Misc
Haider Inam
4/3/2020
Last updated: 2020-04-20
Checks: 7 0
Knit directory: duplex_sequencing_screen/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200402) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: analysis/grant_fig.pdf
Untracked: analysis/grant_fig_v2.pdf
Untracked: data/Combined_data_frame_IC_Mutprob_abundance.csv
Untracked: data/IC50HeatMap.csv
Untracked: data/Twinstrand/
Untracked: data/gfpenrichmentdata.csv
Untracked: data/heatmap_concat_data.csv
Untracked: grant_fig.pdf
Untracked: grant_fig_v2.pdf
Untracked: output/archive/
Untracked: output/ic50data_all_conc.csv
Untracked: shinyapp/
Unstaged changes:
Deleted: data/README.md
Modified: output/twinstrand_maf_merge.csv
Modified: output/twinstrand_simple_melt_merge.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2bba93e | haiderinam | 2020-04-20 | wflow_publish(“analysis/*.Rmd“) |
| html | c3e9499 | haiderinam | 2020-04-10 | Build site. |
| Rmd | 7a5e2ff | haiderinam | 2020-04-10 | wflow_publish(“analysis/misc.Rmd”) |
| html | e477777 | haiderinam | 2020-04-06 | Build site. |
| Rmd | 0a6e9cb | haiderinam | 2020-04-06 | wflow_publish(files = “analysis/misc.Rmd”) |
Here I include some miscellaneous, pre-prod analyses
net_gr_wodrug=0.055
conc_for_predictions=0.8
########################Reading IC50 Data########################
ic50data_all=read.csv("data/IC50HeatMap.csv",header = T,stringsAsFactors = F)
# ic50data_all=read.csv("../data/IC50HeatMap.csv",header = T,stringsAsFactors = F)
twinstrand_maf_merge=read.csv("output/twinstrand_maf_merge.csv",header = T,stringsAsFactors = F)
# twinstrand_maf_merge=read.csv("../output/twinstrand_maf_merge.csv",header = T,stringsAsFactors = F)
# twinstrand_simple_melt_merge=read.csv("../output/twinstrand_simple_melt_merge.csv",header = T,stringsAsFactors = F)
twinstrand_simple_melt_merge=read.csv("output/twinstrand_simple_melt_merge.csv",header = T,stringsAsFactors = F)
# ic50data_long=read.csv("../output/ic50data_all_conc.csv",header = T,stringsAsFactors = F)
ic50data_long=read.csv("output/ic50data_all_conc.csv",header = T,stringsAsFactors = F)
ic50data_long$netgr_pred=net_gr_wodrug-ic50data_long$drug_effectModeling out confidence intervals on growth rate predictions from IC50s
Sources of error: 1. Sampling error (reads). Will be estimated by * Multinomial distributions based on observed coverages. * Sampling error in observed reads may be eliminated by removing mutants with <=1 depth of coverage
- Dose response variability
- Well known observation that IC50s can vary by as much as 50%. Will incorporate this into predictions. One direct, albeit simple, solution is to just take the 95% confidence intervals of the IC50s that fell in the dose response range.
- Dose variability. i.e. error between your expected dose and dose response.
Getting the dose responses for all mutants with the errors.
