BCRABL_FunctionalKinaseAnalysis
Haider Inam
2022-11-02
Last updated: 2022-11-12
Checks: 7 0
Knit directory: duplex_sequencing_screen/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200402) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5aea8c2. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/Consensus_Data/.Rhistory
Ignored: data/Consensus_Data/Novogene_lane13/sample8/variant_caller_outputs/
Ignored: data/Consensus_Data/Novogene_lane14/sample13/
Ignored: data/Consensus_Data/Novogene_lane14/sample14b/
Ignored: data/Consensus_Data/Novogene_lane14/sample1_combined/
Ignored: data/Consensus_Data/Novogene_lane14/sample7/variant_caller_outputs/duplex/
Ignored: data/Consensus_Data/Novogene_lane14/sample8/variant_caller_outputs/
Ignored: data/Consensus_Data/Novogene_lane2/abl_ref/kraken2-master/newdir/
Ignored: data/Consensus_Data/Novogene_lane4/.ipynb_checkpoints/
Ignored: data/Consensus_Data/Novogene_lane4/GalaxyData/initialanalysis/
Ignored: data/Consensus_Data/Novogene_lane4/output/
Ignored: data/Consensus_Data/Novogene_lane6/
Ignored: data/Consensus_Data/Novogene_lane7/
Ignored: data/Consensus_Data/Ranomics_Pooled/RP4/
Ignored: data/Consensus_Data/Ranomics_Pooled/RP5/
Ignored: data/Consensus_Data/novogene_lane15/sample_5/duplex/variant_caller_outputs/
Ignored: data/Consensus_Data/novogene_lane15/sample_6/duplex/variant_caller_outputs/
Ignored: data/Consensus_Data/novogene_lane15/sample_7/duplex/variant_caller_outputs/
Ignored: data/Consensus_Data/novogene_lane15/sample_8/duplex/
Ignored: data/Consensus_Data/novogene_lane15/sample_8/sscs/
Untracked files:
Untracked: code/variants_distribution_plotter.R
Untracked: data/Consensus_Data/novogene_lane15/sample_3/duplex/
Untracked: data/Consensus_Data/novogene_lane15/sample_3/sscs/
Untracked: data/Consensus_Data/novogene_lane15/sample_4/duplex/.DS_Store
Untracked: data/Consensus_Data/novogene_lane15/sample_4/duplex/duplex_sorted_filtered.tsv
Untracked: data/Consensus_Data/novogene_lane15/sample_4/duplex/variant_caller_outputs/
Untracked: data/Consensus_Data/novogene_lane15/sample_4/sscs/.DS_Store
Untracked: data/Consensus_Data/novogene_lane15/sample_4/sscs/sscs_sorted_filtered.tsv
Untracked: data/Consensus_Data/novogene_lane15/sample_4/sscs/variant_caller_outputs/
Untracked: data/Consensus_Data/novogene_lane15/sample_5/duplex/.DS_Store
Untracked: data/Consensus_Data/novogene_lane15/sample_5/duplex/duplex_sorted_filtered.tsv
Untracked: data/Consensus_Data/novogene_lane15/sample_5/sscs/.DS_Store
Untracked: data/Consensus_Data/novogene_lane15/sample_5/sscs/sscs_sorted_filtered.tsv
Untracked: data/Consensus_Data/novogene_lane15/sample_5/sscs/variant_caller_outputs/
Untracked: data/Consensus_Data/novogene_lane15/sample_6/duplex/.DS_Store
Untracked: data/Consensus_Data/novogene_lane15/sample_6/sscs/.DS_Store
Untracked: data/Consensus_Data/novogene_lane15/sample_6/sscs/sscs_sorted_filtered.tsv
Untracked: data/Consensus_Data/novogene_lane15/sample_6/sscs/variant_caller_outputs/
