Differential gene expression analysis using edgeR
2024-10-22
Last updated: 2024-10-22
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version f7369ce. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/pbmc3k.csv
Ignored: data/pbmc3k.csv.gz
Ignored: data/pbmc3k/
Ignored: r_packages_4.4.0/
Untracked files:
Untracked: rsem.merged.gene_counts.tsv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/edger_de.Rmd) and HTML
(docs/edger_de.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | f7369ce | Dave Tang | 2024-10-22 | Differential gene expression analysis using edgeR |
edgeR carries out:
Differential expression analysis of RNA-seq expression profiles with biological replication. Implements a range of statistical methodology based on the negative binomial distributions, including empirical Bayes estimation, exact tests, generalized linear models and quasi-likelihood tests. As well as RNA-seq, it be applied to differential signal analysis of other types of genomic data that produce read counts, including ChIP-seq, ATAC-seq, Bisulfite-seq, SAGE and CAGE.
Installation
Install using BiocManager::install().
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("edgeR")Data
https://zenodo.org/records/13968456
download.file(url = "https://zenodo.org/records/13968456/files/rsem.merged.gene_counts.tsv?download=1", destfile = "rsem.merged.gene_counts.tsv")
gene_counts <- read_tsv("rsem.merged.gene_counts.tsv", show_col_types = FALSE)
head(gene_counts)# A tibble: 6 × 8
gene_id `transcript_id(s)` ERR160122 ERR160123 ERR164473 ERR164550 ERR164551
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 ENSG0000… ENST00000373020,E… 2 6 374 1637 650
2 ENSG0000… ENST00000373031,E… 19 40 0 1 0
3 ENSG0000… ENST00000371582,E… 268. 274. 489 637 879
4 ENSG0000… ENST00000367770,E… 360. 449. 363. 606. 709.
5 ENSG0000… ENST00000286031,E… 156. 185. 85.4 312. 239.
6 ENSG0000… ENST00000374003,E… 24 23 1181 423 3346
# ℹ 1 more variable: ERR164552 <dbl>Metadata.
tibble::tribble(
~sample, ~run_id, ~group,
"C2_norm", "ERR160122", "normal",
"C3_norm", "ERR160123", "normal",
"C1_norm", "ERR164473", "normal",
"C1_cancer", "ERR164550", "cancer",
"C2_cancer", "ERR164551", "cancer",
"C3_cancer", "ERR164552", "cancer"
) -> my_metadata
my_metadata$group <- factor(my_metadata$group, levels = c('normal', 'cancer'))DGEList
The input to edgeR is the DGEList object.
The required inputs for creating a DGEList object is the
count table and a grouping factor.
Filtering to remove low counts
Remove genes that are lowly expressed.
keep <- rowSums(cpm(y) > 0.5) >= 2
y <- y[keep, , keep.lib.sizes=FALSE]
yAn object of class "DGEList"
$counts
C2_norm C3_norm C1_norm C1_cancer C2_cancer C3_cancer
ENSG00000000003 2 6 374 1637 650 1015
ENSG00000000005 19 40 0 1 0 0
ENSG00000000419 268 273 489 637 879 1157
ENSG00000000457 360 449 362 605 708 632
ENSG00000000460 155 184 85 312 239 147
35001 more rows ...