ic50data_all=ic50data_all%>%filter(species%in%c("Wt","V299L_H","E355A","D276G_maxi","H396R","F317L","F359I","E459K","G250E","F359C","F359V","M351T","L248V","E355G_maxi","Q252H_maxi","Y253F","F486S_maxi","H396P_maxi","E255K","Y253H","T315I","E255V"))
ic50data_all=ic50data_all%>%
mutate(species=case_when(species=="F486S_maxi"~"F486S",
species=="H396P_maxi"~"H396P",
species=="Q252H_maxi"~"Q252H",
species=="E355G_maxi"~"E355G",
species=="D276G_maxi"~"D276G",
species=="V299L_H" ~ "V299L",
TRUE ~as.character(species)))
########################Getting Confidence Intervals########################
#will just start of with one dose at 1.25uM
ic50data_all2=data.frame(cbind(ic50data_all$species,ic50data_all$X1.25))
# ic50data_all2=data.frame(cbind(ic50data_all$species,ic50data_all$X0.625))
colnames(ic50data_all2)=c("species","dose_response")
ic50data_all2$dose_response=as.numeric(as.character(ic50data_all2$dose_response))
#Calculating confidence limit and standard deviations
ic50data_all_sum=ic50data_all2%>%group_by(species)%>%summarise(dr_mean=mean(dose_response),dr_ci_ul=dr_mean+1.96*sd(dose_response)*sqrt(n()),dr_ci_ll=dr_mean-1.96*sd(dose_response)*sqrt(n()),dr_sd_ul=dr_mean+sd(dose_response),dr_sd_ll=dr_mean-sd(dose_response))
#Since some mutants have 0% alive at the lower limit of the confidence interval, I'm converting those to 0
ic50data_all_sum=ic50data_all_sum%>%mutate(dr_ci_ll=case_when(dr_ci_ll<=0~0,
TRUE~dr_ci_ll),
dr_sd_ll=case_when(dr_sd_ll<=0~0,
TRUE~dr_sd_ll))
#Dose response here is essentially y. aka %alive
#Converting y to drug effect on growth rate aka alpha value
ic50data_all_sum=ic50data_all_sum%>%mutate(netgr_pred_mean=net_gr_wodrug-(-log(dr_mean)/72),netgr_pred_ci_ul=net_gr_wodrug-(-log(dr_ci_ul)/72),netgr_pred_ci_ll=net_gr_wodrug-(-log(dr_ci_ll)/72),netgr_pred_sd_ul=net_gr_wodrug-(-log(dr_sd_ul)/72),netgr_pred_sd_ll=net_gr_wodrug-(-log(dr_sd_ll)/72))
########################Plotting Count with CIs########################
twinstrand_maf_merge=twinstrand_maf_merge%>%mutate(MAF=AltDepth/Depth)
twinstrand_maf_merge=merge(twinstrand_maf_merge%>%