Untracked: data/Consensus_Data/novogene_lane15/sample_7/sscs/.DS_Store
Untracked: data/Consensus_Data/novogene_lane15/sample_7/sscs/sscs_sorted_filtered.tsv
Untracked: data/Consensus_Data/novogene_lane15/sample_7/sscs/variant_caller_outputs/
Unstaged changes:
Modified: .DS_Store
Modified: BCRABL_Imatinib_Scores_Resmuts.pdf
Modified: BCRABL_Imatinib_Scores_Resresids.pdf
Modified: BCRABL_imatinib_D2.pdf
Modified: BCRABL_imatinib_D2_resistantresidues.pdf
Modified: analysis/lane14_comparisons.Rmd
Modified: analysis/variant_caller_2022.Rmd
Modified: data/.DS_Store
Modified: data/Consensus_Data/.DS_Store
Modified: data/Consensus_Data/Novogene_lane11/.DS_Store
Modified: data/Consensus_Data/Novogene_lane11/sample6/.DS_Store
Modified: data/Consensus_Data/Novogene_lane11/sample7/.DS_Store
Modified: data/Consensus_Data/Novogene_lane12/.DS_Store
Modified: data/Consensus_Data/Novogene_lane12/sample1/.DS_Store
Modified: data/Consensus_Data/Novogene_lane12/sample3/.DS_Store
Modified: data/Consensus_Data/Novogene_lane12/sample9/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/sample1/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/sample10/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/sample3/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/sample4/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/sample5/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/sample7/.DS_Store
Modified: data/Consensus_Data/Novogene_lane13/sample9/.DS_Store
Modified: data/Consensus_Data/Novogene_lane14/.DS_Store
Modified: data/Consensus_Data/Novogene_lane14/sample12/.DS_Store
Modified: data/Consensus_Data/Novogene_lane14/sample2_combined/.DS_Store
Modified: data/Consensus_Data/Novogene_lane14/sample7/.DS_Store
Modified: data/Consensus_Data/Novogene_lane2/.DS_Store
Modified: data/Consensus_Data/Novogene_lane2/abl_ref/.DS_Store
Modified: data/Consensus_Data/R01Figure/.DS_Store
Modified: data/Consensus_Data/archive/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_1/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_2/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_3/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_4/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_5/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_6/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_7/.DS_Store
Modified: data/Consensus_Data/novogene_lane15/sample_8/.DS_Store
Modified: data/Consensus_Data/sscs_dcs_comparisons/.DS_Store
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/BCRABL_FunctionalKinaseAnalysis.Rmd) and HTML
(docs/BCRABL_FunctionalKinaseAnalysis.html) files. If
you’ve configured a remote Git repository (see
?wflow_git_remote), click on the hyperlinks in the table
below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 5aea8c2 | haiderinam | 2022-11-12 | wflow_publish("analysis/BCRABL_FunctionalKinaseAnalysis.Rmd") |
| Rmd | b794806 | haiderinam | 2022-11-11 | Plotting imatinib enrichment scores distributions etc |
| html | efcc61d | haiderinam | 2022-11-04 | Build site. |
