L6-population-structure
Hae Kyung Im
2020-01-26
Last updated: 2020-01-27
Checks: 5 1
Knit directory: hgen471/
This reproducible R Markdown analysis was created with workflowr (version 1.3.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200105) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: docs/.DS_Store
Untracked files:
Untracked: analysis/L6-download-data.Rmd
Untracked: analysis/L6-population-structure.Rmd
Untracked: docs/figure/L6-population-structure.Rmd/
Untracked: output/igrowth-adjPC.assoc.linear
Untracked: output/igrowth-adjPC.log
Unstaged changes:
Modified: analysis/L3-multiple-testing.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
library(tidyverse)Registered S3 methods overwritten by 'ggplot2':
method from
[.quosures rlang
c.quosures rlang
print.quosures rlang── Attaching packages ─────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.2 ✔ dplyr 0.8.1
✔ tidyr 0.8.3 ✔ stringr 1.4.0
✔ readr 1.3.1 ✔ forcats 0.4.0── Conflicts ────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()work.dir ="~/Downloads/hapmap/"
## qqunif function
source("https://gist.githubusercontent.com/hakyim/38431b74c6c0bf90c12f/raw/21fbae9a48dc475f42fa60f0ef5509d071dea873/qqunif")*What’s the population composition?**
popinfo = read_tsv(paste0(work.dir,"relationships_w_pops_051208.txt"))Parsed with column specification:
cols(
FID = col_character(),
IID = col_character(),
dad = col_character(),
mom = col_character(),
sex = col_double(),
pheno = col_double(),
population = col_character()
)popinfo %>% count(population)# A tibble: 11 x 2
population n
<chr> <int>
1 ASW 90
2 CEU 180
3 CHB 90
4 CHD 100
5 GIH 100
6 JPT 91
7 LWK 100
8 MEX 90
9 MKK 180
10 TSI 100
11 YRI 180samdata = read_tsv(paste0(work.dir,"phase3_corrected.psam"),guess_max = 2500) Parsed with column specification:
cols(
`#IID` = col_character(),
PAT = col_character(),
MAT = col_character(),
SEX = col_double(),
SuperPop = col_character(),
Population = col_character()
)superpop = samdata %>% select(SuperPop,Population) %>% unique()
superpop = rbind(superpop, data.frame(SuperPop=c("EAS","HIS","AFR"),Population=c("CHD","MEX","MKK")))Effect of population structure in Hardy Weinberg
## what happens if we calculate HWE with this mixed population?
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --hardy --out {work.dir}output/allhwe"))
allhwe = read.table(glue::glue("{work.dir}output/allhwe.hwe"),header=TRUE,as.is=TRUE)
hist(allhwe$P)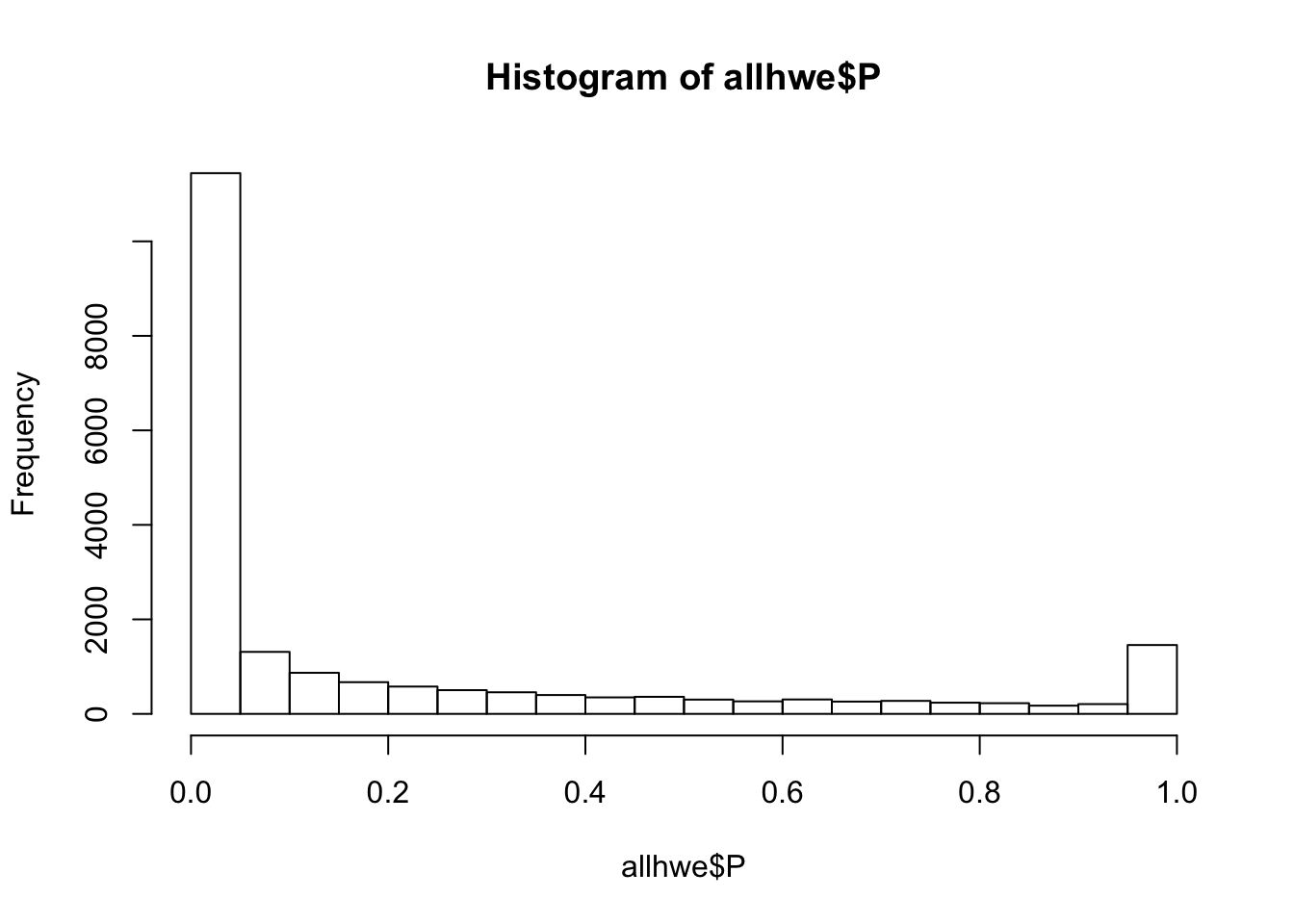
qqunif(allhwe$P,main='HWE HapMap3 All Pop')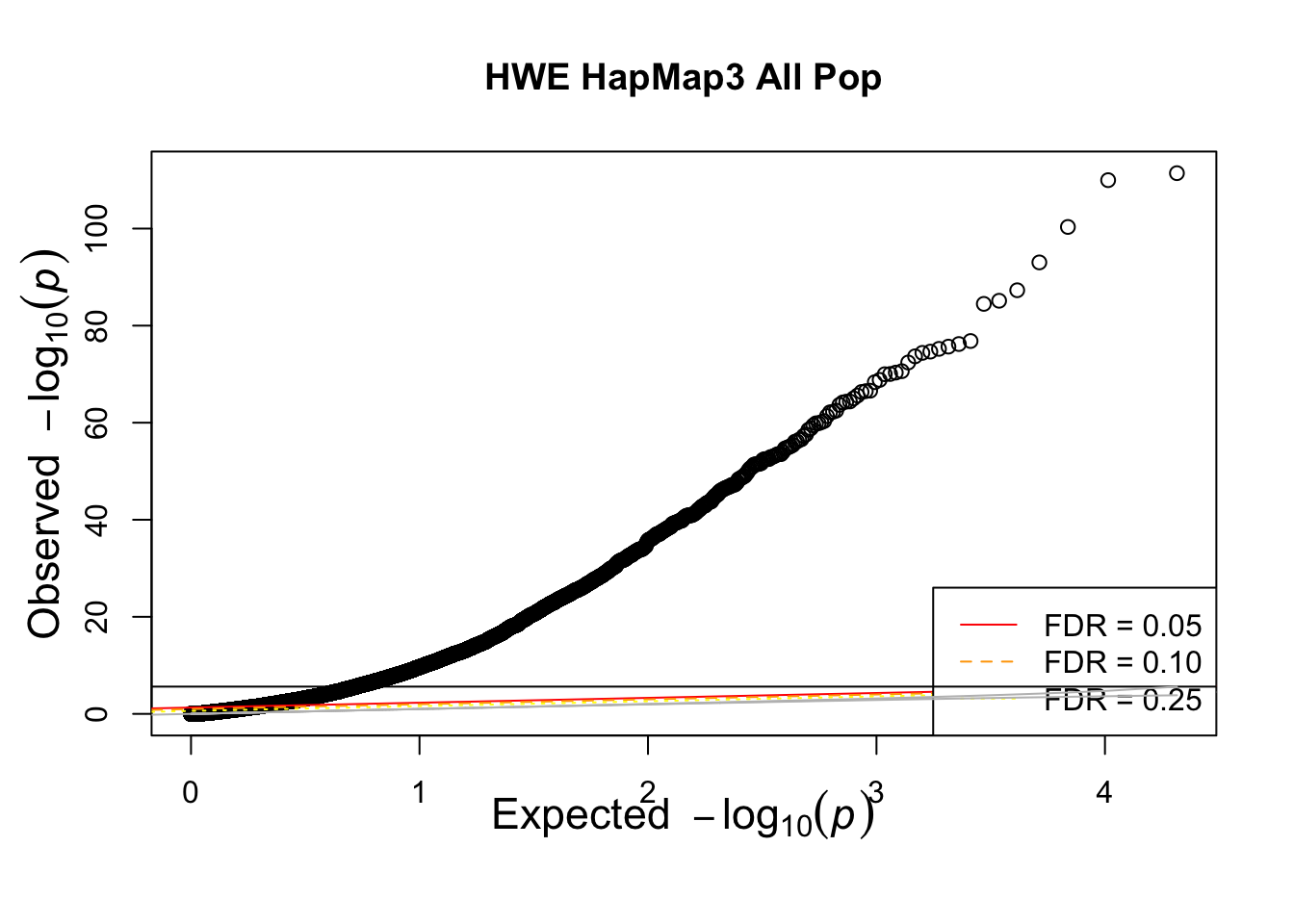
What if we calculate with single population?
pop = "CHB"
pop = "CEU"
pop = "YRI"