filter(experiment=="M3",tki_resistant_mutation=="True",!mutant%in%c("D276G",NA)),ic50data_all_sum%>%
dplyr::select(species,netgr_pred_mean,netgr_pred_ci_ul,netgr_pred_ci_ll,netgr_pred_sd_ul,netgr_pred_sd_ll),by.x="mutant",by.y="species")
twinstrand_simple=twinstrand_maf_merge%>%dplyr::select(AltDepth,Depth,tki_resistant_mutation,mutant,experiment,Spike_in_freq,time_point,totalcells,totalmutant,MAF,netgr_pred_mean,netgr_pred_ci_ul,netgr_pred_ci_ll,netgr_pred_sd_ul,netgr_pred_sd_ll)
twinstrand_merge_forplot=melt(twinstrand_simple,id.vars = c("AltDepth","Depth","tki_resistant_mutation","mutant","experiment","Spike_in_freq","time_point","totalcells","netgr_pred_mean","netgr_pred_ci_ul","netgr_pred_ci_ll","netgr_pred_sd_ul","netgr_pred_sd_ll"),variable.name = "count_type",value.name = "count")
# twinstrand_merge_forplot=merge(twinstrand_maf_merge%>%filter(experiment=="M3",tki_resistant_mutation=="True",!mutant%in%c("D276G",NA)),ic50data_all_sum%>%dplyr::select(species,netgr_pred_mean,netgr_pred_ci_ul,netgr_pred_ci_ll,netgr_pred_sd_ul,netgr_pred_sd_ll),by.x="mutant",by.y="species")
#Basically making an extra column with the D0 total mutant counts for each mutant
twinstrand_merge_forplot=merge(twinstrand_merge_forplot,twinstrand_merge_forplot%>%filter(time_point=="D0")%>%dplyr::select(mutant,count_type,count_D0=count),by=c("mutant","count_type"))
#########Here, figure out why twinstrand_merge_forplot is having two rows for each mutant after being merged with a D0 version of itself. This is leading to weird plotting artifacts
# a=twinstrand_merge_forplot%>%filter(count_type=="totalmutant",mutant=="E255K",time_point=="D0")
############
twinstrand_merge_forplot=twinstrand_merge_forplot%>%mutate(time=case_when(time_point=="D0"~0,
time_point=="D3"~72,
time_point=="D6"~144),
ci_mean=count_D0*exp(netgr_pred_mean*time),
ci_ul=count_D0*exp(netgr_pred_ci_ul*time),
ci_ll=count_D0*exp(netgr_pred_ci_ll*time),
sd_ul=count_D0*exp(netgr_pred_sd_ul*time),
sd_ll=count_D0*exp(netgr_pred_sd_ll*time),)