| Rmd | 261bf5f | haiderinam | 2022-11-04 | wflow_publish("analysis/BCRABL_FunctionalKinaseAnalysis.Rmd") |
| Rmd | 3a2f887 | haiderinam | 2022-11-04 | Added analyses of IL3 independence |
| Rmd | 6f54acf | haiderinam | 2022-11-04 | Added analyses of SSCS vs DCS error rates |
Plotting the correlation of allele frequencies of stuff that I see in sample 2 vs in sample 7
samplex=read.csv("data/Consensus_Data/Novogene_lane14/sample15/sscs/variant_caller_outputs/variants_unique_ann.csv",header=T,stringsAsFactors = F)
samplex=samplex%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
samplex=samplex%>%mutate(maf=ct/depth)
samplex_simple=samplex%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
sampley=read.csv("data/Consensus_Data/Novogene_lane14/sample17/sscs/variant_caller_outputs/variants_unique_ann.csv",header=T,stringsAsFactors = F)
sampley=sampley%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
sampley=sampley%>%mutate(maf=ct/depth)
sampley_simple=sampley%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
samples_xy=merge(samplex_simple%>%filter(consequence_terms%in%"missense_variant"),sampley_simple%>%filter(consequence_terms%in%"missense_variant"),by=c("ref_aa","protein_start","alt_aa","ref","alt","alt_start_pos","consequence_terms"),all = T)
plotly=ggplot(samples_xy%>%filter(protein_start>=242,protein_start<=492),aes(x=maf.x,y=maf.y))+
geom_point(color="black",shape=21,aes(fill=ct.x))+
scale_x_continuous(trans="log")+
scale_y_continuous(trans="log")+
geom_abline()+
theme_bw()
ggplotly(plotly)Warning in L$marker$color[idx] <- aes2plotly(data, params, "fill")[idx]: number
of items to replace is not a multiple of replacement length# cor(samples_xy$maf.x,samples_xy$maf.y)Background samples: Lane13 Sample 7, Lane 14 Sample 10, 11? D2 Post iL3 withdrawal: Lane 13 Sample 9,10 D4 Post iL3 withdrawal: Lane 13 Sample 11,12 D2 Post Imatinib treatment: Lane 14 Sample 12
source("code/merge_samples.R")
# il3all=merge_samples("Novogene_lane14/Sample10_combined","Novogene_lane13/sample7")
# il3all=merge_samples(il3all,"Novogene_lane14/sample11")
# il3all=merge_samples(il3all,"Novogene_lane15/sample_3/sscs")
# il3all=merge_samples(il3all,"Novogene_lane13/Sample10")
# # il3all=merge_samples(il3all,"Novogene_lane13/Sample9")
# # a=il3all%>%filter(protein_start%in%c(242:494),consequence_terms%in%"missense_variant")
il3D0=merge_samples("Novogene_lane14/Sample10_combined","Novogene_lane13/sample7")
il3D0=merge_samples(il3D0,"Novogene_lane14/sample11")
il3D0=merge_samples(il3D0,"Novogene_lane15/sample_3/sscs")
il3D0=il3D0%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
il3D0=il3D0%>%mutate(maf=ct/depth)
il3D0_simple=il3D0%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
# a=il3D0%>%filter(protein_start%in%c(242:494),consequence_terms%in%"missense_variant")
il3D2=merge_samples("Novogene_lane13/Sample9","Novogene_lane13/Sample10")
il3D2=merge_samples(il3D2,"Novogene_lane15/Sample_4/sscs")
il3D2=il3D2%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
