tcga_luad_data_parser_egfr
Haider Inam
2/20/2019
Last updated: 2019-02-20
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20190211)The command
set.seed(20190211)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 6fec622
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .Rhistory Ignored: .Rproj.user/ Untracked files: Untracked: code/alldata_compiler.R Untracked: code/contab_maker.R Untracked: code/mut_excl_genes_datapoints.R Untracked: code/mut_excl_genes_generator.R Untracked: code/quadratic_solver.R Untracked: code/simresults_generator.R Untracked: data/All_Data_V2.csv Untracked: data/all_data.csv Untracked: data/tcga_luad_expression/ Untracked: data/tcga_skcm_expression/ Untracked: docs/figure/Filteranalysis.Rmd/ Untracked: output/alkati_filtercutoff_allfilters.csv Untracked: output/alkati_luad_exonimbalance.pdf Untracked: output/alkati_mtn_pval_fig2B.pdf Untracked: output/alkati_skcm_exonimbalance.pdf Untracked: output/all_data_luad.csv Untracked: output/all_data_luad_egfr.csv Untracked: output/all_data_skcm.csv Untracked: output/egfr_luad_exonimbalance.pdf Untracked: output/fig2b2_filtercutoff_atinras_totalalk.pdf Untracked: output/fig2b_filtercutoff_atibraf.pdf Untracked: output/fig2b_filtercutoff_atinras.pdf Untracked: output/luad_alk_exon_expression.csv Untracked: output/luad_egfr_exon_expression.csv Untracked: output/skcm_alk_exon_expression.csv
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 6fec622 | haiderinam | 2019-02-20 | Added EGFR Expression Analysis |
This code is to look at the EGFR exon imbalance. Therefore, we need the count data, rsem data, and exon data
LUAD Genes RSEM
rsemdatanormalized=read.table("data/tcga_luad_expression/luadrsemdata/gdac.broadinstitute.org_LUAD.Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data.Level_3.2016012800.0.0/LUAD.rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data.data.txt",sep = "\t",header = T,stringsAsFactors = F)
egfr_rsem=data.frame(t(rsemdatanormalized[grepl("^egfr\\|",rsemdatanormalized$Hybridization.REF,ignore.case = T),])[-1,])
#410 of the 577 patients have an RSEM higher than 410
colnames(egfr_rsem)[1]="RSEM_normalized"
egfr_rsem$Patid=rownames(egfr_rsem)
#Standardizing Patid Names
egfr_rsem$Patid=substring(egfr_rsem$Patid,first = 1,last = 12)
egfr_rsem$Patid=gsub("\\.","-",egfr_rsem$Patid)
# # As Character
egfr_rsem[colnames(egfr_rsem)] <- lapply(egfr_rsem[colnames(egfr_rsem)],as.character)
# # As Numeric: Converting from list to numeric
egfr_rsem$RSEM_normalized=unlist(egfr_rsem$RSEM_normalized)
egfr_rsem$RSEM_normalized=as.numeric(egfr_rsem$RSEM_normalized)LUAD Count data:
#Non-normalized:
gene_expression_data=read.table("data/tcga_luad_expression/luadgeneexpression/gdac.broadinstitute.org_LUAD.Merge_rnaseq__illuminahiseq_rnaseq__unc_edu__Level_3__gene_expression__data.Level_3.2016012800.0.0/LUAD.rnaseq__illuminahiseq_rnaseq__unc_edu__Level_3__gene_expression__data.data.txt",sep = "\t",header = T,stringsAsFactors = F)
#Normalized
# gene_expression_data=read.table(,sep = "\t",header = T,stringsAsFactors = F)
#Finding egfr
egfr_gene_exp=rbind(gene_expression_data[1,],gene_expression_data[grepl("^egfr\\|",gene_expression_data$Hybridization.REF,ignore.case = T),])
#Removing Columns for Median_length_normalized and RPKM
t_egfr_gene_exp=data.frame(t(egfr_gene_exp[,grepl("raw_count",egfr_gene_exp[1,])]))
#Counting patients with raw reads >500
# sum(as.numeric(as.numeric(as.character(t_egfr_gene_exp$X580))>=500))
#ONLY 6 PATIENTS HAVE RAW COUNTS OF >500LUAD Exon RPKM This creates a .csv file and only needs to be run once.