twinstrand_merge_forplot=twinstrand_merge_forplot%>%mutate(ci_ll=case_when(ci_ll=="NaN"~0,
TRUE~ci_ll))
####Since the more sensitive mutants were appearing to grow fast if I take the raw IC50 predicted growth rates, I am going to instead take the predicted growth rates from the IC50s that were fit on a 4-parameter logistic. To get standard deviations, I will just add/subtract the standard deviations from the regular plots.
twinstrand_merge_forplot=merge(twinstrand_merge_forplot,ic50data_long%>%filter(conc==conc_for_predictions)%>%dplyr::select(mutant,netgr_pred_model=netgr_pred),by = "mutant")
twinstrand_merge_forplot=twinstrand_merge_forplot%>%mutate(netgr_pred_model_sd_ul=netgr_pred_model+(netgr_pred_mean-netgr_pred_sd_ll),netgr_pred_model_sd_ll=netgr_pred_model-(netgr_pred_mean-netgr_pred_sd_ll))
twinstrand_merge_forplot=twinstrand_merge_forplot%>%mutate(
sd_mean_model=count_D0*exp(netgr_pred_model*time),
sd_ul_model=count_D0*exp(netgr_pred_model_sd_ul*time),
sd_ll_model=count_D0*exp(netgr_pred_model_sd_ll*time))
twinstrand_merge_forplot=twinstrand_merge_forplot%>%mutate(ci_ll=case_when(ci_ll=="NaN"~0,
TRUE~ci_ll))
###########
#Factoring the mutants from more to less resistant
twinstrand_merge_forplot$mutant=factor(twinstrand_merge_forplot$mutant,levels = as.character(unique(twinstrand_merge_forplot$mutant[order((twinstrand_merge_forplot$netgr_pred_mean),decreasing = T)])))
getPalette = colorRampPalette(brewer.pal(9, "Spectral"))
####In the plots below, the dashed line is the mean prediction form the IC50s. Points are what we see in the spike-in experiment
#Plotting with 95% confidence intervals
# %>%filter(count_type=="totalmutant")
ggplot(twinstrand_merge_forplot,aes(x=time/24,y=count,fill=factor(mutant)))+geom_point()+
facet_grid(count_type~mutant)+
geom_line(aes(y=ci_mean),linetype="dashed")+
geom_ribbon(aes(ymin=ci_ll,ymax=ci_ul,alpha=.3))+
scale_y_continuous(trans="log2")+
cleanup+
scale_fill_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))Warning: Transformation introduced infinite values in continuous y-axis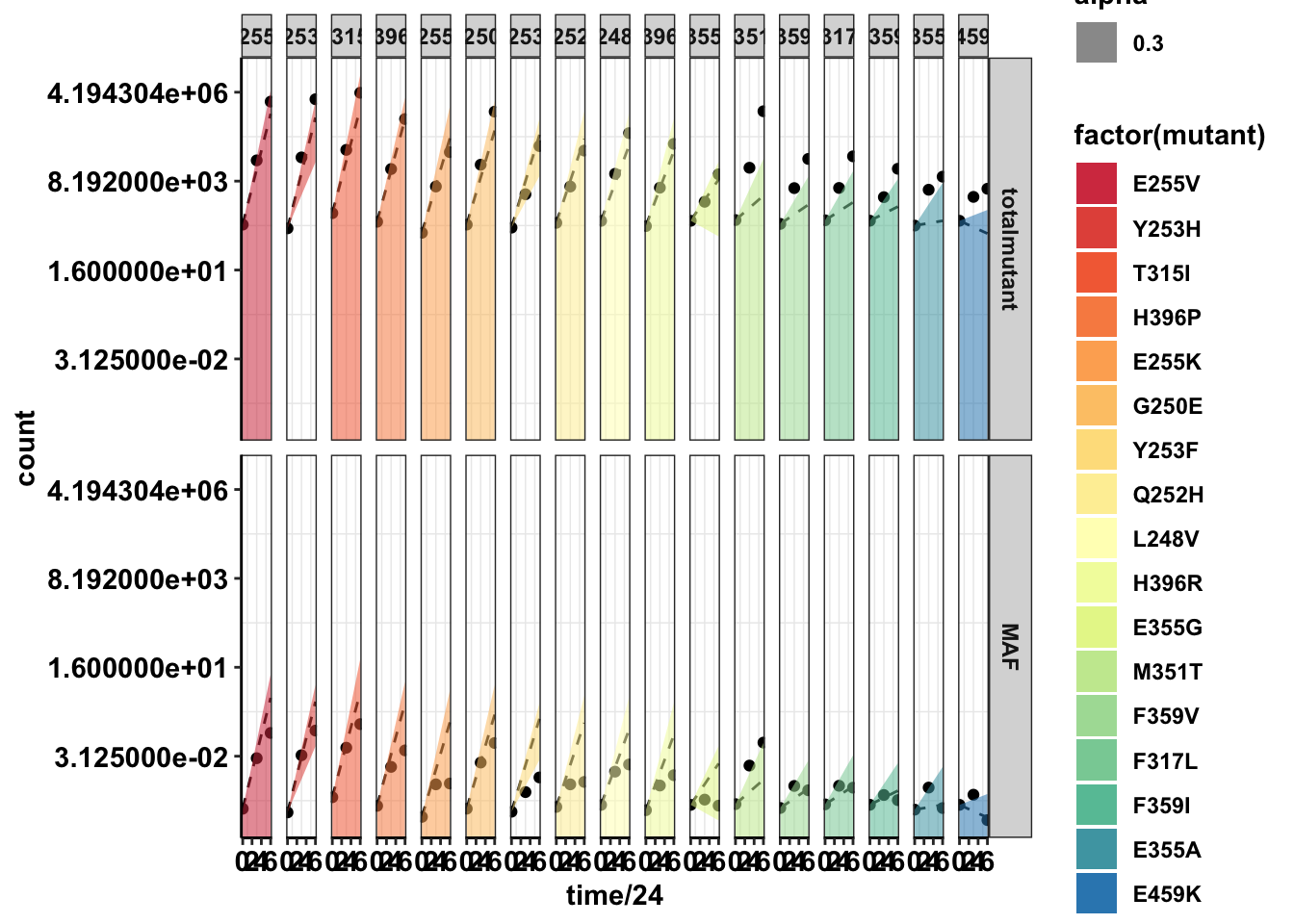