il3D2=il3D2%>%mutate(maf=ct/depth)
il3D2_simple=il3D2%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
# imatD2=read.csv("data/Consensus_Data/Novogene_lane14/sample12/variant_caller_outputs/variants_unique_ann.csv",stringsAsFactors = F)
imatD2=merge_samples("Novogene_lane14/sample12","Novogene_lane15/sample_6/sscs")
# imatD2=merge_samples(imatD2,"Novogene_lane15/Sample4")
imatD2=imatD2%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
imatD2=imatD2%>%mutate(maf=ct/depth)
imatD2_simple=imatD2%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
il3D0.D2=merge(il3D0_simple%>%filter(consequence_terms%in%"missense_variant"),il3D2_simple%>%filter(consequence_terms%in%"missense_variant"),by=c("ref_aa","protein_start","alt_aa","ref","alt","alt_start_pos","consequence_terms"),all.x = T)
il3D0.D2[il3D0.D2$ct.y%in%NA,"ct.y"]=0
il3D0.D2$alt_aa=factor(il3D0.D2$alt_aa,levels=c("P","G","Y","W","F","V","L","I","A","T","S","Q","N","M","C","E","D","R","K","H"))
il3D0.D2[il3D0.D2$ct.y%in%NA,"ct.y"]=0
il3D0.D2=il3D0.D2%>%mutate(score=log2(maf.y/maf.x))
il3D0.D2[il3D0.D2$score%in%NA,"score"]=-6
imatD0.D2=merge(il3D0_simple%>%filter(consequence_terms%in%"missense_variant"),imatD2_simple%>%filter(consequence_terms%in%"missense_variant"),by=c("ref_aa","protein_start","alt_aa","ref","alt","alt_start_pos","consequence_terms"),all.x = T)
imatD0.D2[imatD0.D2$ct.y%in%NA,"ct.y"]=0
imatD0.D2=imatD0.D2%>%mutate(score=log2(maf.y/maf.x))
imatD0.D2[imatD0.D2$score%in%NA,"score"]=-6
# imatD0.D2$conserved=F
# imatD0.D2[imatD0.D2$protein_start%in%c(271,381,382,383),"conserved"]=T
imatD0.D2$alt_aa=factor(il3D0.D2$alt_aa,levels=c("P","G","Y","W","F","V","L","I","A","T","S","Q","N","M","C","E","D","R","K","H"))
######Plotting imat D0 D2 Heatmap####
ggplot(imatD0.D2%>%filter(nchar(as.character(alt_aa))%in%1,protein_start>=242,protein_start<=494),aes(x=protein_start,y=alt_aa,fill=score))+
geom_tile()+
theme(panel.background=element_rect(fill="white", colour="black"))+
scale_fill_gradient2(low ="blue",midpoint=-1,mid="white", high ="red",name="Enrichment Score")+
scale_color_manual(values=c("black"))+scale_x_continuous(name="Residue on the ABL Kinase",limits=c(242,493),expand=c(0,0),breaks = c(250,253,255,276,299,315,317,351,355,359,396,459,486))+
ylab("Mutant Amino Acid")Warning: Removed 4 rows containing missing values (geom_tile).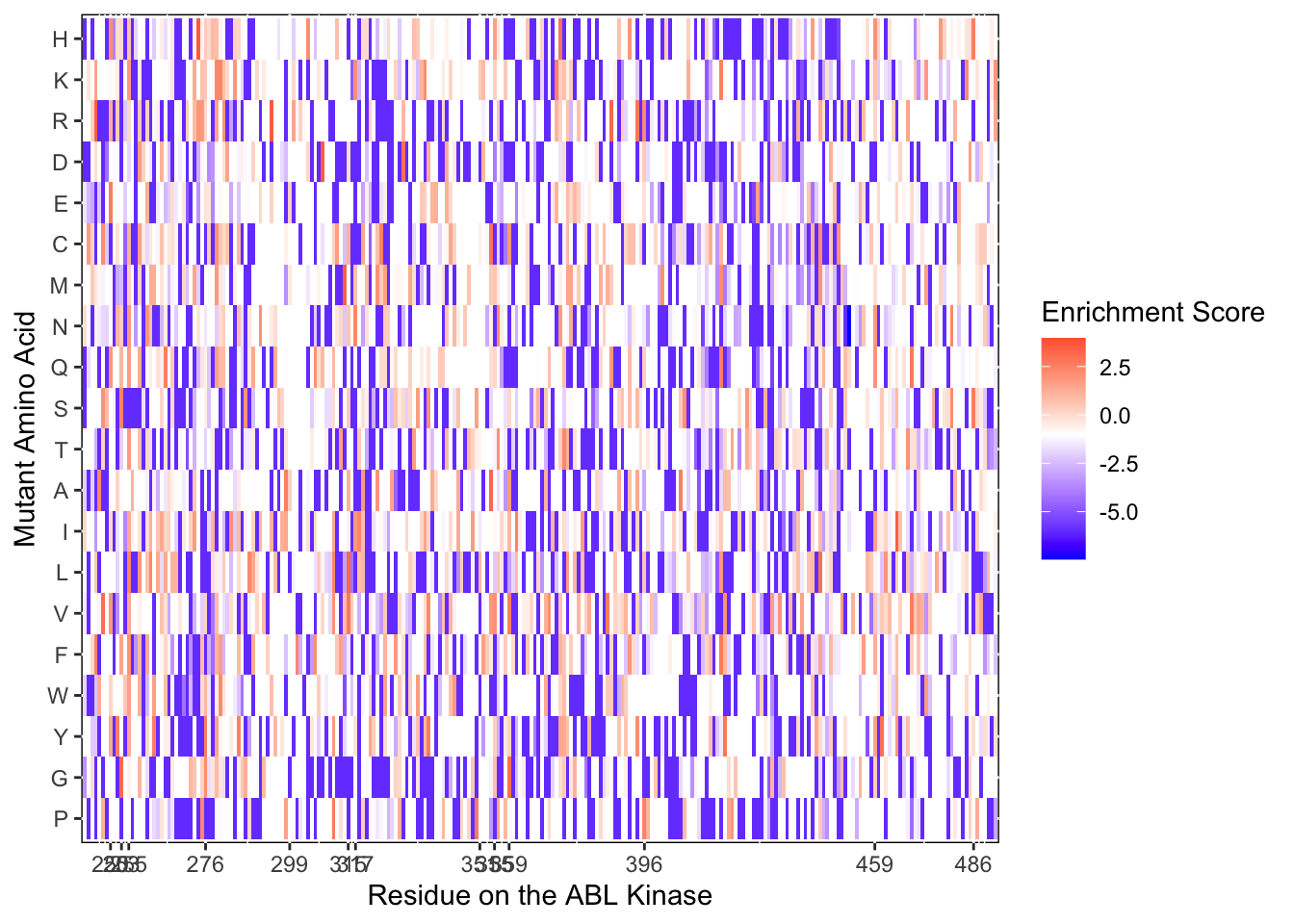
| Version | Author | Date |
|---|---|---|
| efcc61d | haiderinam | 2022-11-04 |
ggsave("BCRABL_imatinib_D2.pdf",height=6,width=24,units="in",useDingbats=F)Warning: Removed 4 rows containing missing values (geom_tile).######Plotting resistant imat D0 D2 Heatmap####
ggplot(imatD0.D2%>%filter(nchar(as.character(alt_aa))%in%1,protein_start%in%c(250,253,255,276,299,315,317,351,355,359,396,459,486)),aes(x=protein_start,y=alt_aa,fill=score))+
geom_tile()+
theme(panel.background=element_rect(fill="white", colour="black"))+
scale_fill_gradient2(low ="blue",midpoint=-1,mid="white", high ="red",name="Enrichment Score")+