# rm(list=ls())#Clears workspace
exondatacomb=read.table("data/tcga_luad_expression/luadexondatacomb/gdac.broadinstitute.org_LUAD.Merge_rnaseq__illuminahiseq_rnaseq__unc_edu__Level_3__exon_expression__data.Level_3.2016012800.0.0/LUAD.rnaseq__illuminahiseq_rnaseq__unc_edu__Level_3__exon_expression__data.data.txt",stringsAsFactors=FALSE,header=TRUE, sep="\t",fill=TRUE)
# head(exondatacomb)
# Obtained EGFR data from here: https://useast.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000146648;r=7:55019021-55211628
#Chromosome 7
exondatachr7=exondatacomb[grep("^chr7:",exondatacomb$Hybridization.REF),] #i.e. it starts with chromosome 7
#EGFR within Chromosome 7
exondatachr7egfr=exondatacomb[c(90511:90542),]
exondatachr7egfr=exondatachr7egfr[grep(":\\+",exondatachr2egfr$Hybridization.REF),] #i.e. it is only on +ive strand
exondatachr7egfr=exondatachr7egfr[c(-4,-16,-17),]
exondatachr7egfr$exon=c(27:1)
alldataegfr=exondatachr7egfr[,c(488,c(2:487))]
#Switching up order
alldataegfr2=alldataegfr[c(27:1),]
#Making the dataframe of a numeric type so that analysis can be carried out on it.
# As Character
alldataegfr[colnames(alldataegfr)] <- lapply(alldataegfr[colnames(alldataegfr)],as.character)
# As Numeric
alldataegfr[colnames(alldataegfr)] <- lapply(alldataegfr[colnames(alldataegfr)],as.numeric)
#Getting the correct column names for alldataegfr
colnames_exondata=exondatacomb[1,]
colnames(colnames_exondata)=colnames(alldataegfr)
alldataegfr=rbind(colnames_exondata,alldataegfr) #Adding first row that contains names of measurements such as RPKM, RSEM, Counts
write.table(alldataegfr,'output/luad_egfr_exon_expression.csv')Obtaining RPKM, Count data from LUAD Exon Data. Followed by merging with RSEM data
Note that we’re not concerned with the mutation data of EGFR so we don’t use it here
alldataegfr=read.csv("output/luad_egfr_exon_expression.csv",stringsAsFactors = F,header = T,sep = "",fill = T)
#Getting Count Data
alldataegfr_count=cbind(alldataegfr$exon,alldataegfr[,grepl("raw_counts",alldataegfr[1,])])[-1,]
# As Character
alldataegfr_count[colnames(alldataegfr_count)] <-lapply(alldataegfr_count[colnames(alldataegfr_count)],as.character)
# As Numeric
alldataegfr_count[colnames(alldataegfr_count)] <- lapply(alldataegfr_count[colnames(alldataegfr_count)],as.numeric)
#Sum exons 1:27
egfr_count_data=data.frame(t(data.frame(lapply(alldataegfr_count[c(1:27),],sum))[,-1])) #Not sure if lapply is the right thing to use here. Really messed up way of summing indices in dataframe
colnames(egfr_count_data)="mRNA_count"
alldataegfr_medianlength=cbind(alldataegfr$exon,alldataegfr[,grepl("median_length",alldataegfr[1,])])