| Version | Author | Date |
|---|---|---|
| c3e9499 | haiderinam | 2020-04-10 |
#Plotting with standard deviations
ggplot(twinstrand_merge_forplot,aes(x=time/24,y=count,fill=factor(mutant),shape=factor(count_type)))+geom_point()+
geom_line(aes(y=ci_mean),linetype="dashed")+
geom_ribbon(aes(ymin=sd_ll,ymax=sd_ul,alpha=.3))+
facet_wrap(~mutant,ncol=4)+
scale_y_continuous(trans="log2")+
cleanup+
scale_fill_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))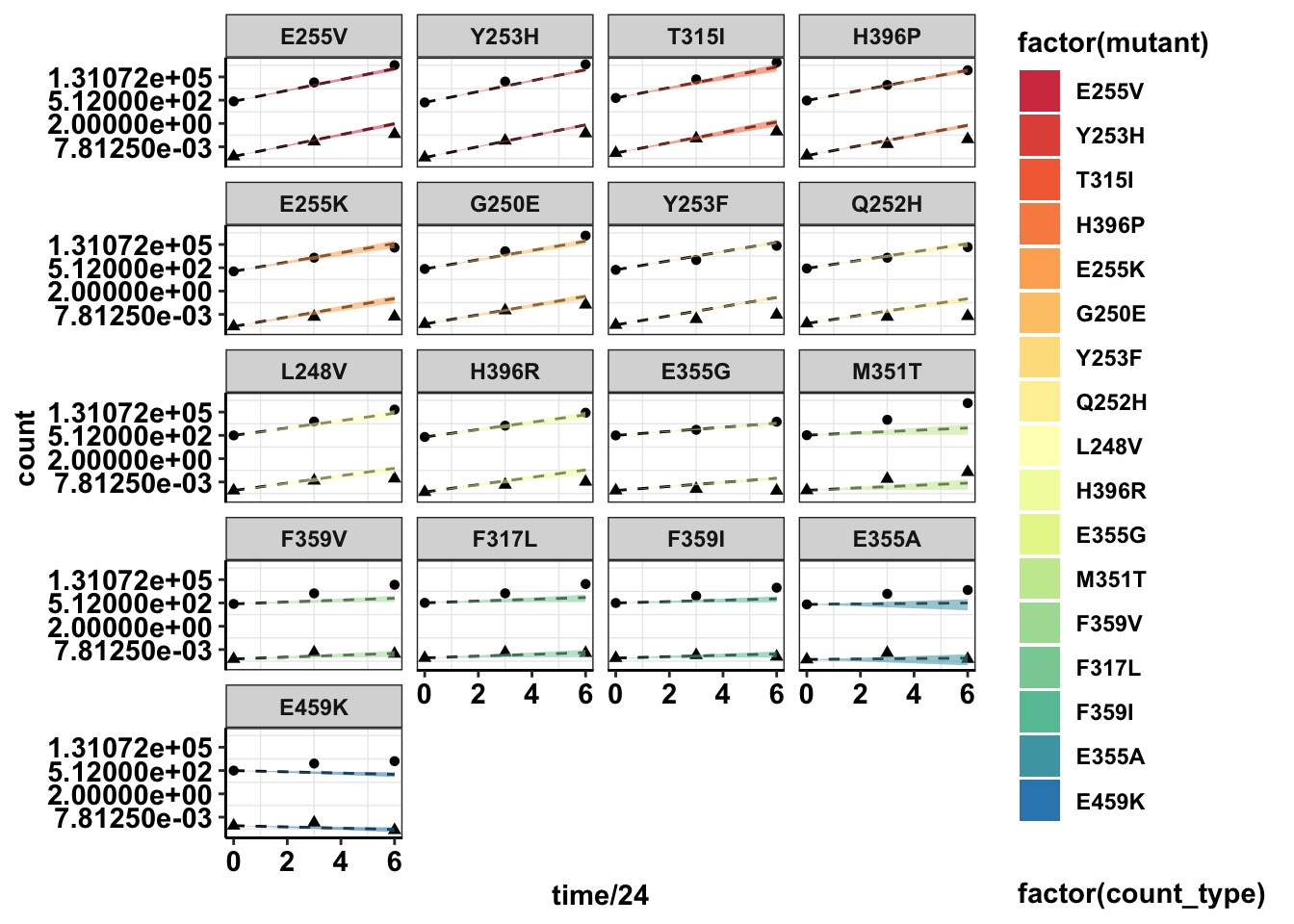
| Version | Author | Date |
|---|---|---|
| c3e9499 | haiderinam | 2020-04-10 |
#Next step: extend for experiments besides M3
plotly=ggplot(twinstrand_merge_forplot%>%filter(count_type=="totalmutant"),aes(x=time/24,y=count,fill=factor(mutant),shape=factor(count_type)))+geom_point()+
geom_line(aes(y=ci_mean),linetype="dashed")+
geom_ribbon(aes(ymin=sd_ll,ymax=sd_ul,alpha=.3))+
facet_wrap(~mutant,ncol=4)+
scale_y_continuous(trans="log2")+
cleanup+
scale_fill_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
ggplotly(plotly)plotly=ggplot(twinstrand_merge_forplot%>%filter(count_type=="MAF"),aes(x=time/24,y=count,fill=factor(mutant),shape=factor(count_type)))+geom_point()+
geom_line(aes(y=ci_mean),linetype="dashed")+
geom_ribbon(aes(ymin=sd_ll,ymax=sd_ul,alpha=.3))+
facet_wrap(~mutant,ncol=4)+
scale_y_continuous(trans="log2")+
cleanup+
scale_fill_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
ggplotly(plotly)#Plotting IC50s form 4 Parameter model
plotly=ggplot(twinstrand_merge_forplot%>%filter(count_type=="totalmutant"),aes(x=time/24,y=count,fill=factor(mutant),shape=factor(count_type)))+geom_point()+
geom_line(aes(y=sd_mean_model),linetype="dashed")+
geom_ribbon(aes(ymin=sd_ll_model,ymax=sd_ul_model,alpha=.3))+
facet_wrap(~mutant,ncol=4)+
scale_y_continuous(trans="log2")+
cleanup+
scale_fill_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
ggplotly(plotly)plotly=ggplot(twinstrand_merge_forplot%>%filter(count_type=="MAF"),aes(x=time/24,y=count,fill=factor(mutant),shape=factor(count_type)))+geom_point()+
geom_line(aes(y=sd_mean_model),linetype="dashed")+