$samples
group lib.size norm.factors
C2_norm normal 4415649 1
C3_norm normal 5326875 1
C1_norm normal 15961581 1
C1_cancer cancer 22314452 1
C2_cancer cancer 29908600 1
C3_cancer cancer 24872257 1Normalisation for composition bias
The
normLibSizes()function normalizes the library sizes in such a way to minimize the log-fold changes between the samples for most genes. The default method for computing these scale factors uses a trimmed mean of M-values (TMM) between each pair of samples. We call the product of the original library size and the scaling factor the effective library size, i.e., the normalized library size. The effective library size replaces the original library size in all downstream analyses
MDS
plotMDS(y, plot = FALSE)$eigen.vectors[, 1:2] |>
as.data.frame() |>
cbind(my_metadata) |>
dplyr::rename(`Eigenvector 1` = V1, `Eigenvector 2` = V2) |>
ggplot(aes(`Eigenvector 1`, `Eigenvector 2`, colour = group, label = sample)) +
geom_point(size = 2) +
geom_text_repel(show.legend = FALSE) +
theme_minimal() +
ggtitle("MDS plot")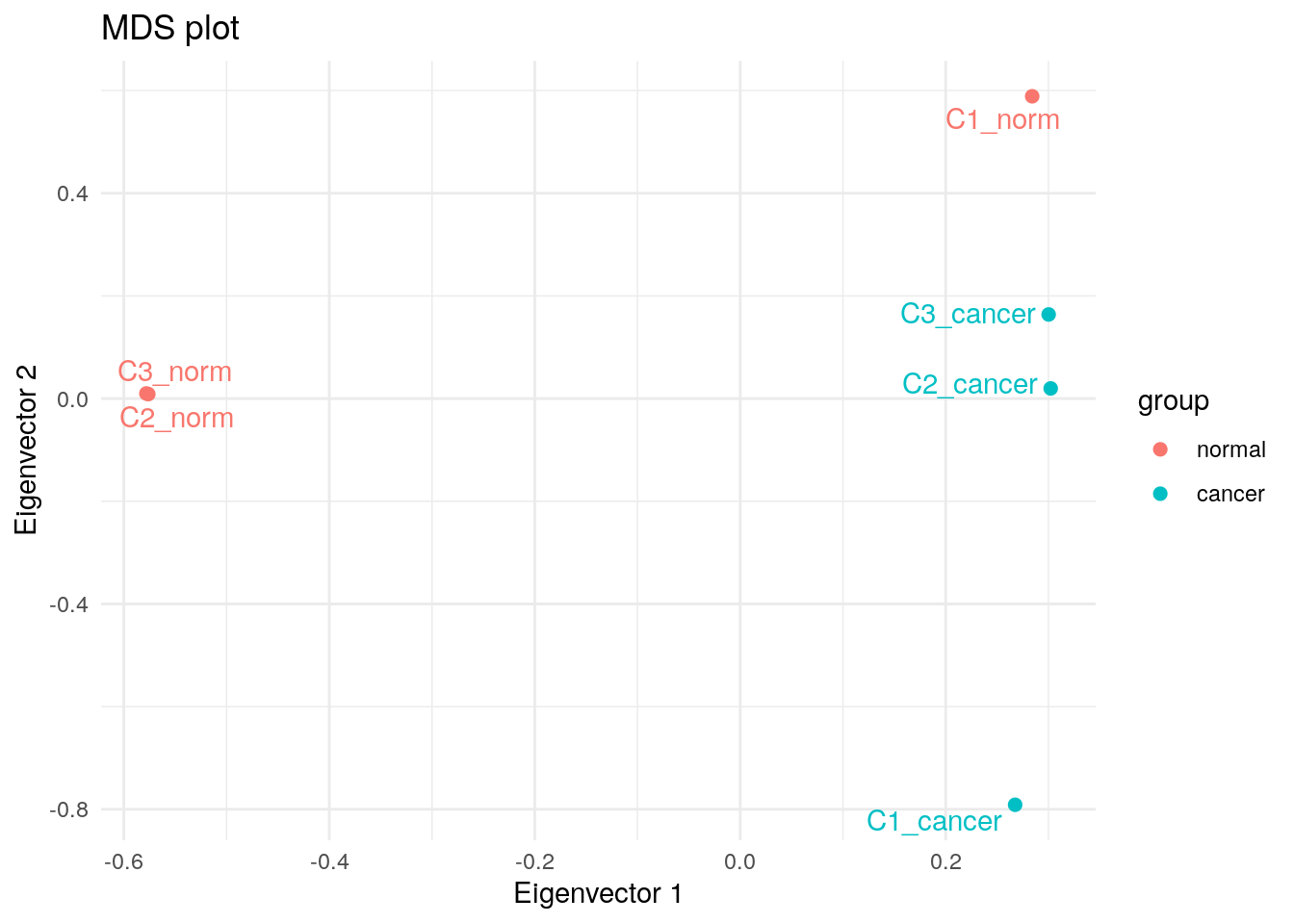
Differential expression
design <- model.matrix(~y$samples$group)
y <- estimateDisp(y, design, robust=TRUE)
fit <- glmQLFit(y, design, robust=TRUE)
res <- glmQLFTest(fit)
topTags(res)Coefficient: y$samples$groupcancer
logFC logCPM F PValue FDR
ENSG00000100985 5.970692 5.990789 73.55648 2.690923e-05 0.07372405
ENSG00000289381 -7.325570 2.197344 47.78707 5.721670e-05 0.07372405
ENSG00000070601 -9.280195 4.481223 41.00090 7.330913e-05 0.07372405
ENSG00000198183 7.916863 5.844195 41.62005 7.841906e-05 0.07372405
