Figure 3.
Carmen Navarro
2021-07-01
Last updated: 2021-07-09
Checks: 7 0
Knit directory: hesc-epigenomics/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210202) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version d50d87a. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/bed/
Ignored: data/bw
Ignored: data/igv/
Ignored: data/liftover/
Ignored: data/peaks
Ignored: data/rnaseq/
Ignored: figures_data/
Untracked files:
Untracked: data/meta/Kumar_2020_bins_panels_design.csv
Untracked: data/meta/Kumar_2020_master_bins_10kb_table_final_raw.tar.gz
Untracked: data/meta/Kumar_2020_master_bins_10kb_table_final_raw.tsv
Untracked: data/meta/Kumar_2020_master_bins_10kb_table_final_raw.zip
Untracked: data/meta/Kumar_2020_master_bins_10kb_table_raw (copy).tsv
Untracked: data/meta/Kumar_2020_master_bins_10kb_table_raw.tsv
Untracked: data/meta/Kumar_2020_master_bins_5kb_table_raw.tsv
Untracked: data/meta/Kumar_2020_master_bins_5kb_table_raw.zip
Untracked: data/meta/Kumar_2020_master_bins_5kb_table_replicates_only.tsv
Untracked: data/meta/Kumar_2020_master_bins_5kb_table_shrunk.tsv
Untracked: data/meta/Kumar_2020_master_bins_5kb_table_shrunk.zip
Untracked: data/meta/Kumar_2020_master_gene_table.zip
Untracked: data/meta/Kumar_2020_master_gene_table_rnaseq_shrunk.tsv
Untracked: data/meta/Kumar_2020_master_gene_table_rnaseq_shrunk_annotated.tsv
Untracked: data/meta/Kumar_2020_master_gene_table_rnaseq_shrunk_plus_annotations.tsv
Untracked: data/meta/Kumar_2020_master_gene_table_rnaseq_shrunk_plus_annotations_2.tsv
Untracked: data/meta/Kumar_2020_promoters_panels_design.csv
Untracked: data/meta/gene_names_bivalent.tsv
Untracked: data/other/Bock_2011/
Untracked: data/other/Cinkornpumin_2020/
Untracked: data/other/Lanner/
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS1_NaivePrimed_Diff.csv
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS1_NaivePrimed_Diff.xls
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS2_IntermediatePop.csv
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS2_IntermediatePop.xls
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS3_GermLayerMarkers.csv
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS3_GermLayerMarkers.xls
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS4_LineageEpiMarkers.csv
Untracked: data/other/Messmer_2019/Messmer_CellRep2019_TableS4_LineageEpiMarkers.xls
Untracked: output/Kumar_2020_H3K27m3_Ni_higher_than_Pr_tss.bed
Untracked: output/Kumar_2020_H3K27m3_Pr_higher_than_Ni_tss.bed
Untracked: output/Kumar_2020_H3K27m3_always_up_tss.bed
Untracked: output/fig2_chr_means_boxplot.svg
Untracked: output/fig3_violin_rnaseq_ratios_naive_higher_than_pr.svg
Untracked: output/fig3_violin_rnaseq_ratios_primed_higher_than_ni.svg
Untracked: output/fig4_lineage_makers_new.png
Untracked: output/fig4_lineage_makers_new.svg
Untracked: output/fig4_lineage_markers_new.png
Untracked: output/fig4_lineage_markers_new.svg
Untracked: output/fig4_lineage_markers_new_cluster.png
Untracked: output/fig4_lineage_markers_new_cluster.svg
Untracked: output/fig4_lineage_markers_new_k4.png
Untracked: output/fig4_lineage_markers_new_k4.svg
Untracked: output/fig4_messmer_top25.png
Untracked: output/fig4_messmer_top25.svg
Untracked: output/fig4_messmer_top25_down.png
Untracked: output/fig4_messmer_top25_down.svg
Untracked: output/fig4_messmer_top25_up.png
Untracked: output/fig4_messmer_top25_up.svg
Untracked: output/fig4_messmer_top50_down.png
Untracked: output/fig4_messmer_top50_down.svg
Untracked: output/fig4_messmer_top50_up.png
Untracked: output/fig4_messmer_top50_up.svg
Untracked: output/fig4_pca_all_plus_dong_data.svg
Untracked: output/fig4_pca_only_our_samples.svg
Untracked: output/fig4_pca_only_our_samples_labeled.svg
Untracked: output/fig4_pluripotency_markers_new.png
Untracked: output/fig4_pluripotency_markers_new.svg
Untracked: output/fig4_pluripotency_markers_new_cluster.png
Untracked: output/fig4_pluripotency_markers_new_cluster.svg
Untracked: output/panel_figures/
Untracked: output/promoter_panels_final/
Unstaged changes:
Modified: .gitignore
Modified: analysis/rnaseq_comparison.Rmd
Modified: code/heatmap_panels.R
Modified: code/heatmaply_functions.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/fig_03_h3k27m3_groups.Rmd) and HTML (docs/fig_03_h3k27m3_groups.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | d50d87a | C. Navarro | 2021-07-09 | wflow_publish(“./analysis/fig_03_h3k27m3_groups.Rmd”, verbose = T) |
| html | daf3d4a | C. Navarro | 2021-07-08 | Build site. |
| Rmd | 4d534a3 | C. Navarro | 2021-07-08 | wflow_publish(“./analysis/fig_03_h3k27m3_groups.Rmd”, verbose = T) |
| html | 7117e7a | C. Navarro | 2021-07-08 | Build site. |
| Rmd | 394d32e | C. Navarro | 2021-07-08 | wflow_publish(“./analysis/fig_03_h3k27m3_groups.Rmd”, verbose = T) |
| html | 67895c5 | C. Navarro | 2021-07-01 | Build site. |
| Rmd | 70ee455 | C. Navarro | 2021-07-01 | wflow_publish(“./analysis/fig_03_h3k27m3_groups.Rmd”) |
Summary
This is the supplementary notebook for figure 3.