scale_color_manual(values=c("black"))+scale_x_continuous(name="Residue on the ABL Kinase",limits=c(242,493),expand=c(0,0),breaks =c(250,253,255,276,299,315,317,351,355,359,396,459,486))+
ylab("Mutant Amino Acid")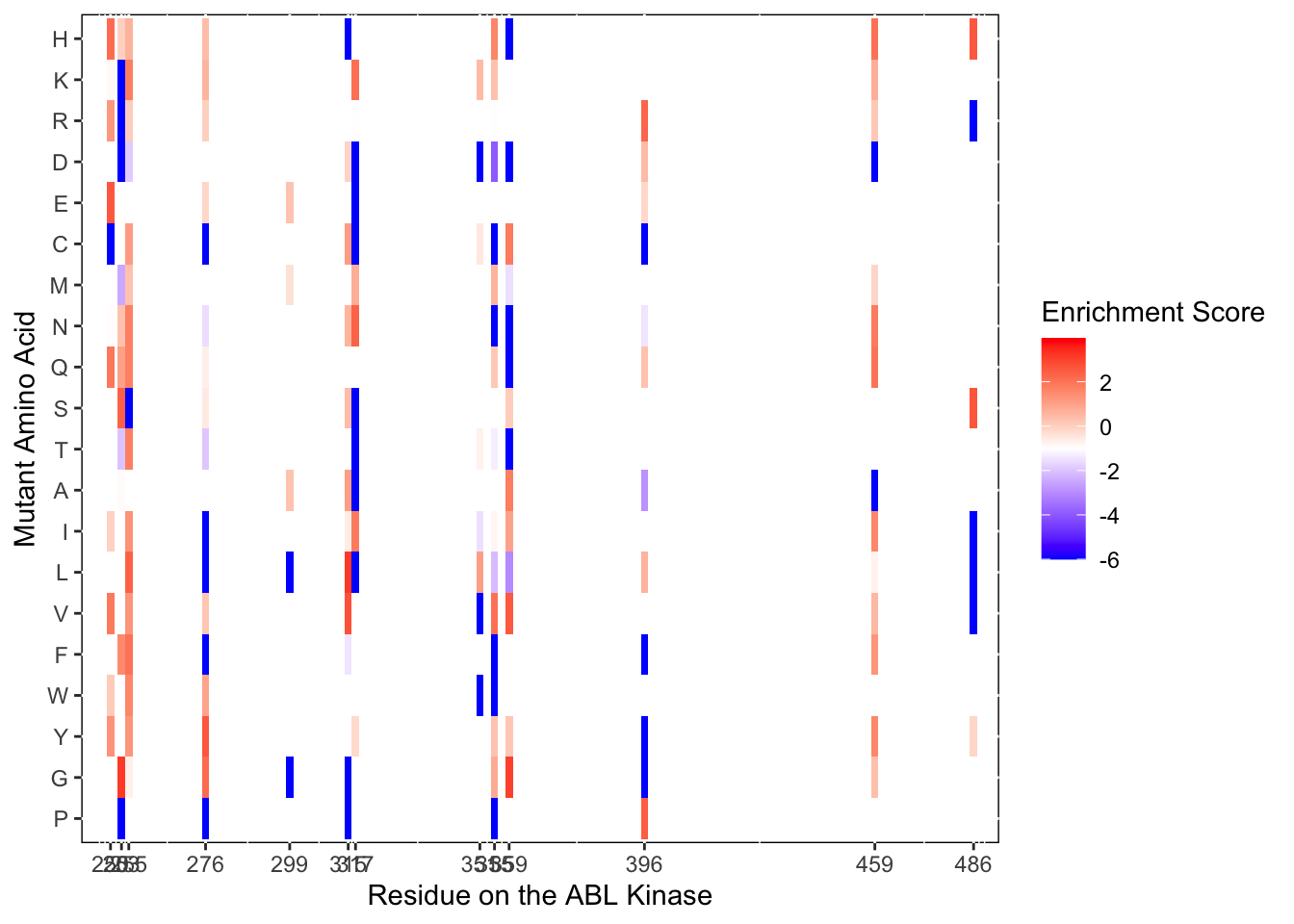
ggsave("BCRABL_imatinib_D2_resistantresidues.pdf",height=6,width=24,units="in",useDingbats=F)
######Plotting iL3 D0 D2 Heatmap####
ggplot(il3D0.D2%>%filter(nchar(as.character(alt_aa))%in%1,protein_start>=242,protein_start<=494),aes(x=protein_start,y=alt_aa,fill=score))+
geom_tile()+
theme(panel.background=element_rect(fill="white", colour="black"))+
scale_fill_gradient2(low ="blue",midpoint=-1,mid="white", high ="red",name="Enrichment Score")+
scale_color_manual(values=c("black"))+scale_x_continuous(name="Residue on the ABL Kinase",limits=c(242,493),expand=c(0,0),breaks = c(248,250,256, 271,275,300,325,350,363,375,381,400,405,425,450,475))+
ylab("Mutant Amino Acid")Warning: Removed 4 rows containing missing values (geom_tile).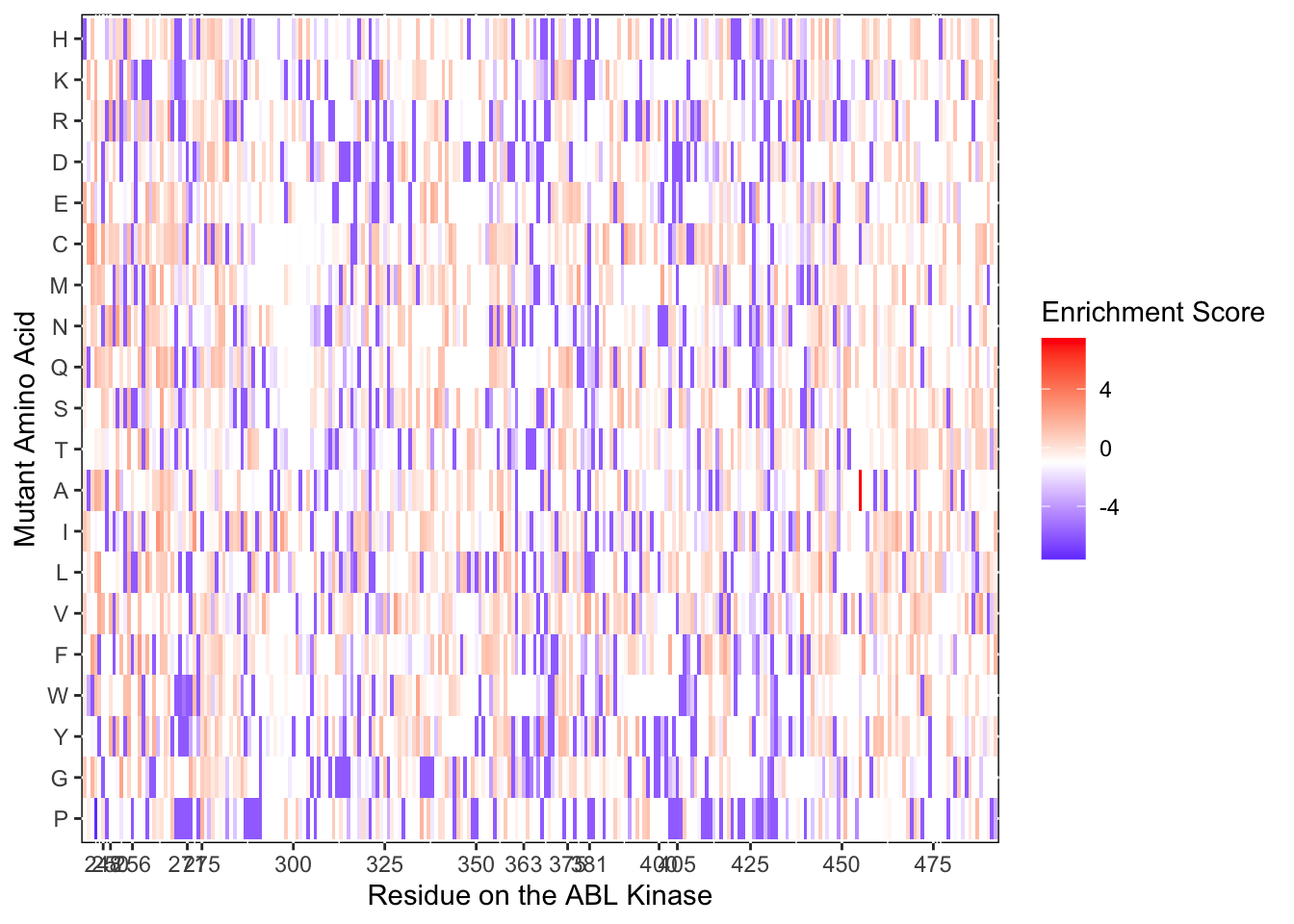
# ggsave("BCRABL_iL3Independence_D2.pdf",height=6,width=24,units="in",useDingbats=F)
# write.csv(il3D0.D2,"BCRABL_Il3Independence_D2.csv")
#####Focusing on conserved residues#####
ggplot(il3D0.D2%>%filter(nchar(as.character(alt_aa))%in%1,protein_start%in%c(271,363,381:383)),aes(x=protein_start,y=alt_aa,fill=score))+
geom_tile()+
theme(panel.background=element_rect(fill="white", colour="black"))+
scale_fill_gradient2(low ="blue",midpoint=-1,mid="white", high ="red",name="Enrichment Score")+
scale_color_manual(values=c("black"))+scale_x_continuous(name="Residue on the ABL Kinase",limits=c(242,493),expand=c(0,0),breaks = c(248,250,256, 271,275,300,325,350,363,375,381,400,405,425,450,475))+
ylab("Mutant Amino Acid")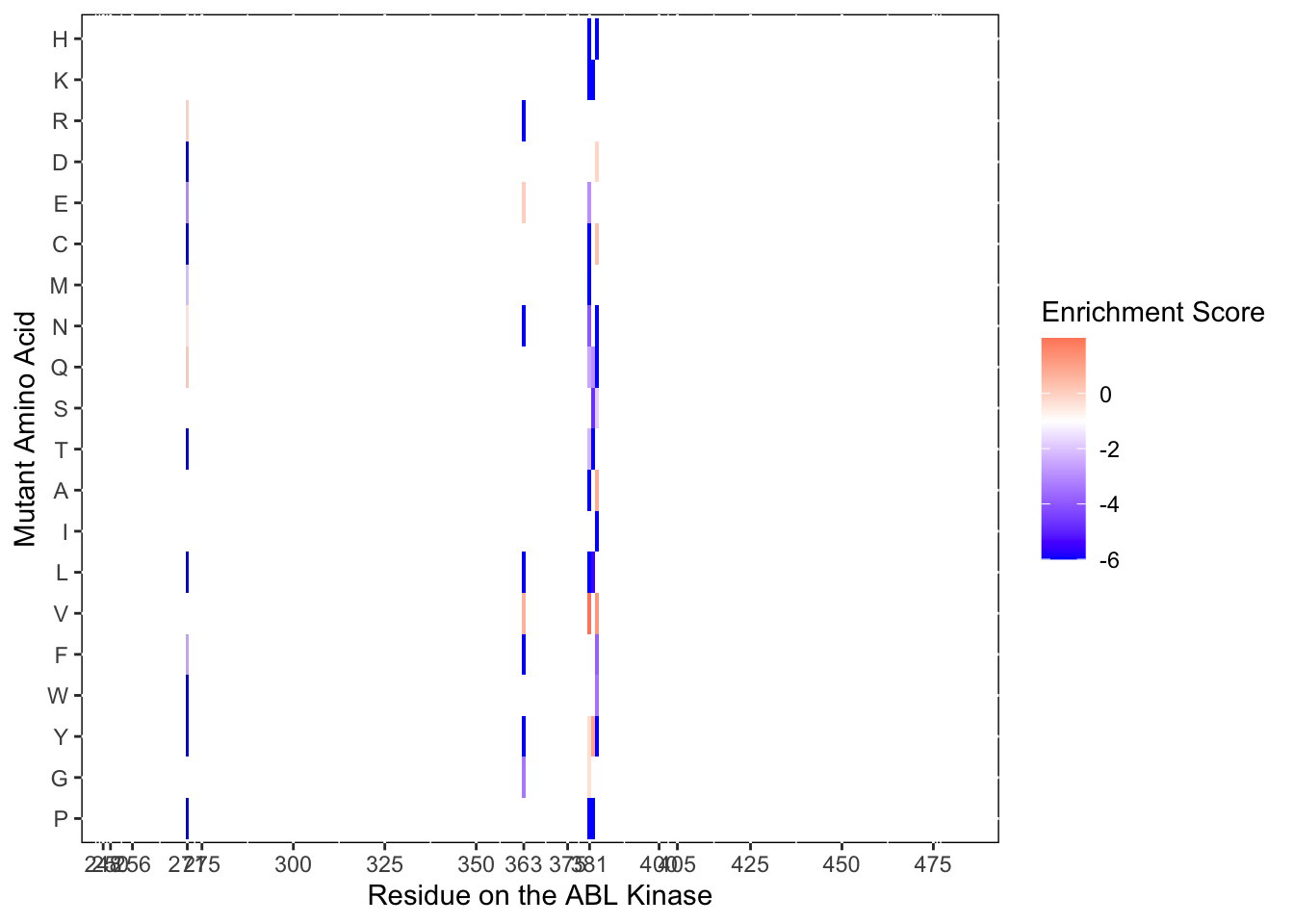
# ggsave("BCRABL_iL3Independence_D2_essential.pdf",height=6,width=24,units="in",useDingbats=F)
#####Graying out unseen residues#####
# df_grid = expand.grid(protein_start = c(242:493),alt_aa = unique(il3D0.D2$alt_aa))
#
# il3D0.D2.merge=merge(df_grid,il3D0.D2,by=c("protein_start","alt_aa"),all=T)
# il3D0.D2.merge=il3D0.D2.merge%>%filter(nchar(as.character(alt_aa))%in%1,protein_start>=242,protein_start<=494)
# il3D0.D2.merge=il3D0.D2.merge%>%mutate(score=case_when(ref_aa==alt_aa~"wt",
# T~"score"))
#
# ggplot(il3D0.D2.merge,aes(x=protein_start,y=alt_aa))+
# geom_tile(data=subset(il3D0.D2.merge,!is.na(score)),aes(fill=score))+
# scale_fill_gradient2(low ="blue",midpoint=-1,mid="white", high ="red",name="Enrichment Score")+
# geom_tile(data=subset(il3D0.D2.merge,is.na(score)),aes(color="white"),linetype = "solid",color="black", fill = "gray", alpha = 0.5)+
# theme(panel.background=element_rect(fill="white", colour="black"))#Next: plot iL3 D2 D4 scores #Correlate Il3 D2 D4 scores with il3 D0 D2 scores
Are any of the enriched mutants in the cosmic somatic database?
# rm(list=ls())
cosmic_data=read.table("data/Cosmic_ABL/ABL_Cosmic_Gene_mutations.tsv",sep="\t",header = T,stringsAsFactors = )