# As Character
alldataegfr_medianlength=alldataegfr_medianlength[-1,] #Removing the first row. May be unnecessary in the future
alldataegfr_medianlength[colnames(alldataegfr_medianlength)] <- lapply(alldataegfr_medianlength[colnames(alldataegfr_medianlength)],as.character)
# As Numeric
alldataegfr_medianlength[colnames(alldataegfr_medianlength)] <- lapply(alldataegfr_medianlength[colnames(alldataegfr_medianlength)],as.numeric)
#Sum exons 1:27
egfr_medianlength_data=data.frame(t(data.frame(lapply(alldataegfr_medianlength[c(1:27),],sum))[,-1])) #Removing sum of exons lol
colnames(egfr_medianlength_data)="medianlength"
#Getting RPKM
alldataegfr_RPKM=cbind(alldataegfr$exon,alldataegfr[,grepl("RPKM",alldataegfr[1,])])
# As Character
alldataegfr_RPKM=alldataegfr_RPKM[-1,] #Removing the first row. May be unnecessary in the future
alldataegfr_RPKM[colnames(alldataegfr_RPKM)] <- lapply(alldataegfr_RPKM[colnames(alldataegfr_RPKM)],as.character)
# As Numeric
alldataegfr_RPKM[colnames(alldataegfr_RPKM)] <- lapply(alldataegfr_RPKM[colnames(alldataegfr_RPKM)],as.numeric)
egfr_RPKM_data=data.frame(cbind(lapply(alldataegfr_RPKM[c(1:17),],mean),lapply(alldataegfr_RPKM[c(18:24),],mean))[-1,])
colnames(egfr_RPKM_data)=c("mean_RPKM_1.17","mean_RPKM_18.24")
# As Character
egfr_RPKM_data[colnames(egfr_RPKM_data)] <- lapply(egfr_RPKM_data[colnames(egfr_RPKM_data)],as.character)
# As Numeric
egfr_RPKM_data[colnames(egfr_RPKM_data)] <- lapply(egfr_RPKM_data[colnames(egfr_RPKM_data)],as.numeric)
# Calculating Ratios of exon RPKM means
egfr_RPKM_data$Ratio18.24=egfr_RPKM_data$mean_RPKM_18.24/egfr_RPKM_data$mean_RPKM_1.17
#Changing rownames (patient_ids) to become the same between each other
rownames(egfr_RPKM_data)=substring(rownames(egfr_RPKM_data),first=1,last=28)
rownames(egfr_medianlength_data)=substring(rownames(egfr_medianlength_data),first=1,last=28)
egfr_RPKM_data$Patid=rownames(egfr_RPKM_data)
egfr_medianlength_data$Patid=rownames(egfr_medianlength_data)
egfr_count_data$Patid=rownames(egfr_count_data)
mergetemp=merge(egfr_RPKM_data,egfr_count_data,by="Patid")
egfr_exon_data=merge(mergetemp,egfr_medianlength_data,by="Patid")
#Transforming the Patids so that they're compatible with the Patids in the mutation data
egfr_exon_data$Patid=substring(egfr_exon_data$Patid,first = 1,last = 12)
#Since the names for exon data are not the same format as the mutation data, we're gonna change that here
egfr_exon_data$Patid=gsub("\\.","-",egfr_exon_data$Patid)
egfr_merged_data=merge(egfr_exon_data,egfr_rsem,by="Patid",all=T)