H3K27m3 promoter volcano plot
genes_tss <- makeGRangesFromDataFrame(genes, keep.extra.columns = T)
court_peaks <- import("./data/bed/Bivalent_Court2017.hg38.bed")
court_tss <- subsetByOverlaps(genes_tss, court_peaks)
court_tss_df <- data.frame(court_tss)
ggplot(genes, aes(x=H3K27m3_DS_Pr_vs_Ni_log2FoldChange, y=-log10(H3K27m3_DS_Pr_vs_Ni_padj))) +
rasterise(geom_point(size = 1, alpha = 0.7, color = "gray"), dpi = 300) +
theme_default(base_size = 12) +
rasterise(geom_point(data = court_tss_df, size = 1, alpha = 0.8, color = "black"), dpi = 300) +
labs(x = "Log2 FC (primed/naive)",
y = "Log10 pval", title = "Promoter H3K27m3 - Court 2017 bivalent in black") +
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = 0) +
coord_cartesian(xlim = c(-6,6), ylim = c(0, 20))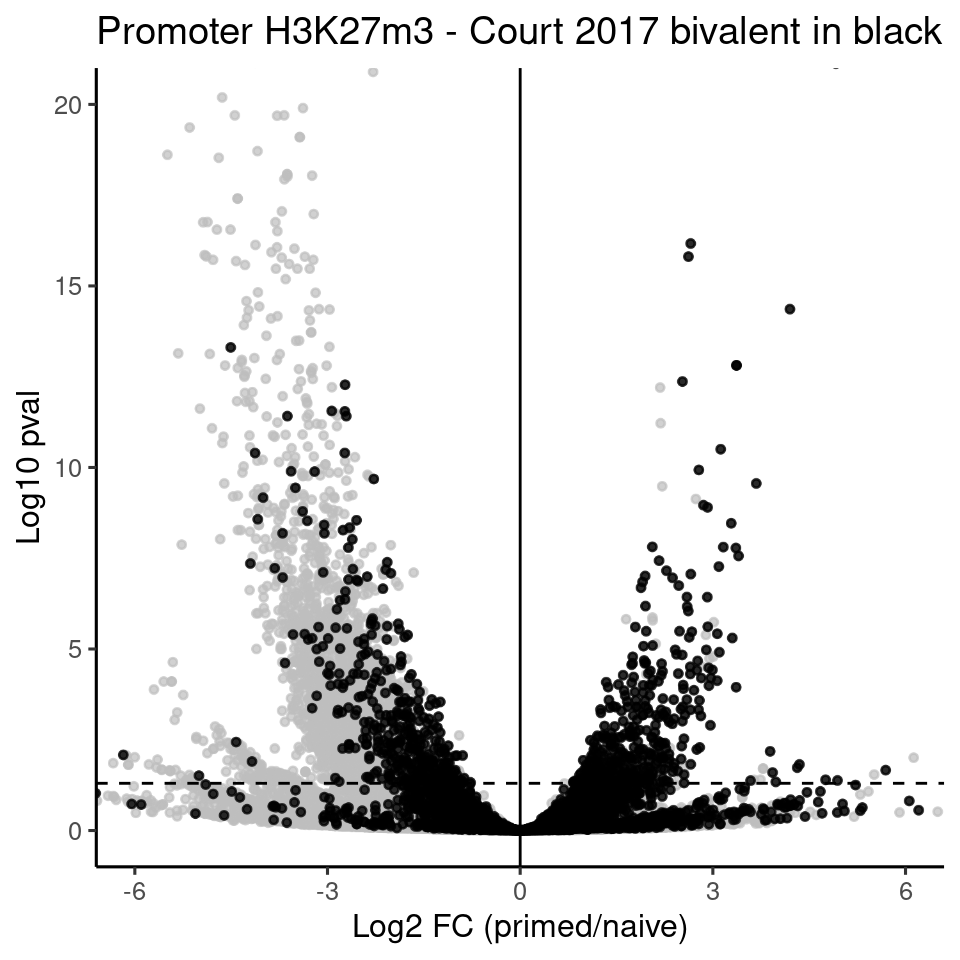
df <- genes[, c("H3K27m3_DS_Pr_vs_Ni_log2FoldChange", "H3K27m3_DS_Pr_vs_Ni_padj")]
df[, "H3K27m3_DS_Pr_vs_Ni_padj"] <- -log10(df[, "H3K27m3_DS_Pr_vs_Ni_padj"])
write.table(df, "./figures_data/fig3_h3k27m3_promoter_volcano_all.tsv", sep = "\t", row.names = F)
df <- court_tss_df[, c("H3K27m3_DS_Pr_vs_Ni_log2FoldChange", "H3K27m3_DS_Pr_vs_Ni_padj")]
df[, "H3K27m3_DS_Pr_vs_Ni_padj"] <- -log10(df[, "H3K27m3_DS_Pr_vs_Ni_padj"])
write.table(df, "./figures_data/fig3_h3k27m3_promoter_volcano_court.tsv", sep = "\t", row.names = F)H3K27m3 groups heatmap panels
H3K27m3 group annotation
select_groups <- function(df, pval_col, fc_col, basemean_col, quantile,
p_cutoff = 0.05, fc_cutoff = 1, basemean_quantile = 0.1) {
# I don't want to discard the NAs as they will go to the unenriched group.
df[is.na(df[[pval_col]]), pval_col] <- 1
min_mean <- quantile(df[[basemean_col]], probs = basemean_quantile)
signif_up_tss <- df %>% filter(.data[[pval_col]] <= p_cutoff & .data[[fc_col]] > fc_cutoff & .data[[basemean_col]] > min_mean)
signif_down_tss <- df %>% filter(.data[[pval_col]] <= p_cutoff & .data[[fc_col]] < -fc_cutoff & .data[[basemean_col]] > min_mean)
not_signif <- df %>% filter(.data[[pval_col]] > p_cutoff)