cosmic_data=cosmic_data%>%mutate(AA.Mutation=gsub("p.","",AA.Mutation))
cosmic_data=cosmic_data[!grepl("ins|del",cosmic_data$CDS.Mutation),]
cosmic_data=cosmic_data[grepl("Missense",cosmic_data$Type),]
cosmic_data=cosmic_data%>%filter(!Type%in%"Substitution - coding silent")
cosmic_data=cosmic_data%>%filter(Position<=500,Position>=64,Count>=2)
# write.csv(cosmic_data,"cosmic_abl.csv")
cosmic_simple=cosmic_data%>%dplyr::select(subs_name=AA.Mutation,cosmic_count=Count)
cosmic_simple=cosmic_simple%>%group_by(subs_name)%>%summarize(cosmic_count=sum(cosmic_count))
cosmic_simple$cosmic_present=Tsource("code/merge_samples.R")
sample10=read.csv(file = "data/Consensus_Data/Novogene_lane14/sample10_combined/variant_caller_outputs/variants_unique_ann.csv",header=T,stringsAsFactors = F)
sample10=sample10%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
sample10=sample10%>%mutate(maf=ct/depth)
sample10_simple=sample10%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
sample12=read.csv(file = "data/Consensus_Data/Novogene_lane14/sample12/variant_caller_outputs/variants_unique_ann.csv",header=T,stringsAsFactors = F)
sample12=sample12%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
sample12=sample12%>%mutate(maf=ct/depth)
sample12_simple=sample12%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
samples_10.12=merge(sample10_simple%>%filter(consequence_terms%in%"missense_variant"),sample12_simple%>%filter(consequence_terms%in%"missense_variant"),by=c("ref_aa","protein_start","alt_aa","ref","alt","alt_start_pos","consequence_terms"))
samples_10.12=samples_10.12%>%mutate(score=log2(maf.y/maf.x),score2=log2(ct.y/ct.x))
samples_10.12_simple=samples_10.12%>%dplyr::select(ref_aa,protein_start,alt_aa,consequence_terms,ct.x,score)
ggplot(samples_10.12,aes(x=score))+geom_density()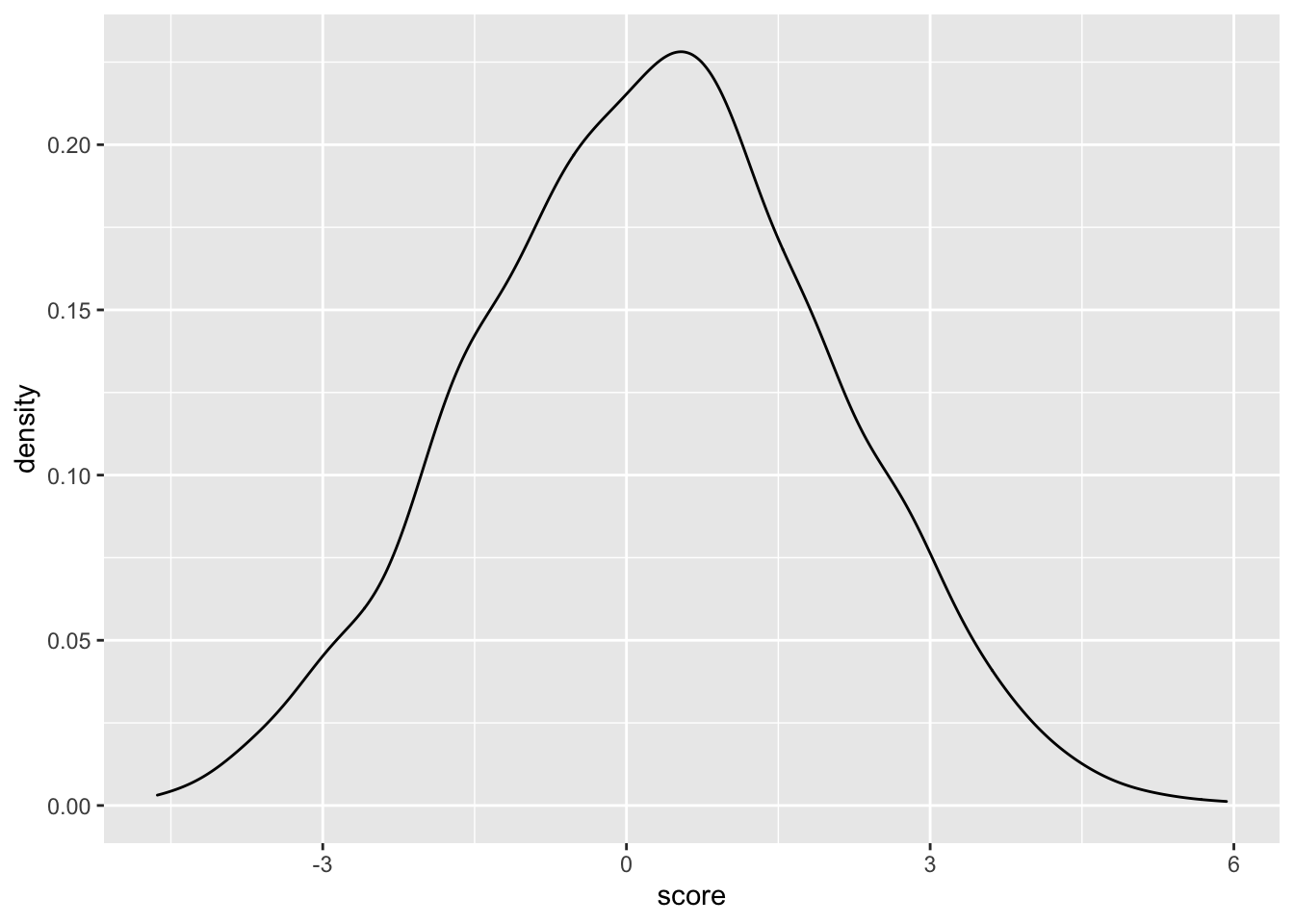
# ggplot(samples_14.16,aes(x=score))+geom_histogram(bins=100)
samples_10.12=samples_10.12%>%mutate(resmuts=case_when(protein_start%in%253&alt_aa%in%"H"~T,
protein_start%in%255&alt_aa%in%"V"~T,
protein_start%in%486&alt_aa%in%"S"~T,
protein_start%in%396&alt_aa%in%"P"~T,
protein_start%in%255&alt_aa%in%"K"~T,
protein_start%in%315&alt_aa%in%"I"~T,
protein_start%in%252&alt_aa%in%"H"~T,
protein_start%in%253&alt_aa%in%"F"~T,
protein_start%in%250&alt_aa%in%"E"~T,
protein_start%in%359&alt_aa%in%"C"~T,
protein_start%in%351&alt_aa%in%"T"~T,
protein_start%in%355&alt_aa%in%"G"~T,
protein_start%in%317&alt_aa%in%"L"~T,
protein_start%in%359&alt_aa%in%"I"~T,
protein_start%in%355&alt_aa%in%"A"~T,
protein_start%in%459&alt_aa%in%"K"~T,
protein_start%in%276&alt_aa%in%"G"~T,
protein_start%in%299&alt_aa%in%"L"~T,
T~F))
samples_10.12=samples_10.12%>%mutate(resresids=case_when(protein_start%in%253~T,
protein_start%in%255~T,
protein_start%in%486~T,
protein_start%in%396~T,
protein_start%in%255~T,
protein_start%in%315~T,
protein_start%in%252~T,
protein_start%in%253~T,
protein_start%in%250~T,
protein_start%in%359~T,
protein_start%in%351~T,
protein_start%in%355~T,