write.csv(egfr_merged_data,"output/all_data_luad_egfr.csv")Making EGFR Expression the plots:
egfr_merged_data=read.csv("output/all_data_luad_egfr.csv")
ggplot(egfr_merged_data,aes(x=mean_RPKM_1.17, y=mean_RPKM_18.24))+
geom_abline(size=1)+
geom_point(size=4)+
scale_x_continuous(trans = "log10",name="Exon 1:17 RPKM",breaks=c(1e-2,1e0,1e2),labels = parse(text = c("10^-2","10^0","10^2")),limits = c(1e-3,1e3))+
scale_y_continuous(trans = "log10",name="Exon 20:24 RPKM",breaks=c(1e-2,1e0,1e2),labels = parse(text = c("10^-2","10^0","10^2")),limits = c(1e-3,1e3))+
cleanup+
theme(plot.title = element_text(hjust=.5),
text = element_text(size=24,face = "bold"),
axis.title = element_text(face="bold",size="24"),
axis.text=element_text(face="bold",size="24",colour = "black"))+
theme(legend.key.size = unit(30,"pt"))Warning: Removed 389 rows containing missing values (geom_point).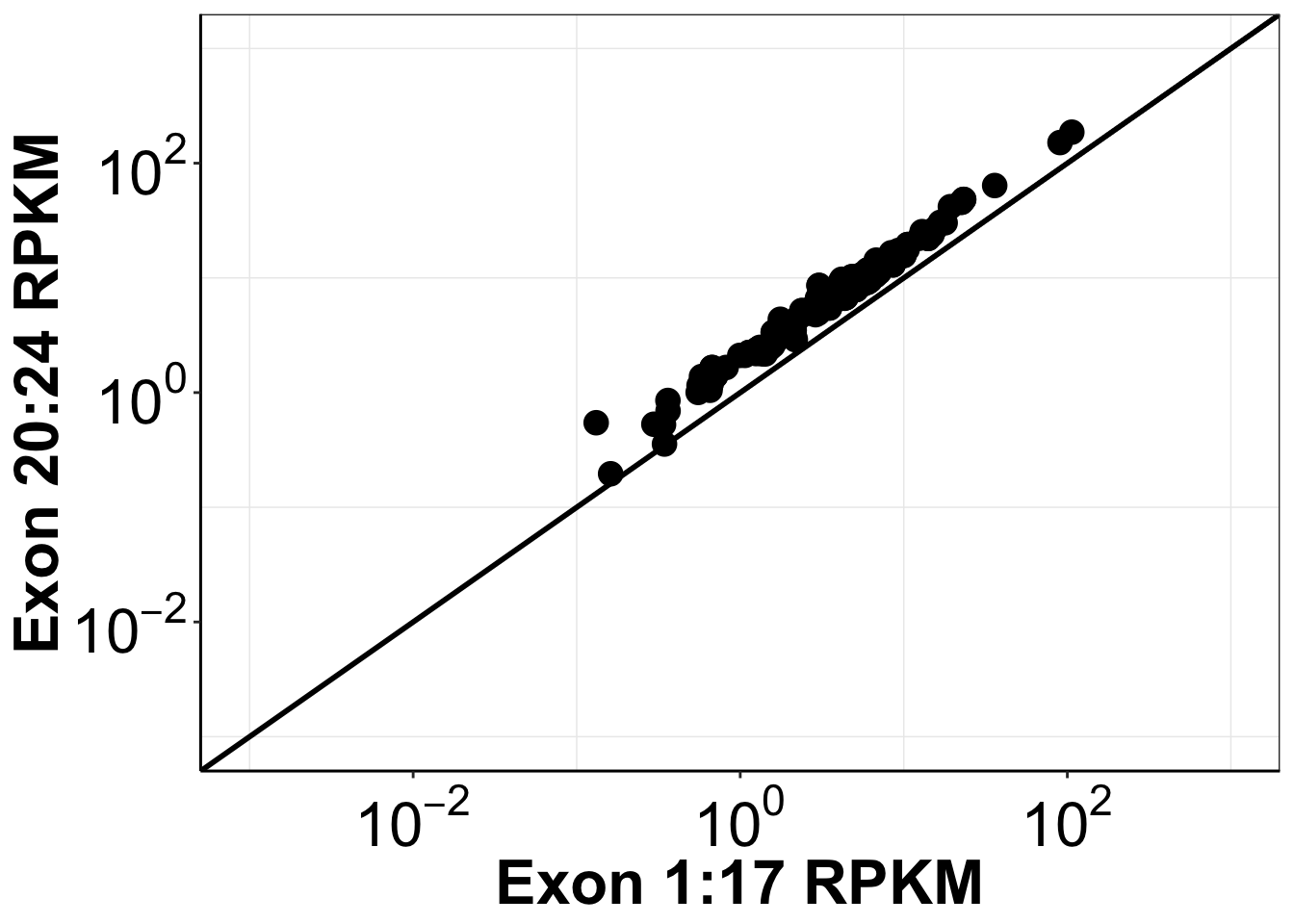
ggsave("output/egfr_luad_exonimbalance.pdf",width =12 ,height =10 ,units = "in",useDingbats=F)Warning: Removed 389 rows containing missing values (geom_point).###We observed an insignificant difference between the distribution for the 1-17 exons and the 20-24 exons.Session information
sessionInfo()R version 3.5.2 (2018-12-20)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.3
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel grid stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] ggsignif_0.4.0 usethis_1.4.0 devtools_2.0.1
[4] RColorBrewer_1.1-2 reshape2_1.4.3 ggplot2_3.1.0
[7] doParallel_1.0.14 iterators_1.0.10 foreach_1.4.4
[10] dplyr_0.7.8 VennDiagram_1.6.20 futile.logger_1.4.3
[13] workflowr_1.1.1 tictoc_1.0 knitr_1.21
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 xfun_0.4 remotes_2.0.2
[4] purrr_0.3.0 colorspace_1.4-0 htmltools_0.3.6
[7] yaml_2.2.0 rlang_0.3.1 pkgbuild_1.0.2
[10] R.oo_1.22.0 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 R.utils_2.7.0 sessioninfo_1.1.1
[16] lambda.r_1.2.3 bindrcpp_0.2.2 bindr_0.1.1
[19] plyr_1.8.4 stringr_1.3.1 munsell_0.5.0
[22] gtable_0.2.0 R.methodsS3_1.7.1 codetools_0.2-16
[25] evaluate_0.12 memoise_1.1.0 callr_3.1.1
[28] ps_1.3.0 Rcpp_1.0.0 backports_1.1.3
[31] scales_1.0.0 formatR_1.5 desc_1.2.0
[34] pkgload_1.0.2 fs_1.2.6 digest_0.6.18
[37] stringi_1.2.4 processx_3.2.1 rprojroot_1.3-2
[40] cli_1.0.1 tools_3.5.2 magrittr_1.5
[43] lazyeval_0.2.1 tibble_2.0.1 futile.options_1.0.1
[46] crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2
[49] prettyunits_1.0.2 assertthat_0.2.0 rmarkdown_1.11
[52] R6_2.3.0 git2r_0.24.0 compiler_3.5.2 This reproducible R Markdown analysis was created with workflowr 1.1.1