This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210202) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 87ff15f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Summary
Data: Used public data from (Collier et al. 2017): H9 embryonic stem cells in Naïve and Primed state, three biological replicates.
Sample info
dataset_id
gsm_id
description
filename
run_id
input
layout
experiment
26
Collier_2017
GSM2449055
hESC_H9_naive_1
hESC_H9_naive_1
SRR5151102
-
single
RNA-seq
27
Collier_2017
GSM2449056
hESC_H9_naive_2
hESC_H9_naive_2
SRR5151103
-
single
RNA-seq
28
Collier_2017
GSM2449057
hESC_H9_naive_3
hESC_H9_naive_3
SRR5151104
-
single
RNA-seq
29
Collier_2017
GSM2449058
hESC_H9_primed_1
hESC_H9_primed_1
SRR5151105
-
single
RNA-seq
30
Collier_2017
GSM2449059
hESC_H9_primed_2
hESC_H9_primed_2
SRR5151106
-
single
RNA-seq
31
Collier_2017
GSM2449060
hESC_H9_primed_3
hESC_H9_primed_3
SRR5151107
-
single
RNA-seq
Primary analysis
RNA-seq data was analysed by a standard RNA-seq pipeline v2.0, available on nf-core (Ewels et al. 2019) with mostly default settings. For more information about the specific steps involved in this analysis, refer to the repository documentation: https://nf-co.re/rnaseq/2.0.
Reference genome
Due to some difficulties in using hg38 with iGenomes, and since the rest of the analysis has been done on such reference (more specifically, GRCh38.p12). An annotation was downloaded from Ensembl and used as matching custom --fasta and --gtf parameters:
RNA-seq analysis was run on Uppmax HPC environment using standard parameters: nextflow run nf-core/rnaseq -profile uppmax -revision 2.0 --public_data_ids ids_file.txt. ids_file.txt contained only the GSM ids as plain text.
This run would produce the corresponding FASTQ files and then a further run with the generated samplesheet_grouped.tsv file, the pipeline was run.
Note on pipeline version
There is a newer RNA-seq version: https://github.com/nf-core/rnaseq/releases/tag/3.0. This affects the quantification, so it may be the case that results are better using Salmon instead of featureCounts. However at this point there is a problem with Singularity on Uppmax and this version cannot be easily run. If necessary, I will re-do this analysis in the future, once these problems are solved.
Downstream analysis
Helper functions
color_list <- c(Naive=global_palette$teal,
Primed=global_palette$coral)
#' Subset a stringtie output table intro high/med/low expr genes
#'
#' @param counts Counts table.
#' @param min_length Gene length cutoff.
#' @param group_size Number of genes per group.
#' @param min_cutoff Minimum threshold to consider.
#' @param field Which field to use (default = TPM).
#'
#' @return Named list top, bottom, med
categorize_genes <- function(counts, min_length, group_size, min_cutoff = 0, field = "TPM") {
counts$length <- (counts$End - counts$Start)
# Filter by gene length
counts <- counts[counts$length > min_length, ]
# Count as "low" genes that have something, instead of none?
counts <- counts[counts[, field] > min_cutoff, ]
# Sort by corresponding column
counts <- counts[order(counts[, field], decreasing = TRUE), ]
total <- nrow(counts)
top <- counts[1:group_size, ]
bottom <- counts[(total-group_size+1):total, ]
med <- counts[((total/2)-(group_size/2)):((total/2)+(group_size/2)-1),]
list(top = top, bottom = bottom, med = med)
}
#' Make a venn diagram out ouf a list of selected gene counts
#'
#' @param lists List of lists of three (top, med, bottom) gene counts.
#' @param field Which group to select (top, med, bottom)-
#' @param title Plot title
#' @param color Base color
#' @return ggplot object
genes_venn <- function(lists, field, title, color) {
field_list <- list(r1 = lists[[1]][[field]]$Gene.Name,
r2 = lists[[2]][[field]]$Gene.Name,
r3 = lists[[3]][[field]]$Gene.Name)
clist <- rep(color, 3)
ggvenn(field_list, fill_color = clist, fill_alpha = 0.5, text_size = 5, stroke_color = "#ffffff") +
ggtitle(title)
}
#' Intersect per category
#'
#' @param group_list List of lists of three (top, med, bottom) gene counts.