# Top
mean_cutoff <- quantile(not_signif[[basemean_col]], quantile)
always_up <- not_signif %>% filter(.data[[basemean_col]] >= mean_cutoff)
rest <- not_signif %>% filter(.data[[basemean_col]] < mean_cutoff)
list(up = signif_up_tss,
down = signif_down_tss,
always_up = always_up,
not_enriched = rest)
}
select_groups_bivalent <- function(df, quantile = 0.8, p_cutoff = 0.05, fc_cutoff = 1, basemean_quantile = 0.1, min_k4 = 2) {
select_k4 <- function(df, min_k4) {
df %>% filter(.data[["H3K4m3_Pr_mean_cov"]] > min_k4 | .data[["H3K4m3_Ni_mean_cov"]] > min_k4)
}
groups <- select_groups(
df,
"H3K27m3_DS_Pr_vs_Ni_padj",
"H3K27m3_DS_Pr_vs_Ni_log2FoldChange",
"H3K27m3_DS_Pr_vs_Ni_baseMean",
quantile,
p_cutoff,
fc_cutoff,
basemean_quantile
)
lapply(groups, select_k4, min_k4 = min_k4)
}
k27_groups <- select_groups_bivalent(genes)
# Annotate our groups
genes$k27_bivalency_grp <- "None"
genes[genes$name %in% k27_groups$up$name, "k27_bivalency_grp"] <- "Pr_higher_than_Ni"
genes[genes$name %in% k27_groups$down$name, "k27_bivalency_grp"] <- "Ni_higher_than_Pr"
genes[genes$name %in% k27_groups$always_up$name, "k27_bivalency_grp"] <- "Always_up"
genes[genes$name %in% k27_groups$not_enriched$name, "k27_bivalency_grp"] <- "K4_only"
# Annotate court
genes_loci <- import("./data/bed/Kumar_2020/Kumar_2020_genes_hg38_UCSC.bed")
genes_tss <- promoters(genes_loci, upstream = 2500, downstream = 2500)
court_bivalent <- rtracklayer::import("./data/bed/Bivalent_Court2017.hg38.bed")
court_biv_genes <- subsetByOverlaps(genes_tss, court_bivalent)
genes$court_bivalent <- "No"
genes[genes$name %in% court_biv_genes$name, "court_bivalent"] <- "Yes"
write.table(genes, "./data/meta/Kumar_2020_master_gene_table_rnaseq_shrunk_annotated.tsv")
k27_groups_loci <- lapply(k27_groups, make_gr_from_table)
rtracklayer::export(k27_groups_loci$up, "./figures_data/Kumar_2020_H3K27m3_Pr_higher_than_Ni_tss.bed")
rtracklayer::export(k27_groups_loci$down, "./figures_data/Kumar_2020_H3K27m3_Ni_higher_than_Pr_tss.bed")