geom_ribbon(aes(ymin=sd_ll_model,ymax=sd_ul_model,alpha=.3))+
facet_wrap(~mutant,ncol=4)+
scale_y_continuous(trans="log2")+
cleanup+
scale_fill_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
ggplotly(plotly)a_sum=twinstrand_merge_forplot%>%group_by(mutant,count_type)%>%summarize(netgr_obs=log(count[time_point=="D6"]/count[time_point=="D3"])/72,netgr_pred=mean(netgr_pred_mean),netgr_pred_ul=mean(netgr_pred_sd_ul),netgr_pred_ll=mean(netgr_pred_sd_ll))
a_sum=merge(a_sum,ic50data_long%>%filter(conc==.8)%>%dplyr::select(mutant,conc,netgr_pred_model=netgr_pred),by = "mutant")
getPalette2 = colorRampPalette(brewer.pal(9, "Spectral"))
###Troubleshooting plotting colors
plotly=ggplot(a_sum%>%filter(count_type=="MAF"),aes(x=netgr_pred,y=netgr_obs,label=mutant,color=factor(mutant)))+geom_text()+geom_abline()+cleanup+scale_x_continuous(limits=c(-.02,.06))+scale_y_continuous(limits=c(-.02,.06))+scale_color_manual(values = getPalette2(length(unique(a_sum$mutant))))
ggplotly(plotly)###
plotly=ggplot(a_sum%>%filter(count_type=="MAF"),aes(x=netgr_pred,y=netgr_obs,label=mutant,color=mutant))+geom_text()+geom_abline()+cleanup+scale_x_continuous(limits=c(-.02,.06))+scale_y_continuous(limits=c(-.02,.06))+scale_color_manual(values = getPalette(length(unique(a_sum$mutant))))
ggplotly(plotly)plotly=ggplot(a_sum%>%filter(count_type=="totalmutant"),aes(x=netgr_pred,y=netgr_obs,label=mutant,color=mutant))+geom_text()+geom_abline()+cleanup+scale_x_continuous(limits=c(-.02,.06))+scale_y_continuous(limits=c(-.02,.06))+scale_color_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
ggplotly(plotly)plotly=ggplot(a_sum%>%filter(count_type=="totalmutant"),aes(x=netgr_pred_model,y=netgr_obs,label=mutant,color=mutant))+geom_text()+geom_abline()+cleanup+scale_color_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
# scale_x_continuous(limits=c(-.02,.06))+
# scale_y_continuous(limits=c(-.02,.06))
ggplotly(plotly)plotly=ggplot(a_sum%>%filter(count_type=="totalmutant"),aes(x=netgr_pred_model,y=netgr_pred,color=mutant))+geom_errorbar(aes(ymin=netgr_pred_ul,ymax=netgr_pred_ll))+geom_point()+geom_abline()+cleanup+
