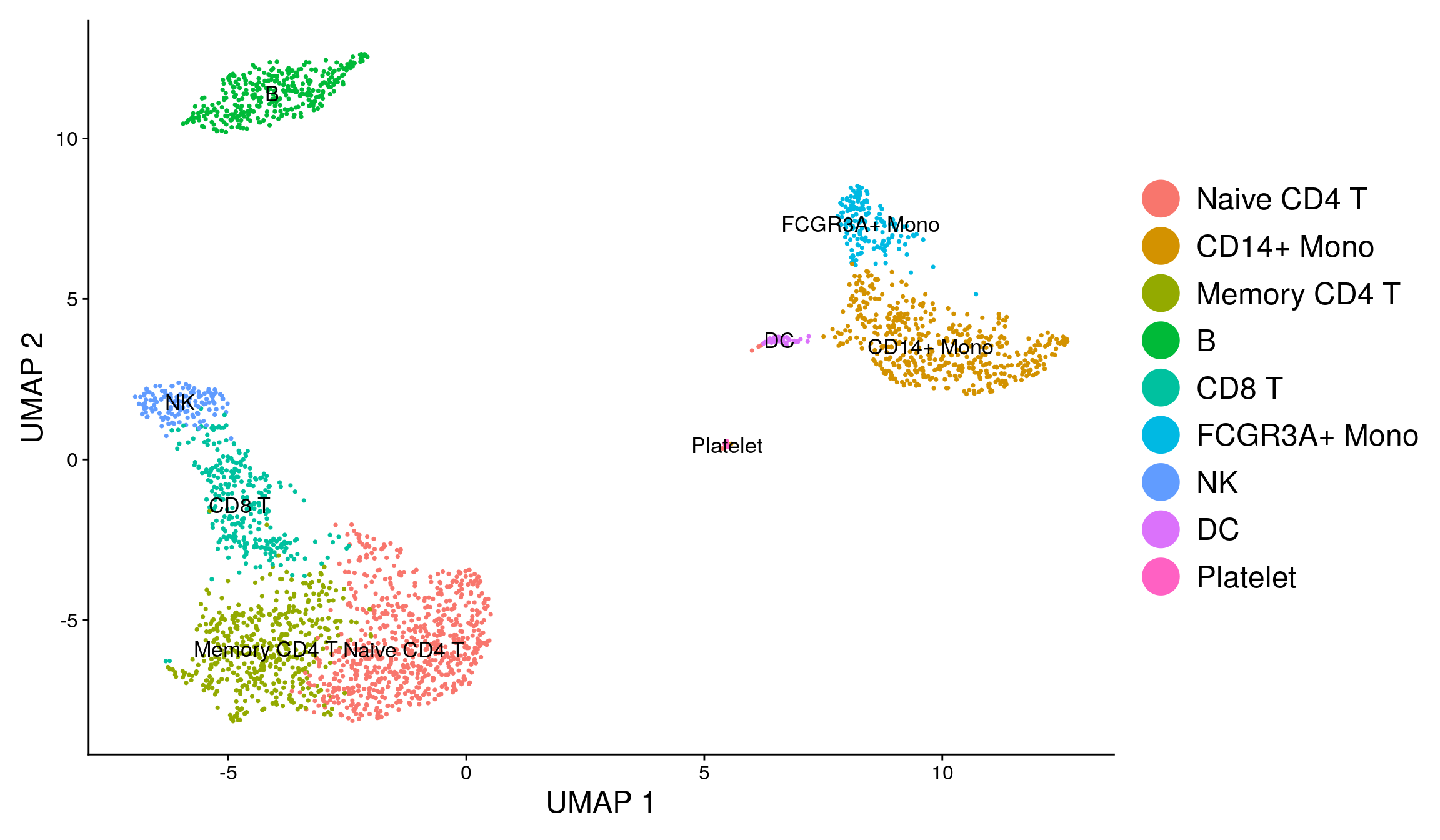
From AnnData
Download 1k PBMCs in AnnData format from Open Problems in Single-Cell Analysis.