rtracklayer::export(k27_groups_loci$always_up, "./figures_data/Kumar_2020_H3K27m3_always_up_tss.bed")
rtracklayer::export(k27_groups_loci$not_enriched, "./figures_data/Kumar_2020_H3K27m3_not_enriched_tss.bed")H3K27m3 group heatmap panels
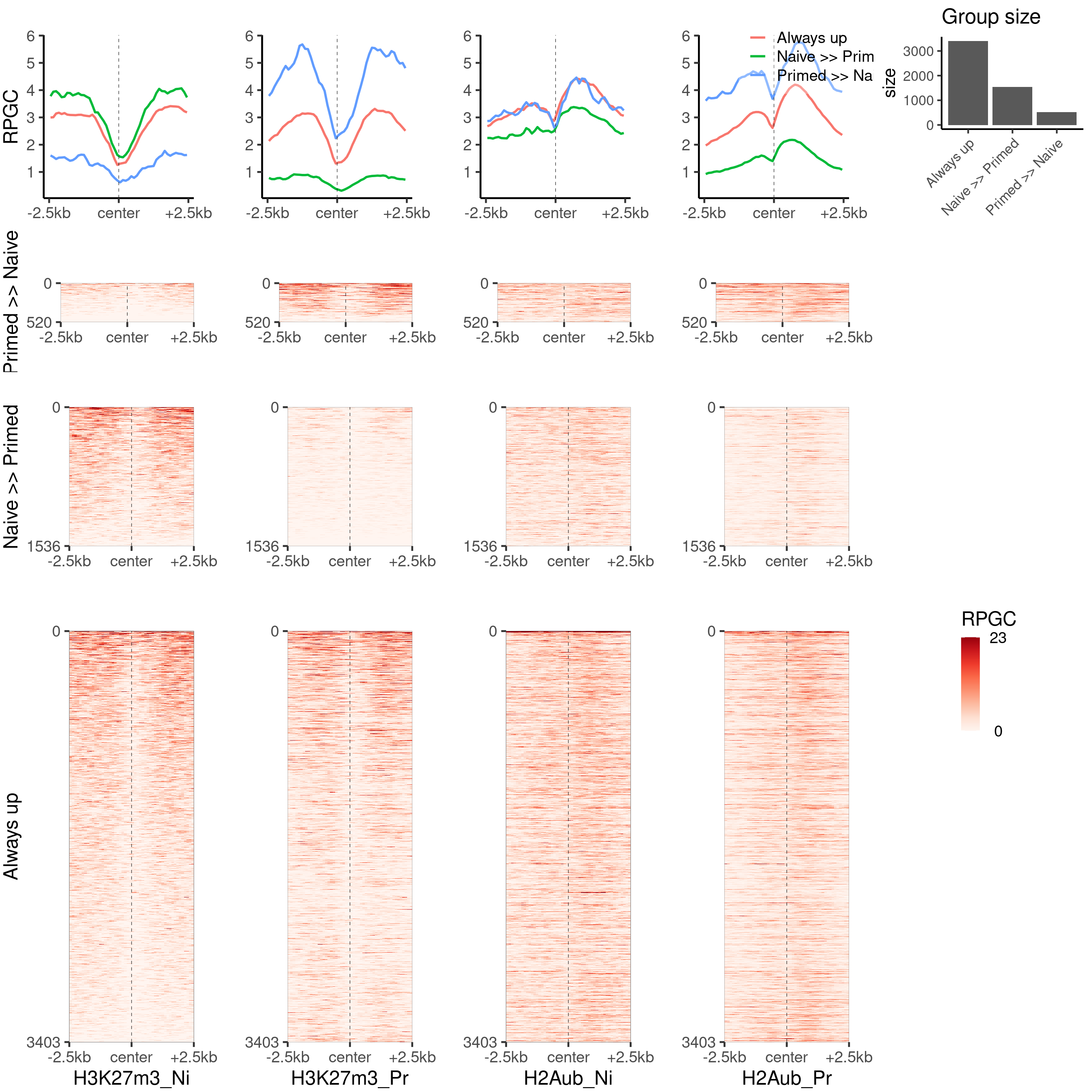
H3K27m3 and H2AUb
plot_bw_heatmap_panel(
c(bwfiles$k27[c(1, 3)], bwfiles$ub[c(1, 3)]),
list(k27_groups_loci$up,
k27_groups_loci$down,
k27_groups_loci$always_up
),
c("H3K27m3_Ni", "H3K27m3_Pr", "H2Aub_Ni", "H2Aub_Pr"),
c("Primed >> Naive", "Naive >> Primed", "Always up"),
global_scale = TRUE,
proportional = TRUE,
mode = "center"
)
| Version | Author | Date |
|---|---|---|
| 67895c5 | C. Navarro | 2021-07-01 |
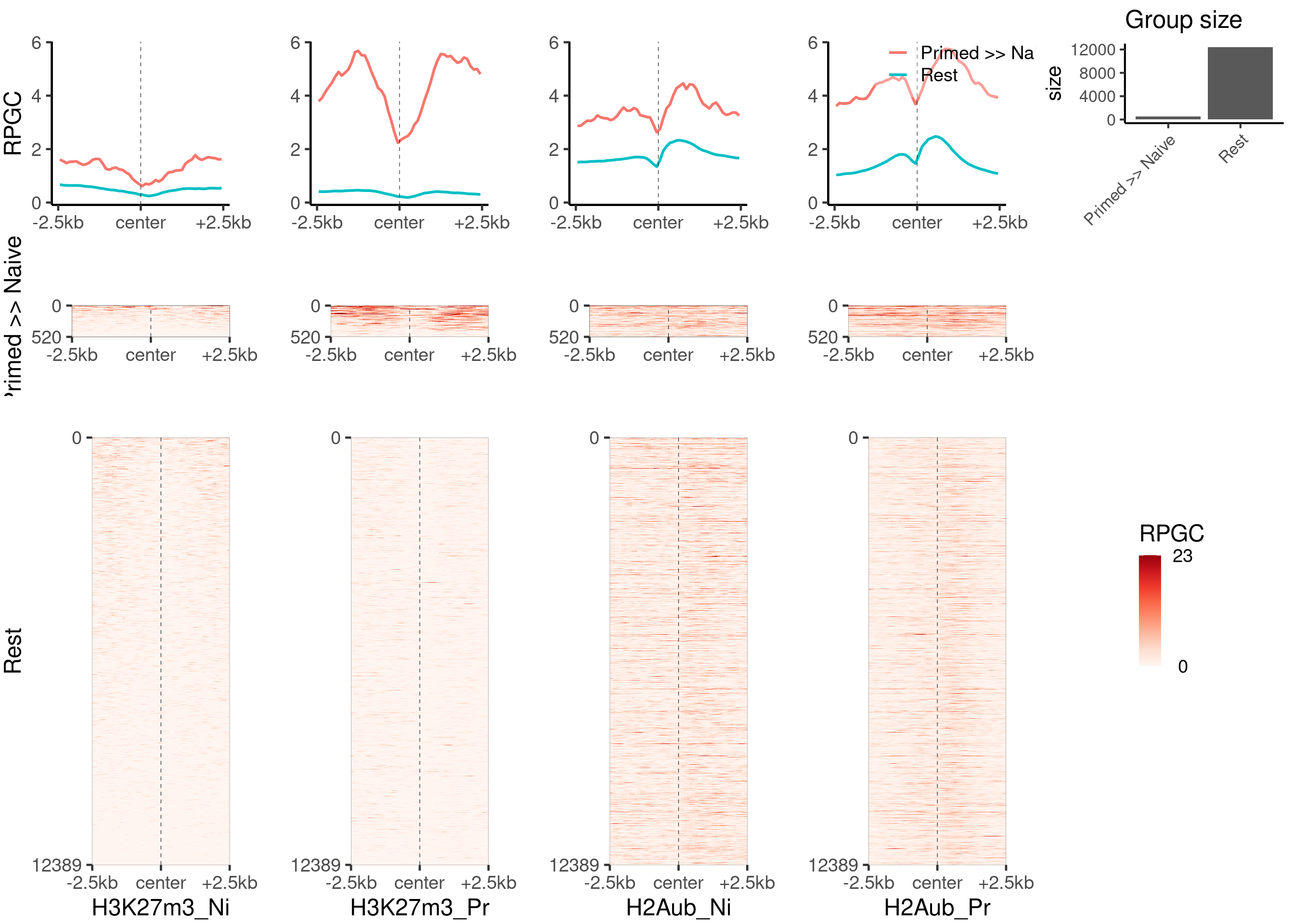
Plot the second part of the heatmap panel.
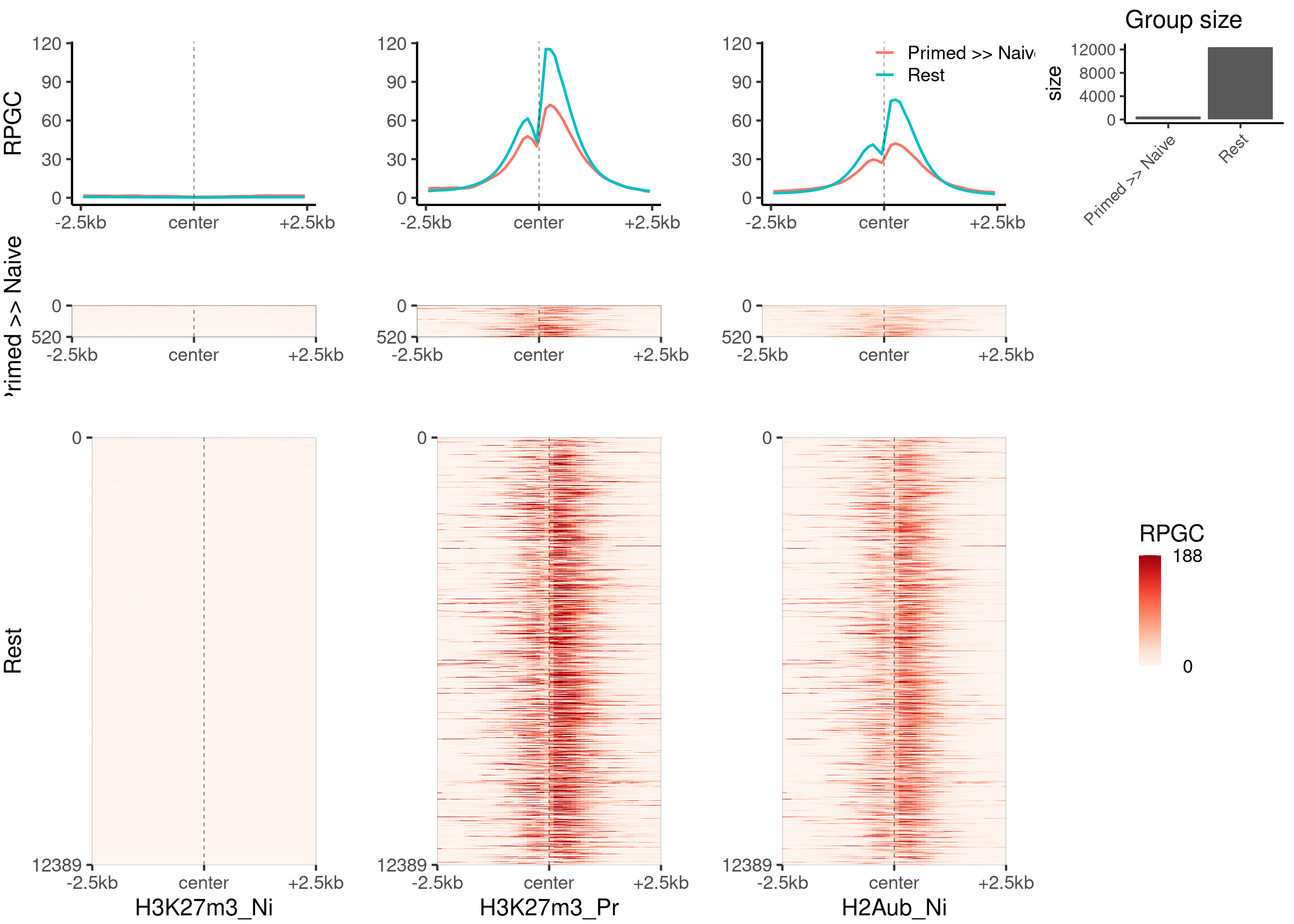
plot_bw_heatmap_panel(
c(bwfiles$k27[c(1, 3)], bwfiles$ub[c(1, 3)]),
list(k27_groups_loci$up, k27_groups_loci$not_enriched),
c("H3K27m3_Ni", "H3K27m3_Pr", "H2Aub_Ni", "H2Aub_Pr"),
c("Primed >> Naive", "Rest"),
global_scale = TRUE,
proportional = TRUE,
mode = "center",
zmin = 0,
zmax = 23
)
| Version | Author | Date |
|---|---|---|
| 67895c5 | C. Navarro | 2021-07-01 |
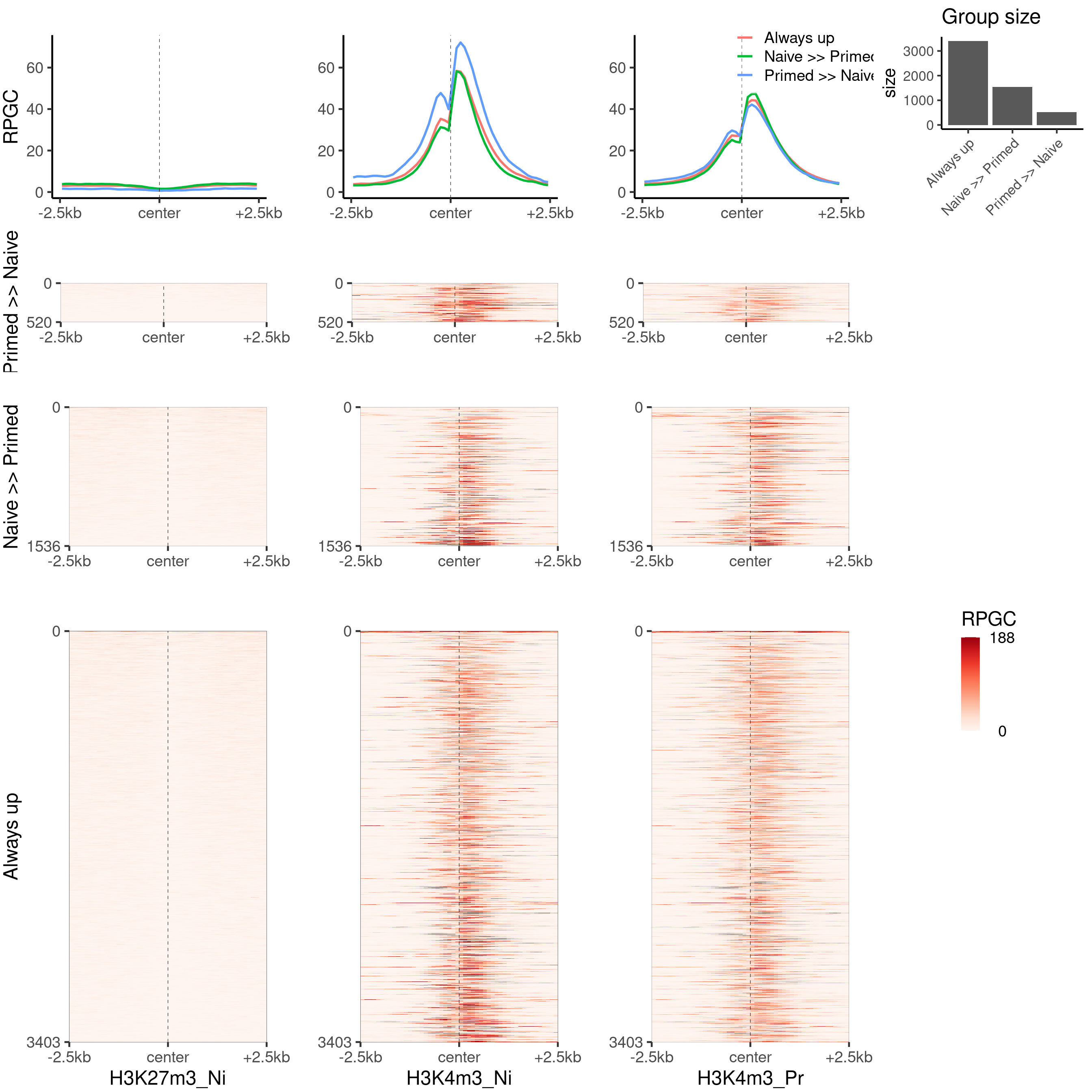
H3K4m3
Plot the H3K4m3 part, sorted by the same reference
plot_bw_heatmap_panel(
c(bwfiles$k27[1], bwfiles$k4[c(1, 3)]),
list(k27_groups_loci$up,
k27_groups_loci$down,
k27_groups_loci$always_up
),
c("H3K27m3_Ni", "H3K4m3_Ni", "H3K4m3_Pr"),
c("Primed >> Naive", "Naive >> Primed", "Always up"),
global_scale = TRUE,
proportional = TRUE,
mode = "center"
)
| Version | Author | Date |
|---|---|---|
| 7117e7a | C. Navarro | 2021-07-08 |
plot_bw_heatmap_panel(
c(bwfiles$k27[1], bwfiles$k4[c(1, 3)]),
list(k27_groups_loci$up, k27_groups_loci$not_enriched),
c("H3K27m3_Ni", "H3K27m3_Pr", "H2Aub_Ni", "H2Aub_Pr"),
c("Primed >> Naive", "Rest"),
global_scale = TRUE,
proportional = TRUE,
mode = "center",
zmin = 0,
zmax = 188
)
RNA-seq ratios at H3K27m3 groups
H327m3 Naive >> Primed
df <-
genes[, c(
"name",