## what if we calculate with single population?
popinfo %>% filter(population==pop) %>% write_tsv(path=glue::glue("{work.dir}{pop}.fam") )
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --hardy --keep {work.dir}{pop}.fam --out {work.dir}output/hwe-{pop}"))
pophwe = read.table(glue::glue("{work.dir}output/hwe-{pop}.hwe"),header=TRUE,as.is=TRUE)
hist(pophwe$P,main=glue::glue("HWE {pop} and founders only"))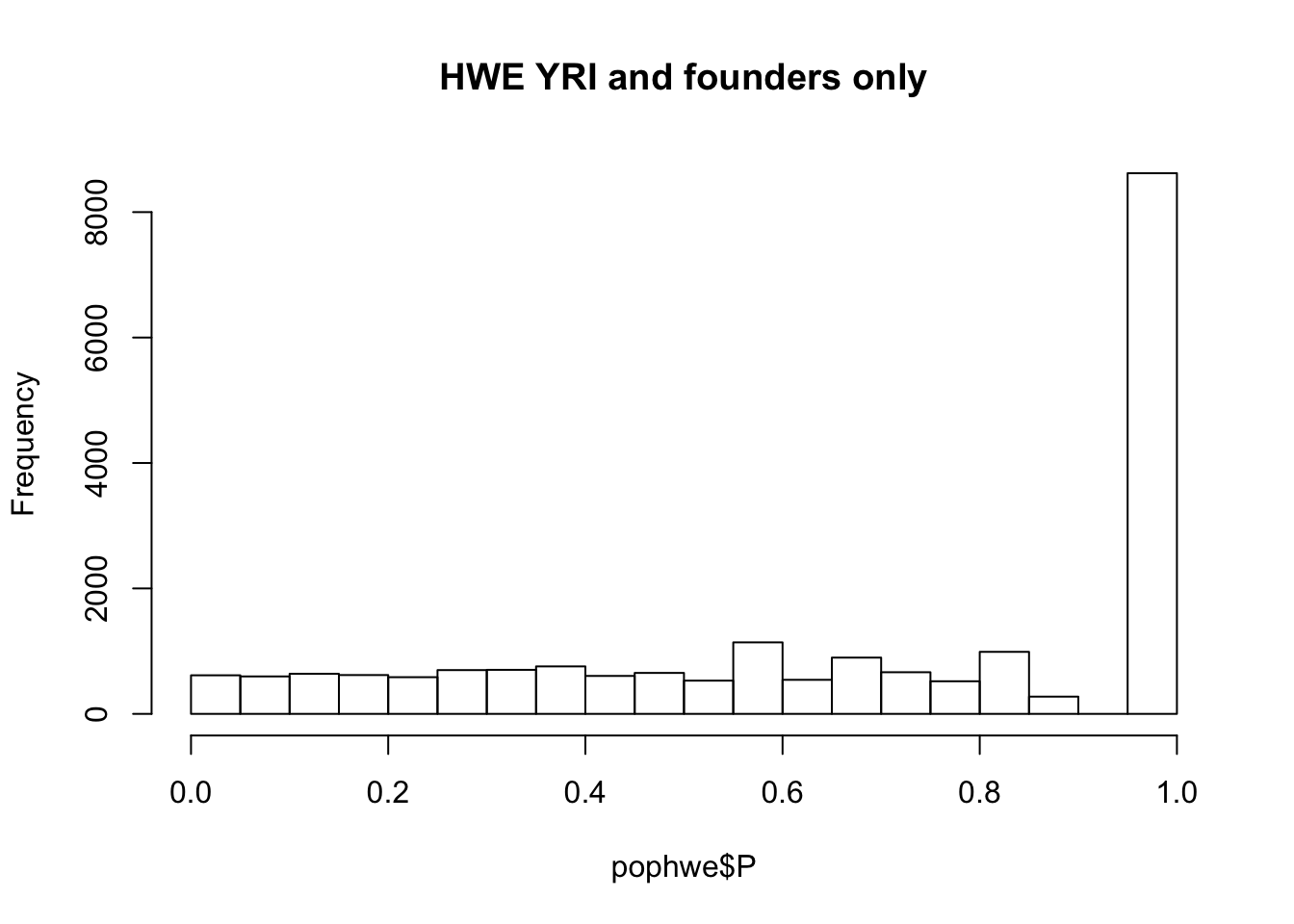
qqunif(pophwe$P,main=glue::glue("HWE {pop} and founders only"))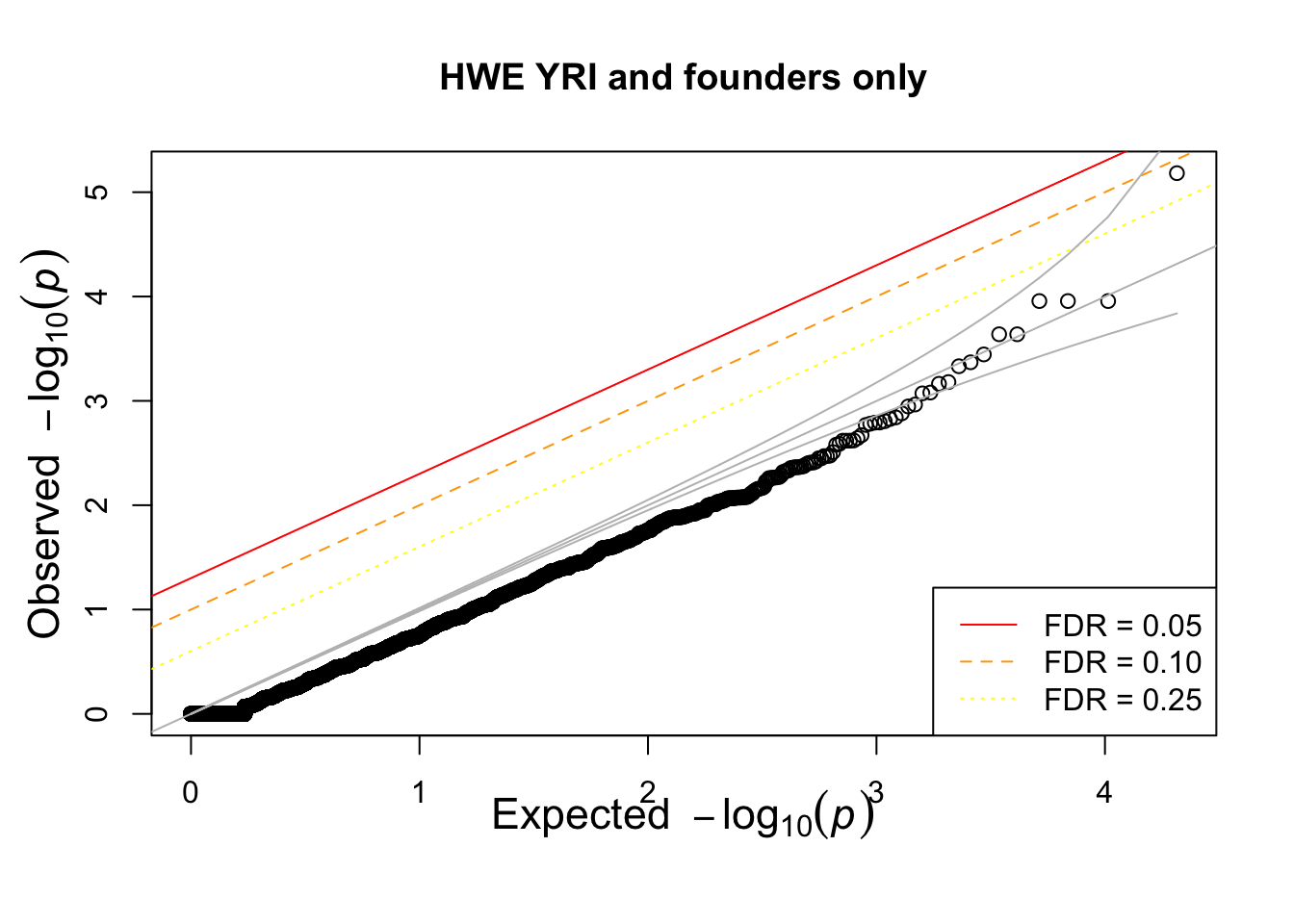
## not much difference when --nonfounders option is addedWhat if we add non founders? Some of the samples in HapMap were recruited from families.