#' @param category Which group to select (top, med, bottom).
#'
#' @return Gene names that intersect
intersect_3 <- function(group_list, category) {
intersect(intersect(group_list[[1]][[category]]$Gene.Name,
group_list[[2]][[category]]$Gene.Name),
group_list[[3]][[category]]$Gene.Name)
}
#' Subset a named granges by name
#'
#' @param names Names list
#' @param granges GRanges to subset.
#'
#' @return GRanges
select_genes <- function(names, granges) {
sort(granges[granges$name %in% names, ])
}
#' Export gene sets out of a list of selected gene counts
#'
#' @param group_list List of lists of three (top, med, bottom) gene counts.
#' @param prefix File name prefix
export_gene_sets <- function(group_list, prefix) {
merged <- map(c("top", "med", "bottom"),
intersect_3,
group_list=group_list)
out_files <- paste0(file.path("./output/Collier_2017/"),
paste(prefix,
c("top.bed", "med.bed", "bottom.bed"), sep="_"))
genes_all <- genes_hg38()
ranges_list <- map(merged, select_genes, granges=genes_all)
file_list <- map2(ranges_list, out_files, export)
}
Gene counts
Gene counts output file from featureCounts are used.
As expected, there are many zero values in all the distributions, and long tails since there is no upper limit in RNA-seq data.