"RNASeq_DS_Pr_vs_Ni_log2FoldChange",
"RNASeq_DS_EZH2i_vs_Ni_log2FoldChange",
"RNASeq_DS_EZH2i_vs_Pr_log2FoldChange"
)]
colnames(df) <- c("name", "Primed vs Naive", "EZH2i vs Naive", "EZH2i vs Primed")
df_k27_up <- df[df$name %in% k27_groups_loci$down$name, ]
df_long <- df %>% pivot_longer(!name, names_to = "group", values_to = "fc")
df_long_k27 <- df_k27_up %>% pivot_longer(!name, names_to = "group", values_to = "fc")
df_long$group <- factor(df_long$group, levels = c("Primed vs Naive", "EZH2i vs Naive", "EZH2i vs Primed"))
df_long_k27$group <- factor(df_long_k27$group, levels = c("Primed vs Naive", "EZH2i vs Naive", "EZH2i vs Primed"))
# Stats subset vs global
ni_pr_test <- wilcox.test(df_k27_up[, "Primed vs Naive"],
df[!df$name %in% df_k27_up$name , "Primed vs Naive"], na.rm = T)
ni_pr_effect <- cohen.d(df_k27_up[, "Primed vs Naive"],
df[!df$name %in% df_k27_up$name , "Primed vs Naive"], na.rm = T)
col <- "EZH2i vs Naive"
ni_ezh2i_test <- wilcox.test(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
ni_ezh2i_effect <- cohen.d(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
col <- "EZH2i vs Primed"
pr_ezh2i_test <- wilcox.test(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
pr_ezh2i_effect <- cohen.d(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
stats_caption <- paste("Wilcoxon global vs. selection",
paste("Ni_Pr:", format(ni_pr_test$p.value, digits=8),
"Cohen D:", round(ni_pr_effect$estimate, digits = 4),
"(", ni_pr_effect$magnitude, ")"),
paste("Ni_EZH2i:", format(ni_ezh2i_test$p.value, digits=8),
"Cohen D:", round(ni_ezh2i_effect$estimate, digits = 4),
"(", ni_ezh2i_effect$magnitude, ")"),
paste("Pr_EZH2i:", format(pr_ezh2i_test$p.value, digits=8),
"Cohen D:", round(pr_ezh2i_effect$estimate, digits = 4),
"(", pr_ezh2i_effect$magnitude, ")"),
sep = "\n")
my_comparisons <- list(c("EZH2i vs Naive", "EZH2i vs Primed"),
c("Primed vs Naive", "EZH2i vs Naive"),
c("Primed vs Naive", "EZH2i vs Primed"))
k27_up_color <- "#009784"
ggplot(data=df_long, aes(x = group, y = fc)) +