scale_x_continuous(limits=c(0,.06))+
scale_y_continuous(limits=c(0,.06))+scale_color_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
ggplotly(plotly)plotly=ggplot(a_sum%>%filter(count_type=="totalmutant"),aes(x=netgr_pred,y=netgr_pred,color=mutant))+geom_errorbar(aes(ymin=netgr_pred_ul,ymax=netgr_pred_ll))+geom_point()+geom_abline()+cleanup+
scale_x_continuous(limits=c(0,.06))+
scale_y_continuous(limits=c(0,.06))+scale_color_manual(values = getPalette(length(unique(twinstrand_merge_forplot$mutant))))
ggplotly(plotly)# a_sum=twinstrand_merge_forplot%>%group_by(mutant,count_type)%>%summarize(netgr_obs=log(count[time_point=="D3"]/count[time_point=="D0"])/72,netgr_pred=mean(netgr_pred_mean))
# ggplot(a_sum%>%filter(count_type=="MAF"),aes(x=netgr_pred,y=netgr_obs,label=mutant))+geom_text()+geom_abline()+cleanup+scale_x_continuous(limits=c(-.02,.06))+scale_y_continuous(limits=c(-.02,.06))
# ggplot(a_sum%>%filter(count_type=="totalmutant"),aes(x=netgr_pred,y=netgr_obs,label=mutant))+geom_text()+geom_abline()+cleanup+scale_x_continuous(limits=c(-.02,.06))+scale_y_continuous(limits=c(-.02,.06))
a=twinstrand_simple_melt_merge%>%
mutate(netgr_obs=case_when(experiment=="M5"~netgr_obs+.015,
experiment%in%c("M6","M3","M5","M4","M7")~netgr_obs))
plotly=ggplot(a%>%filter(experiment%in%c("M3","M4","M5","M6","M7"),duration=="d3d6"),aes(x=netgr_pred,y=netgr_obs,color=factor(experiment),label=factor(mutant)))+geom_text()+geom_abline()+cleanup+scale_x_continuous(trans="log10")+scale_y_continuous(trans="log10")
ggplotly(plotly)Warning in self$trans$transform(x): NaNs producedWarning: Transformation introduced infinite values in continuous y-axisFigure out why your less resistant mutants appear to be more resistant than the IC50 predictions in these plots. Especially because I don’t see that in the observed vs expected plots. Answer: IC50 values tend to underestimate the growth rate of the more sensitive variants. When I fix this with a 4-parameter logistic fit, it looks much better.