protein_start%in%317~T,
protein_start%in%359~T,
protein_start%in%355~T,
protein_start%in%459~T,
protein_start%in%276~T,
protein_start%in%299~T,
T~F))
highscore=samples_10.12%>%filter(!ct.x%in%1,protein_start>=242,protein_start<=494)
highscore=highscore%>%mutate(subs_name=paste(ref_aa,protein_start,alt_aa,sep = ""))
ggplot(highscore,aes(x=reorder(subs_name,-score),y=score,fill=resmuts))+geom_col()+theme(axis.text.x=element_text(angle=90, hjust=1))+scale_y_continuous(name="Enrithcment Score")+scale_x_discrete(name="Mutant")+guides(fill = guide_legend(title = "Clinically\n Observed \n Resmut"))+scale_fill_manual(values=c("gray90","red"))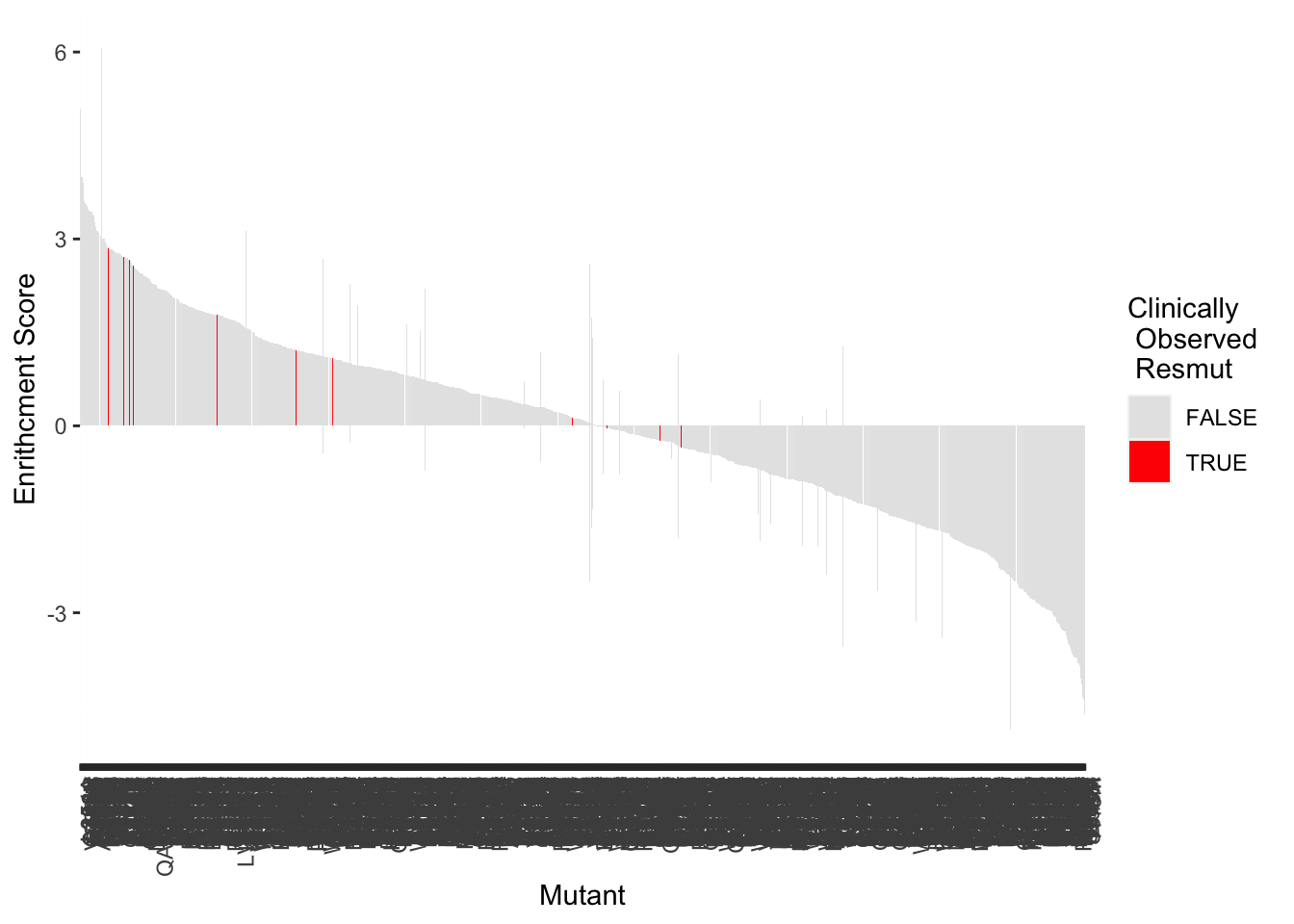
highscore2=highscore%>%group_by(alt_aa,protein_start,subs_name,resmuts,resresids)%>%summarize(score=mean(score))`summarise()` has grouped output by 'alt_aa', 'protein_start', 'subs_name', 'resmuts'. You can override using the `.groups` argument.ggplot(highscore2,aes(x=reorder(subs_name,-score),y=score,fill=resmuts))+geom_col()+theme(axis.text.x=element_blank(),axis.ticks.x=element_blank())+scale_y_continuous(name="Enrithcment Score")+scale_x_discrete(name="Mutant")+guides(fill = guide_legend(title = "Clinically\n Observed \n Resistant Mutant"))+scale_fill_manual(values=c("gray90","red"))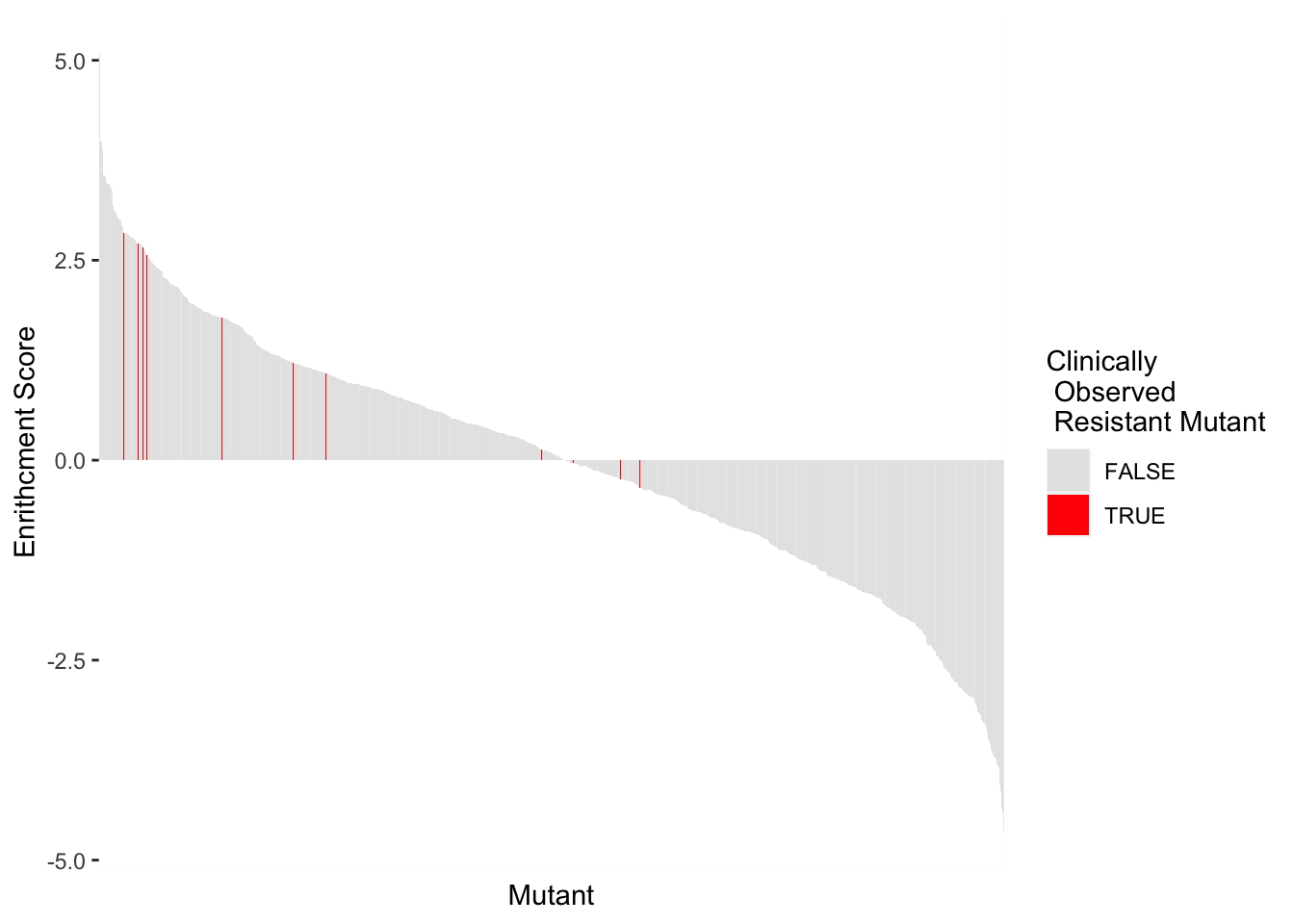
ggsave("BCRABL_Imatinib_Scores_Resmuts.pdf",height=4,width=8,units="in",useDingbats=F)
ggplot(highscore2,aes(x=reorder(subs_name,-score),y=score,fill=resresids))+geom_col()+theme(axis.text.x=element_blank(),axis.ticks.x=element_blank())+scale_y_continuous(name="Enrithcment Score")+scale_x_discrete(name="Mutant")+guides(fill = guide_legend(title = "Clinically\n Observed \n Resistant Residue"))+scale_fill_manual(values=c("gray90","red"))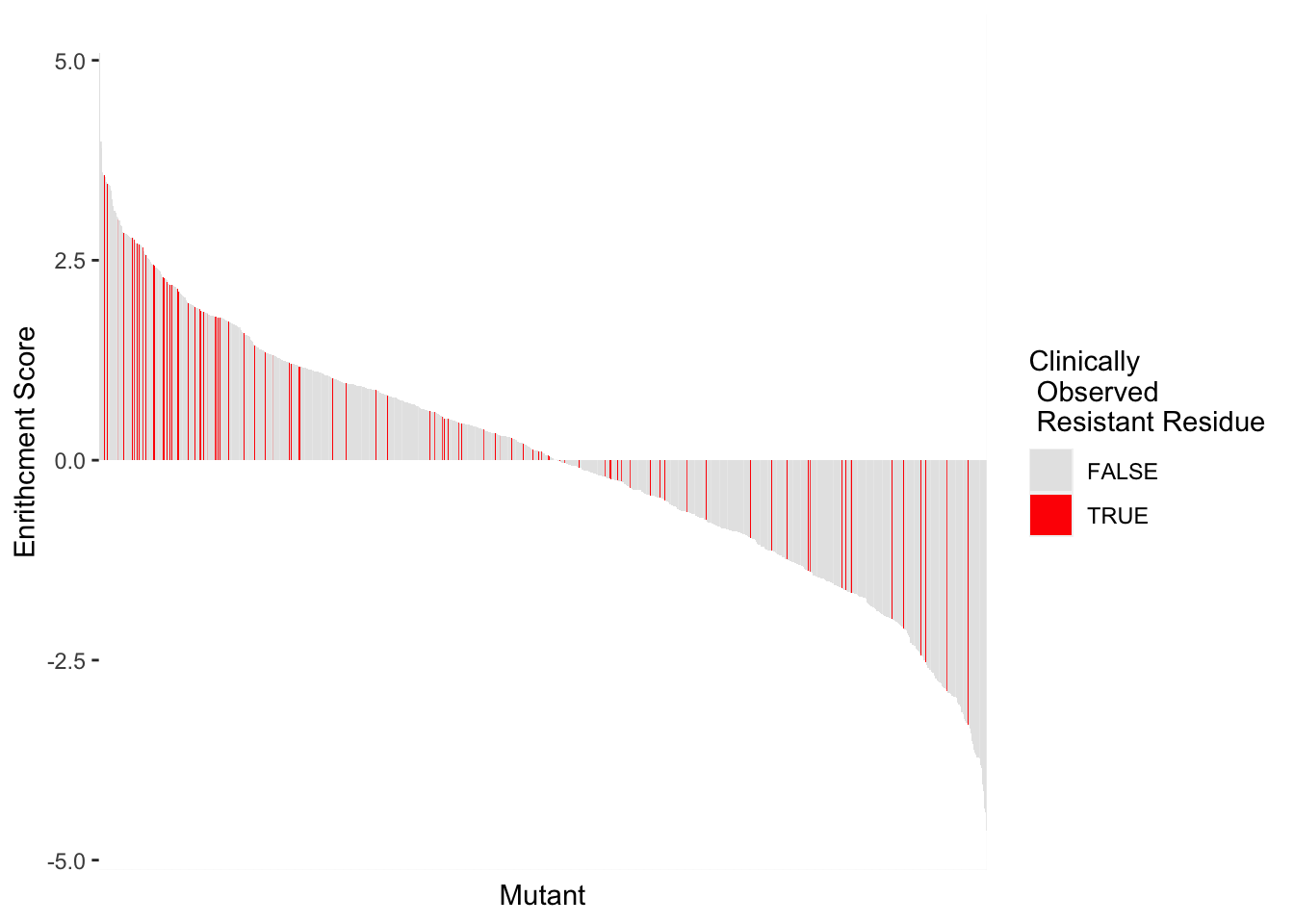
# ggsave("BCRABL_Imatinib_Scores_Resresids.pdf",height=4,width=8,units="in",useDingbats=F)
###Merging cosmic data and highscore
# highscore$cosmic_present=F
highscore_cosmic=merge(highscore,cosmic_simple,by="subs_name",all.x = T)
highscore_cosmic[highscore_cosmic$cosmic_present%in%NA,"cosmic_present"]=F
highscore_cosmic[highscore_cosmic$cosmic_count%in%NA,"cosmic_count"]=0
plotly=ggplot(highscore_cosmic,aes(x=reorder(subs_name,-score),y=score,fill=cosmic_present))+geom_col()+theme(axis.text.x=element_text(angle=90, hjust=1))+scale_y_continuous(name="Enrichment Score")+scale_x_discrete(name="Mutant")+guides(fill = guide_legend(title = "Cosmic\n Observed"))
ggplotly(plotly)a=cosmic_data%>%filter(AA.Mutation%in%"L248V")
a=highscore%>%filter(subs_name%in%"M388L")
plotly=ggplot(highscore_cosmic%>%filter(cosmic_present%in%T),aes(x=reorder(subs_name,-score),y=score,fill=cosmic_count))+geom_col()+theme_bw()
ggplotly(plotly)# a=highscore_cosmic%>%filter(cosmic_present%in%T)Looking at the evenness of the K562 Library
sample1=read.csv("data/Consensus_Data/Novogene_lane14/sample9/variant_caller_outputs/variants_unique_ann.csv")
sample1$sample="sample1"
sample1=sample1%>%mutate(region=case_when(protein_start<250&protein_start>=242~1,