geom_violin(size = 0.8) +
stat_compare_means(data=df_long_k27,
comparisons = my_comparisons, method = "wilcox.test", paired = TRUE) +
rasterize(
geom_jitter(data=df_long_k27, color = k27_up_color, alpha = 0.7, size = 0.1),
dpi = 300) +
geom_hline (yintercept = 0, linetype = "dashed") +
coord_cartesian(ylim = c(-13, 24)) +
theme_default(base_size = 14) +
labs(x = "Condition",
y = "Log2 FC",
title = "RNASeq expression changes",
subtitle = "Global vs H3K27m3 Naïve >> Primed TSS",
caption = stats_caption)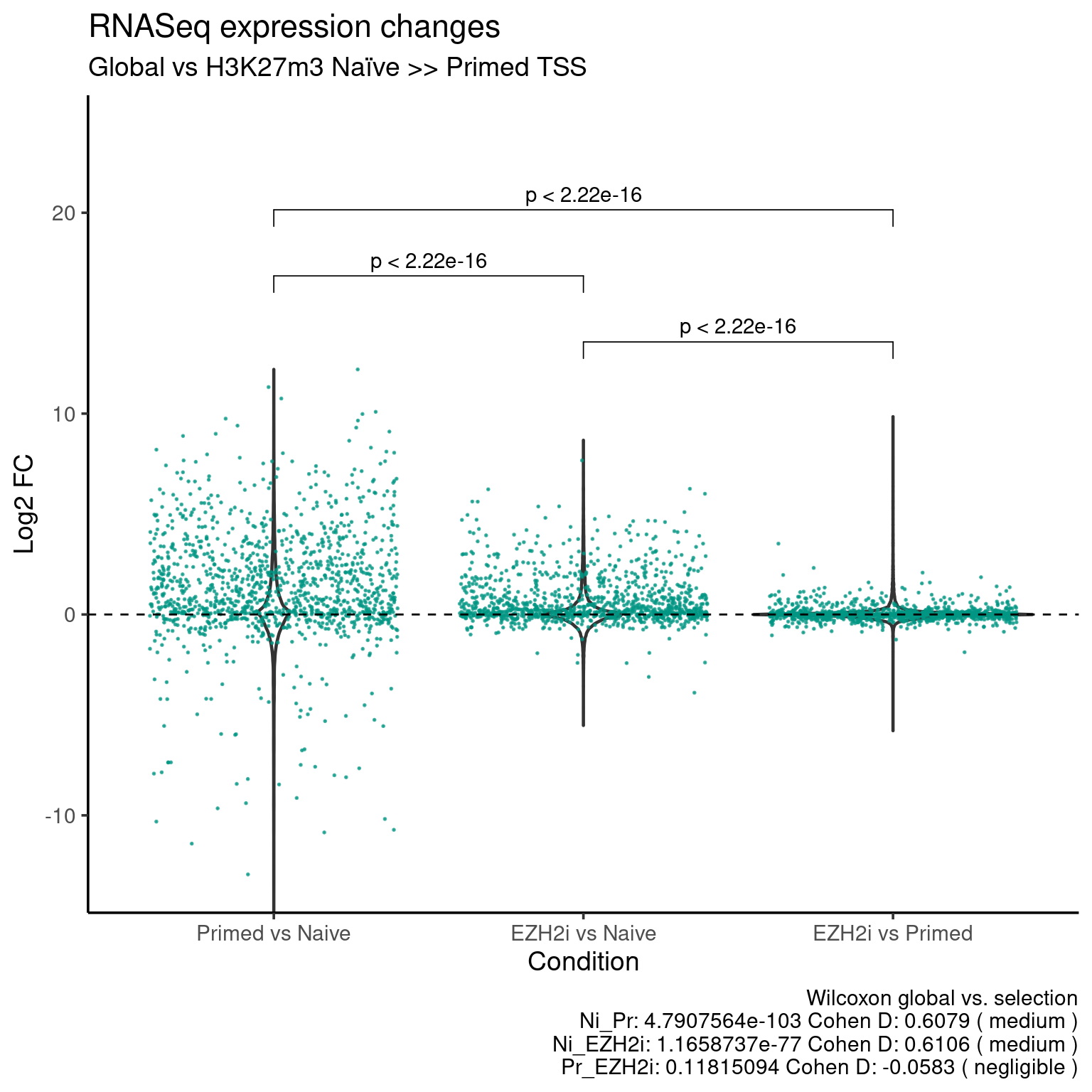
| Version | Author | Date |
|---|---|---|
| 67895c5 | C. Navarro | 2021-07-01 |
Download values: Distribution, K27 higher naive points,
H327m3 Primed >> Naive
df <-
genes[, c(
"name",
"RNASeq_DS_Pr_vs_Ni_log2FoldChange",
"RNASeq_DS_EZH2i_vs_Ni_log2FoldChange",
"RNASeq_DS_EZH2i_vs_Pr_log2FoldChange"
)]
colnames(df) <- c("name", "Primed vs Naive", "EZH2i vs Naive", "EZH2i vs Primed")
df_k27_up <- df[df$name %in% k27_groups_loci$up$name, ]
df_k27_up$`Primed vs Naive` <- -df_k27_up$`Primed vs Naive`
df$`Primed vs Naive` <- -df$`Primed vs Naive`
df_long <- df %>% pivot_longer(!name, names_to = "group", values_to = "fc")
df_long_k27 <- df_k27_up %>% pivot_longer(!name, names_to = "group", values_to = "fc")
df_long$group <- factor(df_long$group, levels = c("Primed vs Naive", "EZH2i vs Naive", "EZH2i vs Primed"))
df_long_k27$group <- factor(df_long_k27$group, levels = c("Primed vs Naive", "EZH2i vs Naive", "EZH2i vs Primed"))
levels(df_long$group) <- c("Naive vs Primed", "EZH2i vs Naive", "EZH2i vs Primed")
levels(df_long_k27$group) <- c("Naive vs Primed", "EZH2i vs Naive", "EZH2i vs Primed")
my_comparisons <- list(c("EZH2i vs Naive", "EZH2i vs Primed"),
c("Naive vs Primed", "EZH2i vs Naive"),
c("Naive vs Primed", "EZH2i vs Primed"))
# Stats subset vs global
ni_pr_test <- wilcox.test(df_k27_up[, "Primed vs Naive"],
df[!df$name %in% df_k27_up$name , "Primed vs Naive"], na.rm = T)
ni_pr_effect <- cohen.d(df_k27_up[, "Primed vs Naive"],
df[!df$name %in% df_k27_up$name , "Primed vs Naive"], na.rm = T)