aws s3 cp --no-sign-request s3://openproblems-data/resources/datasets/openproblems_v1/tenx_1k_pbmc/l1_sqrt/dataset.h5ad .
sessionInfo()R version 4.3.3 (2024-02-29)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggplot2_3.5.0 presto_1.0.0 data.table_1.15.4 Rcpp_1.0.12
[5] Seurat_5.0.3 SeuratObject_5.0.1 sp_2.1-3 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 rstudioapi_0.16.0 jsonlite_1.8.8
[4] magrittr_2.0.3 spatstat.utils_3.0-4 farver_2.1.1
[7] rmarkdown_2.26 fs_1.6.3 vctrs_0.6.5
[10] ROCR_1.0-11 spatstat.explore_3.2-7 htmltools_0.5.8.1
[13] sass_0.4.9 sctransform_0.4.1 parallelly_1.37.1
[16] KernSmooth_2.23-22 bslib_0.7.0 htmlwidgets_1.6.4
[19] ica_1.0-3 plyr_1.8.9 plotly_4.10.4
[22] zoo_1.8-12 cachem_1.0.8 whisker_0.4.1
[25] igraph_2.0.3 mime_0.12 lifecycle_1.0.4
[28] pkgconfig_2.0.3 Matrix_1.6-5 R6_2.5.1
[31] fastmap_1.1.1 fitdistrplus_1.1-11 future_1.33.2
[34] shiny_1.8.1.1 digest_0.6.35 colorspace_2.1-0
[37] patchwork_1.2.0 ps_1.7.6 rprojroot_2.0.4
[40] tensor_1.5 RSpectra_0.16-1 irlba_2.3.5.1
[43] labeling_0.4.3 progressr_0.14.0 fansi_1.0.6
[46] spatstat.sparse_3.0-3 httr_1.4.7 polyclip_1.10-6
[49] abind_1.4-5 compiler_4.3.3 withr_3.0.0
[52] fastDummies_1.7.3 highr_0.10 MASS_7.3-60.0.1
[55] tools_4.3.3 lmtest_0.9-40 httpuv_1.6.15
[58] future.apply_1.11.2 goftest_1.2-3 glue_1.7.0
[61] callr_3.7.6 nlme_3.1-164 promises_1.3.0
[64] grid_4.3.3 Rtsne_0.17 getPass_0.2-4
[67] cluster_2.1.6 reshape2_1.4.4 generics_0.1.3
[70] gtable_0.3.4 spatstat.data_3.0-4 tidyr_1.3.1
[73] utf8_1.2.4 spatstat.geom_3.2-9 RcppAnnoy_0.0.22
[76] ggrepel_0.9.5 RANN_2.6.1 pillar_1.9.0
[79] stringr_1.5.1 spam_2.10-0 RcppHNSW_0.6.0
[82] later_1.3.2 splines_4.3.3 dplyr_1.1.4
[85] lattice_0.22-5 survival_3.5-8 deldir_2.0-4
[88] tidyselect_1.2.1 miniUI_0.1.1.1 pbapply_1.7-2
[91] knitr_1.46 git2r_0.33.0 gridExtra_2.3
[94] scattermore_1.2 xfun_0.43 matrixStats_1.2.0
[97] stringi_1.8.3 lazyeval_0.2.2 yaml_2.3.8
[100] evaluate_0.23 codetools_0.2-19 tibble_3.2.1
[103] cli_3.6.2 uwot_0.1.16 xtable_1.8-4
[106] reticulate_1.35.0 munsell_0.5.1 processx_3.8.4
[109] jquerylib_0.1.4 globals_0.16.3 spatstat.random_3.2-3
[112] png_0.1-8 parallel_4.3.3 dotCall64_1.1-1
[115] listenv_0.9.1 viridisLite_0.4.2 scales_1.3.0
[118] ggridges_0.5.6 leiden_0.4.3.1 purrr_1.0.2
[121] rlang_1.1.3 cowplot_1.1.3