Stringtie output
stringtie(Kovaka et al. 2019)https://ccb.jhu.edu/software/stringtie/) is a tool for quantitation of full-length transcripts (taking variants into account) per gene.
This output is also provided as standard output for RNA-seq pipeline v2.0.
There seems to be some overlap in the Naïve-only vs Primed-only and our groups, when looking at functional annotation, at least. Being more stringent in the top expressed genes:
min_length_cutoff <- 10000
group_size <- 2000
min_expr_cutoff <- 5
deciding_stat <- "TPM"
# Set a cutoff for the low expressed genes, so there is still some signal
# In there. When intersecting otherwise, I get few overlap.
groups_naive <- lapply(counts_naive,
categorize_genes,
min_length = min_length_cutoff,
group_size = group_size,
min_cutoff = min_expr_cutoff,
field = deciding_stat)
counts_primed <- lapply(stringtie_files$primed,
read.csv,
header = T,
sep = "\t")
groups_primed <- lapply(counts_primed,
categorize_genes,
min_length = min_length_cutoff,
group_size = group_size,
min_cutoff = min_expr_cutoff,
field = deciding_stat)
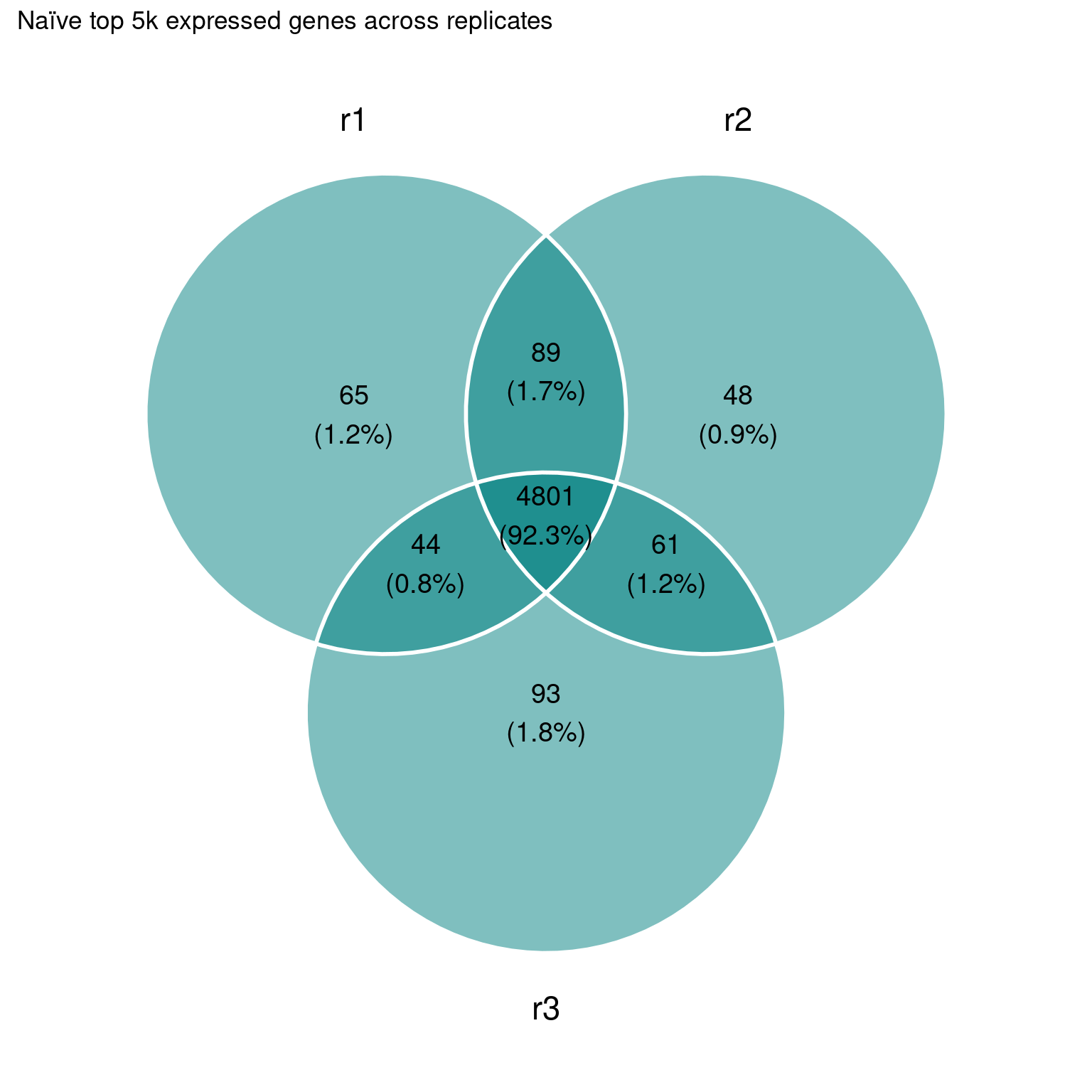
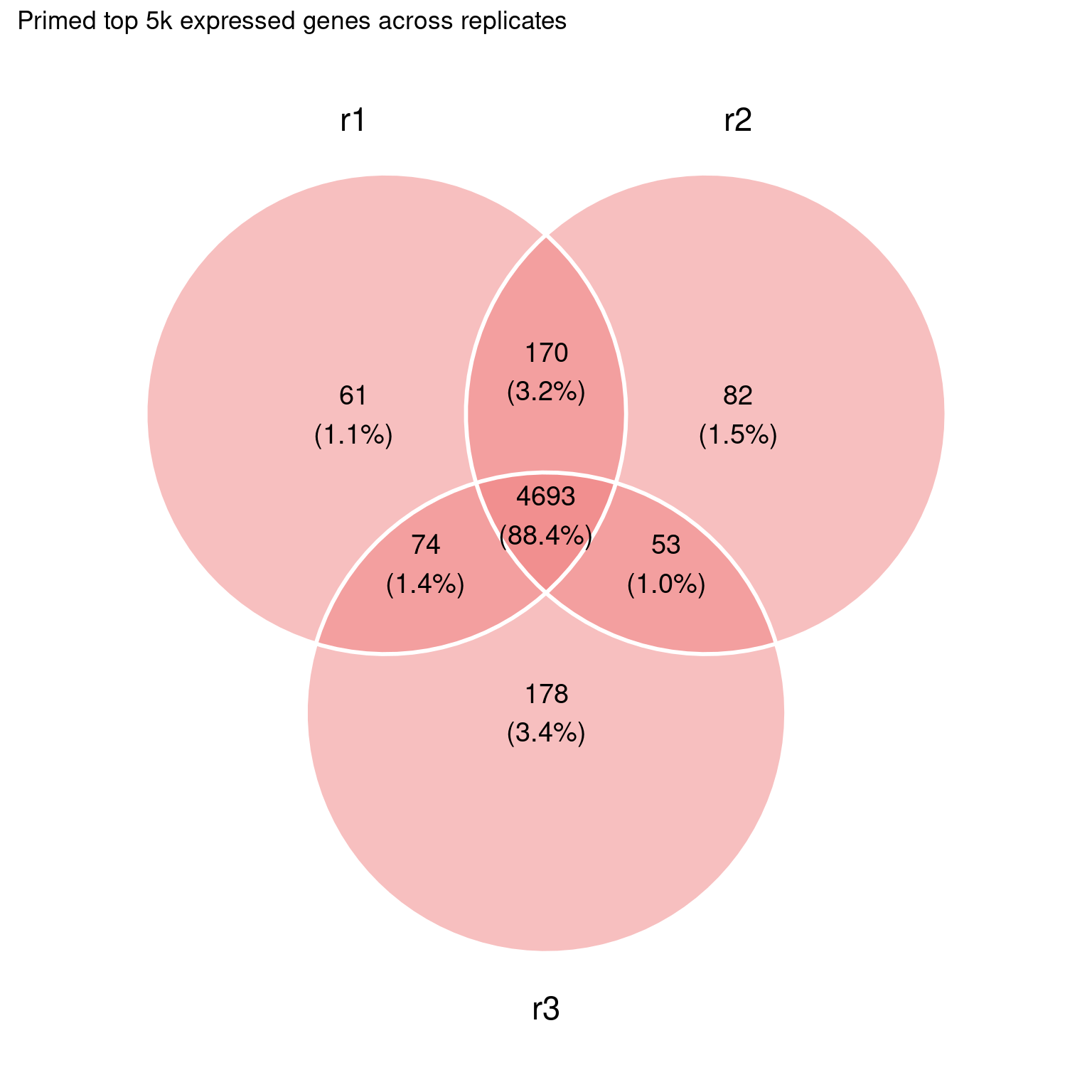
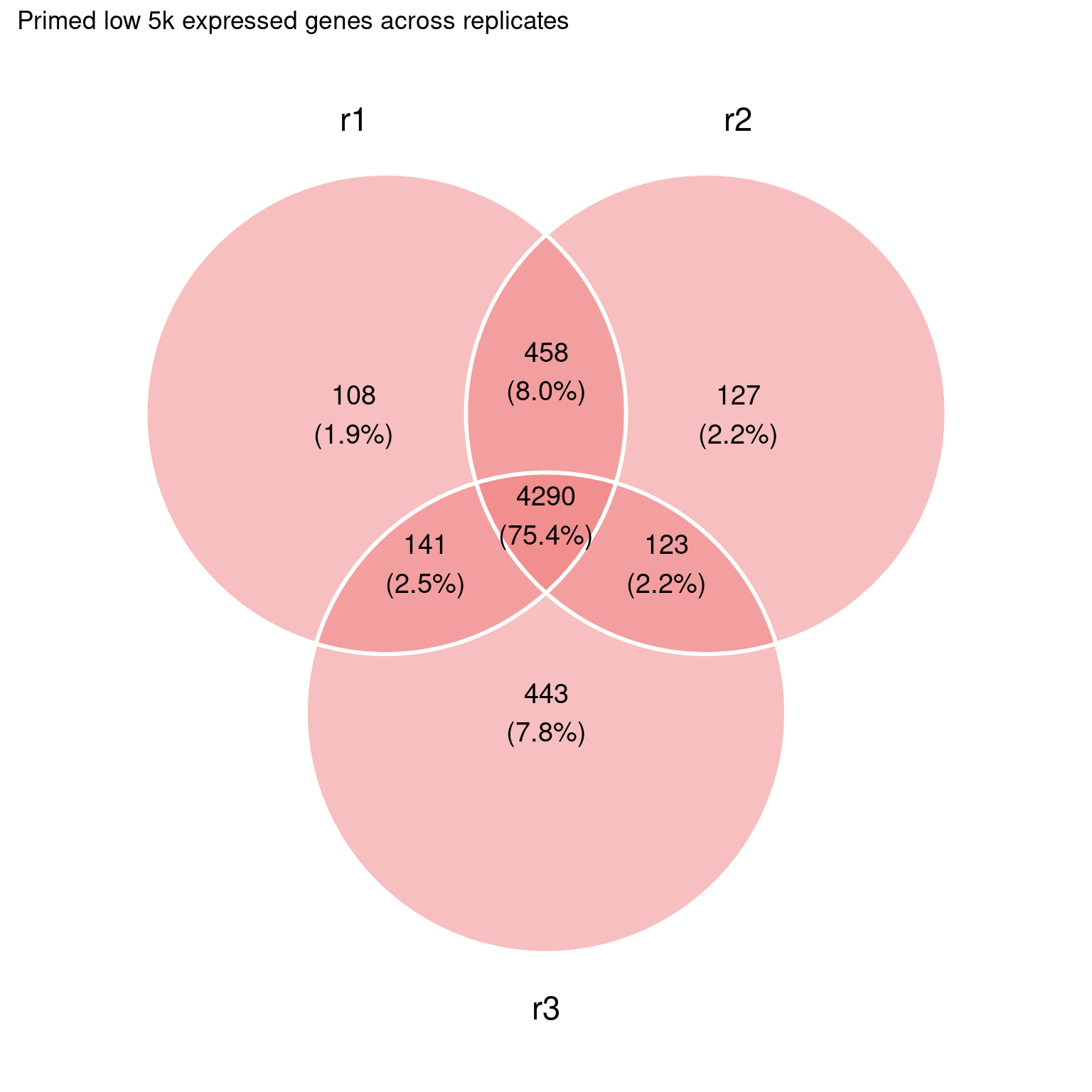
Still quite some overlap among the highest expressed genes:
This is a bit too much at the moment, we can probably refine this.
Annotation of gene groups
Now I annotate the intersection of these sets in the reference genome, to be able to look at chromatin marks there.
genes_hg38 <- function() {
# This matches gene IDs to symbols
x <- org.Hs.egSYMBOL
# Get the gene names that are mapped to an entrez gene identifier
mapped_genes <- mappedkeys(x)
# Convert to a list
xx <- as.list(x[mapped_genes])
# Apparently there are some pathway crap that puts NAs in the list
xx <- xx[!is.na(xx)]
gene_id_to_name <- unlist(xx)
genes <- genes(TxDb.Hsapiens.UCSC.hg38.knownGene)
genes$name <- gene_id_to_name[genes$gene_id]
genes
}
min_length_cutoff <- 10000
group_size <- 5000
min_expr_cutoff <- 5
deciding_stat <- "TPM"
# Set a cutoff for the low expressed genes, so there is still some signal
# In there. When intersecting otherwise, I get few overlap.
groups_naive <- lapply(counts_naive,
categorize_genes,
min_length = min_length_cutoff,
group_size = group_size,
min_cutoff = min_expr_cutoff,
field = deciding_stat)
export_gene_sets(groups_naive, "Naive")
1613 genes were dropped because they have exons located on both strands
of the same reference sequence or on more than one reference sequence,