col <- "EZH2i vs Naive"
ni_ezh2i_test <- wilcox.test(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
ni_ezh2i_effect <- cohen.d(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
col <- "EZH2i vs Primed"
pr_ezh2i_test <- wilcox.test(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
pr_ezh2i_effect <- cohen.d(df_k27_up[, col],
df[!df$name %in% df_k27_up$name , col], na.rm = T)
stats_caption <- paste("Wilcoxon global vs. selection",
paste("Ni_Pr:", format(ni_pr_test$p.value, digits=8),
"Cohen D:", round(ni_pr_effect$estimate, digits = 4),
"(", ni_pr_effect$magnitude, ")"),
paste("Ni_EZH2i:", format(ni_ezh2i_test$p.value, digits=8),
"Cohen D:", round(ni_ezh2i_effect$estimate, digits = 4),
"(", ni_ezh2i_effect$magnitude, ")"),
paste("Pr_EZH2i:", format(pr_ezh2i_test$p.value, digits=8),
"Cohen D:", round(pr_ezh2i_effect$estimate, digits = 4),
"(", pr_ezh2i_effect$magnitude, ")"),
sep = "\n")
k27_up_color <- "#ff4b40"
ggplot(data=df_long, aes(x = group, y = fc)) +
geom_violin(size = 0.8) +
stat_compare_means(
data=df_long_k27, comparisons = my_comparisons,
method = "wilcox.test", paired = TRUE) +
rasterize(
geom_jitter(data=df_long_k27, color = k27_up_color, alpha = 0.7, size = 0.1),
dpi = 300) +
geom_hline (yintercept = 0, linetype = "dashed") +
coord_cartesian(ylim = c(-13, 24)) +
theme_default(base_size = 14) +
labs(x = "Condition",
y = "Log2 FC",
title = "RNASeq expression changes",
subtitle = "Global vs H3K27m3 Primed >> Naïve TSS",
caption = stats_caption)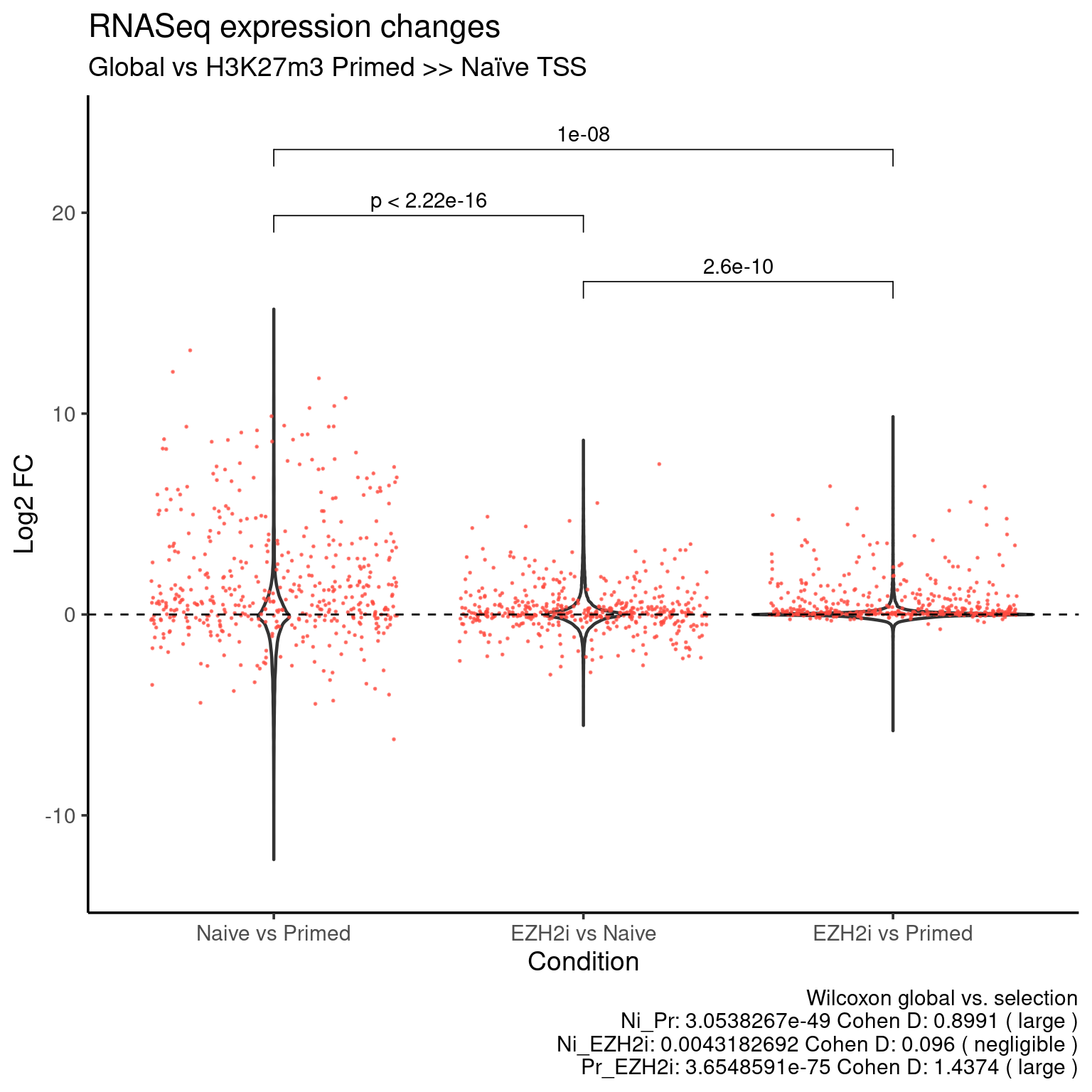
| Version | Author | Date |
|---|---|---|
| 67895c5 | C. Navarro | 2021-07-01 |
write.table(df_long[!is.na(df_long$fc), ],
file = "./figures_data/fig3_violin_k27_pr_higher_violin.tsv",
col.names = T, sep = "\t", quote = F, row.names = F)
write.table(df_long_k27[!is.na(df_long_k27$fc), ],
file = "./figures_data/fig3_violin_k27_pr_higher_jitter_k27_values.tsv",
col.names = T, sep = "\t", quote = F, row.names = F)Download values: K27 primed higher points.