For Shendure, instead of plotting on the same plot, plot the fold change values over time. Where are you getting the predictions from? Would it be possible to make the predictions based off of the MAFs?
Next step: Is our method better than the Shendure method? 1. Can they make accurate predicitons? Can you use just mutant allele frequency, without count data? I.e. does that data match expected growth? 2. Does their method still work for measuring gain of funciton phenotypes? 3. Do they have enough coverage given a detection efficiency of 10,000? Assume you make 1,000 mutants Given a detection efficiency of 1 in 10,000, for mutants treated with drug for a specified amount of time, how many mutants will you get enough coverage for?
sessionInfo()R version 3.5.2 (2018-12-20)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.15.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel grid stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] drc_3.0-1 MASS_7.3-51.5 BiocManager_1.30.10
[4] plotly_4.9.1 ggsignif_0.6.0 devtools_2.2.1
[7] usethis_1.5.1 RColorBrewer_1.1-2 reshape2_1.4.3
[10] ggplot2_3.2.1 doParallel_1.0.15 iterators_1.0.12
[13] foreach_1.4.7 dplyr_0.8.4 VennDiagram_1.6.20
[16] futile.logger_1.4.3 tictoc_1.0 knitr_1.27
[19] workflowr_1.6.0
loaded via a namespace (and not attached):
[1] fs_1.3.1 httr_1.4.1 rprojroot_1.3-2
[4] tools_3.5.2 backports_1.1.5 R6_2.4.1
[7] lazyeval_0.2.2 colorspace_1.4-1 withr_2.1.2
[10] tidyselect_1.0.0 prettyunits_1.1.1 processx_3.4.1
[13] curl_4.3 compiler_3.5.2 git2r_0.26.1
[16] cli_2.0.1 formatR_1.7 sandwich_2.5-1
[19] desc_1.2.0 labeling_0.3 scales_1.1.0
[22] mvtnorm_1.0-12 callr_3.4.1 stringr_1.4.0
[25] digest_0.6.23 foreign_0.8-75 rmarkdown_2.1
[28] rio_0.5.16 pkgconfig_2.0.3 htmltools_0.4.0
[31] sessioninfo_1.1.1 plotrix_3.7-7 fastmap_1.0.1
[34] htmlwidgets_1.5.1 rlang_0.4.4 readxl_1.3.1
[37] shiny_1.4.0 farver_2.0.3 zoo_1.8-7
[40] jsonlite_1.6 crosstalk_1.0.0 gtools_3.8.1
[43] zip_2.0.4 car_3.0-6 magrittr_1.5
[46] Matrix_1.2-18 Rcpp_1.0.3 munsell_0.5.0
[49] fansi_0.4.1 abind_1.4-5 lifecycle_0.1.0
[52] multcomp_1.4-12 stringi_1.4.5 whisker_0.4
[55] yaml_2.2.1 carData_3.0-3 pkgbuild_1.0.6
[58] plyr_1.8.5 promises_1.1.0 forcats_0.4.0
[61] crayon_1.3.4 lattice_0.20-38 splines_3.5.2
[64] haven_2.2.0 hms_0.5.3 ps_1.3.0
[67] pillar_1.4.3 codetools_0.2-16 pkgload_1.0.2
[70] futile.options_1.0.1 glue_1.3.1 evaluate_0.14
[73] lambda.r_1.2.4 data.table_1.12.8 remotes_2.1.0
[76] vctrs_0.2.2 httpuv_1.5.2 testthat_2.3.1
[79] cellranger_1.1.0 gtable_0.3.0 purrr_0.3.3
[82] tidyr_1.0.2 assertthat_0.2.1 xfun_0.12
[85] openxlsx_4.1.4 mime_0.8 xtable_1.8-4
[88] later_1.0.0 survival_3.1-8 viridisLite_0.3.0
[91] tibble_2.1.3 memoise_1.1.0 TH.data_1.0-10
[94] ellipsis_0.3.0