so cannot be represented by a single genomic range.
Use 'single.strand.genes.only=FALSE' to get all the genes in a
GRangesList object, or use suppressMessages() to suppress this message.
1613 genes were dropped because they have exons located on both strands
of the same reference sequence or on more than one reference sequence,
so cannot be represented by a single genomic range.
Use 'single.strand.genes.only=FALSE' to get all the genes in a
GRangesList object, or use suppressMessages() to suppress this message.
Collier, Amanda J, Sarita P Panula, John Paul Schell, Peter Chovanec, Alvaro Plaza Reyes, Sophie Petropoulos, Anne E Corcoran, et al. 2017. “Comprehensive Cell Surface Protein Profiling Identifies Specific Markers of Human Naive and Primed Pluripotent States.”Cell Stem Cell 20 (6): 874–90.
Ewels, Philip A, Alexander Peltzer, Sven Fillinger, Johannes Alneberg, Harshil Patel, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso, and Sven Nahnsen. 2019. “Nf-Core: Community Curated Bioinformatics Pipelines.”BioRxiv, 610741.
Kovaka, Sam, Aleksey V Zimin, Geo M Pertea, Roham Razaghi, Steven L Salzberg, and Mihaela Pertea. 2019. “Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2.”Genome Biology 20 (1): 1–13.