protein_start<258&protein_start>=250~2,
protein_start<266&protein_start>=258~3,
protein_start<274&protein_start>=266~4,
protein_start<282&protein_start>=274~5,
protein_start<290&protein_start>=282~6,
protein_start<298&protein_start>=290~7,
protein_start<306&protein_start>=298~8,
protein_start<314&protein_start>=306~9,
protein_start<322&protein_start>=314~10,
protein_start<330&protein_start>=322~11,
protein_start<338&protein_start>=330~12,
protein_start<346&protein_start>=338~13,
protein_start<354&protein_start>=346~14,
protein_start<362&protein_start>=354~15,
protein_start<370&protein_start>=362~16,
protein_start<378&protein_start>=370~17,
protein_start<386&protein_start>=378~18,
protein_start<394&protein_start>=386~19,
protein_start<402&protein_start>=394~20,
protein_start<410&protein_start>=402~21,
protein_start<418&protein_start>=410~22,
protein_start<426&protein_start>=418~23,
protein_start<434&protein_start>=426~24,
protein_start<442&protein_start>=434~25,
protein_start<450&protein_start>=442~26,
protein_start<458&protein_start>=450~27,
protein_start<466&protein_start>=458~28,
protein_start<474&protein_start>=466~29,
protein_start<482&protein_start>=474~30,
protein_start<490&protein_start>=482~31,
protein_start<498&protein_start>=490~32,
T~0))
library(RColorBrewer)
getPalette = colorRampPalette(brewer.pal(33, "Set2"))Warning in brewer.pal(33, "Set2"): n too large, allowed maximum for palette Set2 is 8
Returning the palette you asked for with that many colorssample1=sample1%>%rowwise()%>%mutate(ID=paste(protein_start,amino_acids,sep=""))
plotly=ggplot(sample1%>%filter(consequence_terms%in%"missense_variant",protein_start>=242,protein_start<=494),aes(x=protein_start,y=ct))+geom_col(color="black",aes(fill=factor(region)))+scale_fill_manual(values=getPalette(33))
ggplotly(plotly)
sessionInfo()R version 4.0.0 (2020-04-24)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] RColorBrewer_1.1-2 doParallel_1.0.15 iterators_1.0.12 foreach_1.5.0
[5] tictoc_1.0 plotly_4.9.2.1 ggplot2_3.3.3 dplyr_1.0.6
[9] stringr_1.4.0
loaded via a namespace (and not attached):
[1] tidyselect_1.1.0 xfun_0.31 bslib_0.3.1 purrr_0.3.4
[5] colorspace_1.4-1 vctrs_0.3.8 generics_0.0.2 htmltools_0.5.2
[9] viridisLite_0.3.0 yaml_2.2.1 utf8_1.1.4 rlang_0.4.11
[13] jquerylib_0.1.4 later_1.0.0 pillar_1.6.1 glue_1.4.1
[17] withr_2.4.2 DBI_1.1.0 lifecycle_1.0.0 munsell_0.5.0
[21] gtable_0.3.0 workflowr_1.6.2 htmlwidgets_1.5.1 codetools_0.2-16
[25] evaluate_0.14 labeling_0.3 knitr_1.28 fastmap_1.1.0
[29] crosstalk_1.1.0.1 httpuv_1.5.2 fansi_0.4.1 Rcpp_1.0.4.6
[33] promises_1.1.0 backports_1.1.7 scales_1.1.1 jsonlite_1.7.2
[37] farver_2.0.3 fs_1.4.1 digest_0.6.25 stringi_1.7.5
[41] rprojroot_1.3-2 grid_4.0.0 tools_4.0.0 magrittr_2.0.1
[45] sass_0.4.1 lazyeval_0.2.2 tibble_3.1.2 crayon_1.4.1
[49] whisker_0.4 tidyr_1.1.3 pkgconfig_2.0.3 ellipsis_0.3.2
[53] data.table_1.12.8 assertthat_0.2.1 rmarkdown_2.14 httr_1.4.2
[57] R6_2.4.1 git2r_0.27.1 compiler_4.0.0