Combined heatmaps
Naive markers
naive_markers <-
c("KLF17",
"DPPA5",
"DNMT3L",
"GATA6",
"TBX3",
"IL6ST",
"DPPA3",
"KLF5",
"KLF4",
"HORMAD1",
"KHDC3L",
"ALPP",
"ALPPL2",
"ZNF729",
"TRIM60"
)
combined_heatmap(genes, naive_markers, rnaseq_limits = c(0, 11.5), k4m3_limits = c(0, 90), k27m3_limits = c(0, 8), ub_limits = c(0, 11)) tpm_cols <- grep("RNASeq_TPM", colnames(genes), value = T)
cols_used <- c("name", "H3K27m3_Ni_mean_cov", "H3K27m3_Pr_mean_cov", "H3K27m3_Ni_EZH2i_mean_cov", "H3K27m3_Pr_EZH2i_mean_cov",
"H3K4m3_Ni_mean_cov", "H3K4m3_Pr_mean_cov", "H3K4m3_Ni_EZH2i_mean_cov", "H3K4m3_Pr_EZH2i_mean_cov",
"H2Aub_Ni_mean_cov", "H2Aub_Pr_mean_cov", "H2Aub_Ni_EZH2i_mean_cov", "H2Aub_Pr_EZH2i_mean_cov",
tpm_cols)
values <- genes[genes$name %in% naive_markers, cols_used]
values[, tpm_cols] <- log2(values[, tpm_cols] + 1)
write.table(values, "./figures_data/fig3_messmer_naive_heatmap_global_scale.tsv")Primed markers
primed_markers <-
c("CD24",
"ZIC2",
"SFRP2",
"OTX2",
"CYTL1",
"HMX2",
"THY1",
"DUSP6",
"PTPRZ1"
)
combined_heatmap(genes, primed_markers, rnaseq_limits = c(0, 11.5), k4m3_limits = c(0, 90), k27m3_limits = c(0, 8), ub_limits = c(0, 11))values <- genes[genes$name %in% primed_markers, cols_used]
values[, tpm_cols] <- log2(values[, tpm_cols] + 1)
write.table(values, "./figures_data/fig3_messmer_primed_heatmap_global_scale.tsv")
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=sv_SE.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=sv_SE.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=sv_SE.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=sv_SE.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] svglite_2.0.0 heatmaply_1.2.1 viridis_0.6.1
[4] viridisLite_0.4.0 plotly_4.9.4.1 wigglescout_0.13.1
[7] cowplot_1.1.1 ggrastr_0.2.3 ggpubr_0.4.0
[10] effsize_0.8.1 forcats_0.5.1 stringr_1.4.0
[13] dplyr_1.0.7 purrr_0.3.4 readr_1.4.0
[16] tidyr_1.1.3 tibble_3.1.2 ggplot2_3.3.5
[19] tidyverse_1.3.1 rtracklayer_1.52.0 GenomicRanges_1.44.0
[22] GenomeInfoDb_1.28.1 IRanges_2.26.0 S4Vectors_0.30.0
[25] BiocGenerics_0.38.0 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] readxl_1.3.1 backports_1.2.1
[3] systemfonts_1.0.2 plyr_1.8.6
[5] lazyeval_0.2.2 crosstalk_1.1.1
[7] BiocParallel_1.26.0 listenv_0.8.0
[9] digest_0.6.27 foreach_1.5.1
[11] htmltools_0.5.1.1 fansi_0.5.0
[13] magrittr_2.0.1 openxlsx_4.2.4
[15] globals_0.14.0 Biostrings_2.60.1
[17] modelr_0.1.8 matrixStats_0.59.0
[19] askpass_1.1 colorspace_2.0-2
[21] rvest_1.0.0 haven_2.4.1
[23] xfun_0.24 crayon_1.4.1
[25] RCurl_1.98-1.3 jsonlite_1.7.2
[27] iterators_1.0.13 glue_1.4.2
[29] registry_0.5-1 gtable_0.3.0
[31] zlibbioc_1.38.0 XVector_0.32.0
[33] webshot_0.5.2 DelayedArray_0.18.0
[35] car_3.0-11 abind_1.4-5
[37] scales_1.1.1 DBI_1.1.1
[39] rstatix_0.7.0 Rcpp_1.0.6
[41] foreign_0.8-81 htmlwidgets_1.5.3
[43] httr_1.4.2 RColorBrewer_1.1-2
[45] ellipsis_0.3.2 pkgconfig_2.0.3
[47] XML_3.99-0.6 farver_2.1.0
[49] sass_0.4.0 dbplyr_2.1.1
[51] utf8_1.2.1 tidyselect_1.1.1
[53] labeling_0.4.2 rlang_0.4.11
[55] reshape2_1.4.4 later_1.2.0
[57] munsell_0.5.0 cellranger_1.1.0
[59] tools_4.1.0 cli_3.0.0
[61] generics_0.1.0 broom_0.7.8
[63] evaluate_0.14 yaml_2.2.1
[65] knitr_1.33 fs_1.5.0
[67] zip_2.2.0 dendextend_1.15.1
[69] future_1.21.0 whisker_0.4
[71] xml2_1.3.2 compiler_4.1.0
[73] rstudioapi_0.13 beeswarm_0.4.0
[75] curl_4.3.2 ggsignif_0.6.2
[77] reprex_2.0.0 bslib_0.2.5.1
[79] stringi_1.6.2 highr_0.9
[81] lattice_0.20-44 Matrix_1.3-4
[83] vctrs_0.3.8 pillar_1.6.1
[85] lifecycle_1.0.0 furrr_0.2.3
[87] jquerylib_0.1.4 data.table_1.14.0
[89] bitops_1.0-7 seriation_1.3.0
[91] httpuv_1.6.1 R6_2.5.0
[93] BiocIO_1.2.0 promises_1.2.0.1
[95] TSP_1.1-10 gridExtra_2.3
[97] rio_0.5.27 vipor_0.4.5
[99] parallelly_1.26.1 codetools_0.2-18
[101] assertthat_0.2.1 SummarizedExperiment_1.22.0
[103] openssl_1.4.4 rprojroot_2.0.2
[105] rjson_0.2.20 withr_2.4.2
[107] GenomicAlignments_1.28.0 Rsamtools_2.8.0
[109] GenomeInfoDbData_1.2.6 hms_1.1.0
[111] grid_4.1.0 rmarkdown_2.9
[113] MatrixGenerics_1.4.0 carData_3.0-4
[115] Cairo_1.5-12.2 git2r_0.28.0
[117] Biobase_2.52.0 lubridate_1.7.10
[119] ggbeeswarm_0.6.0 restfulr_0.0.13