ENSG00000229894 -8.996482 4.718492 32.85812 1.377647e-04 0.07372405
ENSG00000151834 -7.764571 3.500301 32.77897 1.391503e-04 0.07372405
ENSG00000241351 5.176813 5.765535 46.46008 1.539133e-04 0.07372405
ENSG00000204961 -9.182288 4.898420 31.83417 1.572426e-04 0.07372405
ENSG00000185972 -6.786526 3.568939 34.93787 1.689653e-04 0.07372405
ENSG00000145934 -8.497381 6.014324 38.79728 1.892544e-04 0.07372405
sessionInfo()R version 4.4.0 (2024-04-24)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggrepel_0.9.5 edgeR_4.2.1 limma_3.60.4 lubridate_1.9.3
[5] forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2
[9] readr_2.1.5 tidyr_1.3.1 tibble_3.2.1 ggplot2_3.5.1
[13] tidyverse_2.0.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] gtable_0.3.5 xfun_0.44 bslib_0.7.0 processx_3.8.4
[5] lattice_0.22-6 callr_3.7.6 tzdb_0.4.0 vctrs_0.6.5
[9] tools_4.4.0 ps_1.7.6 generics_0.1.3 parallel_4.4.0
[13] fansi_1.0.6 highr_0.11 pkgconfig_2.0.3 lifecycle_1.0.4
[17] farver_2.1.2 compiler_4.4.0 git2r_0.33.0 statmod_1.5.0
[21] munsell_0.5.1 getPass_0.2-4 httpuv_1.6.15 htmltools_0.5.8.1
[25] sass_0.4.9 yaml_2.3.8 later_1.3.2 pillar_1.9.0
[29] crayon_1.5.2 jquerylib_0.1.4 whisker_0.4.1 cachem_1.1.0
[33] tidyselect_1.2.1 locfit_1.5-9.9 digest_0.6.37 stringi_1.8.4
[37] splines_4.4.0 labeling_0.4.3 rprojroot_2.0.4 fastmap_1.2.0
[41] grid_4.4.0 colorspace_2.1-0 cli_3.6.3 magrittr_2.0.3
[45] utf8_1.2.4 withr_3.0.1 scales_1.3.0 promises_1.3.0
[49] bit64_4.0.5 timechange_0.3.0 rmarkdown_2.27 httr_1.4.7
[53] bit_4.0.5 hms_1.1.3 evaluate_0.24.0 knitr_1.47
[57] rlang_1.1.4 Rcpp_1.0.12 glue_1.7.0 rstudioapi_0.16.0
[61] vroom_1.6.5 jsonlite_1.8.8 R6_2.5.1 fs_1.6.4