## what if we add nonfounders?
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --hardy --keep {work.dir}CEU.fam --nonfounders --out {work.dir}output/CEUhwe_nf"))
CEUhwe_nf = read.table(glue::glue("{work.dir}output/CEUhwe_nf.hwe"),header=TRUE,as.is=TRUE)
hist(CEUhwe_nf$P,main="HWE CEU + non founders")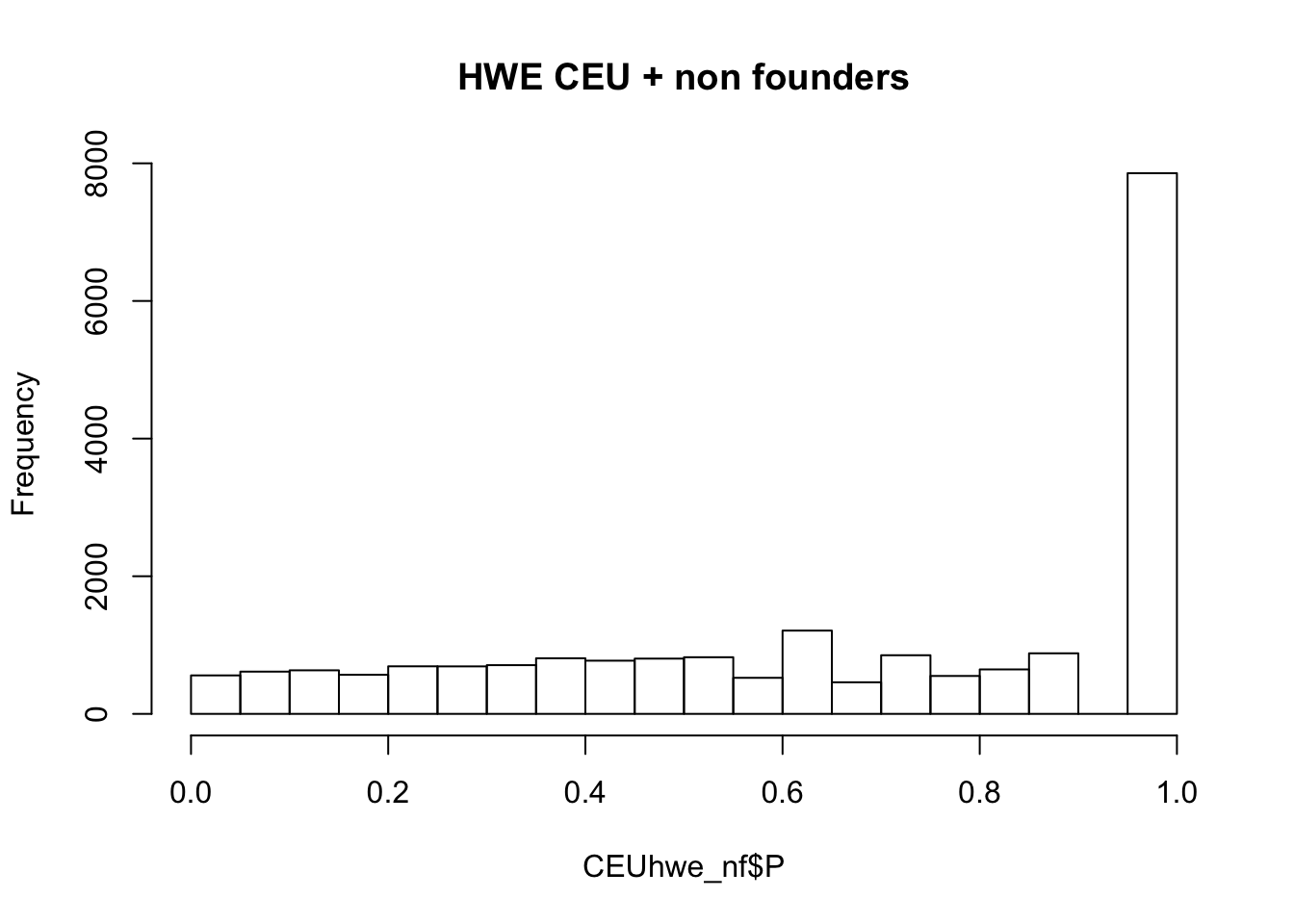
qqunif(CEUhwe_nf$P,main="HWE CEU + non founders")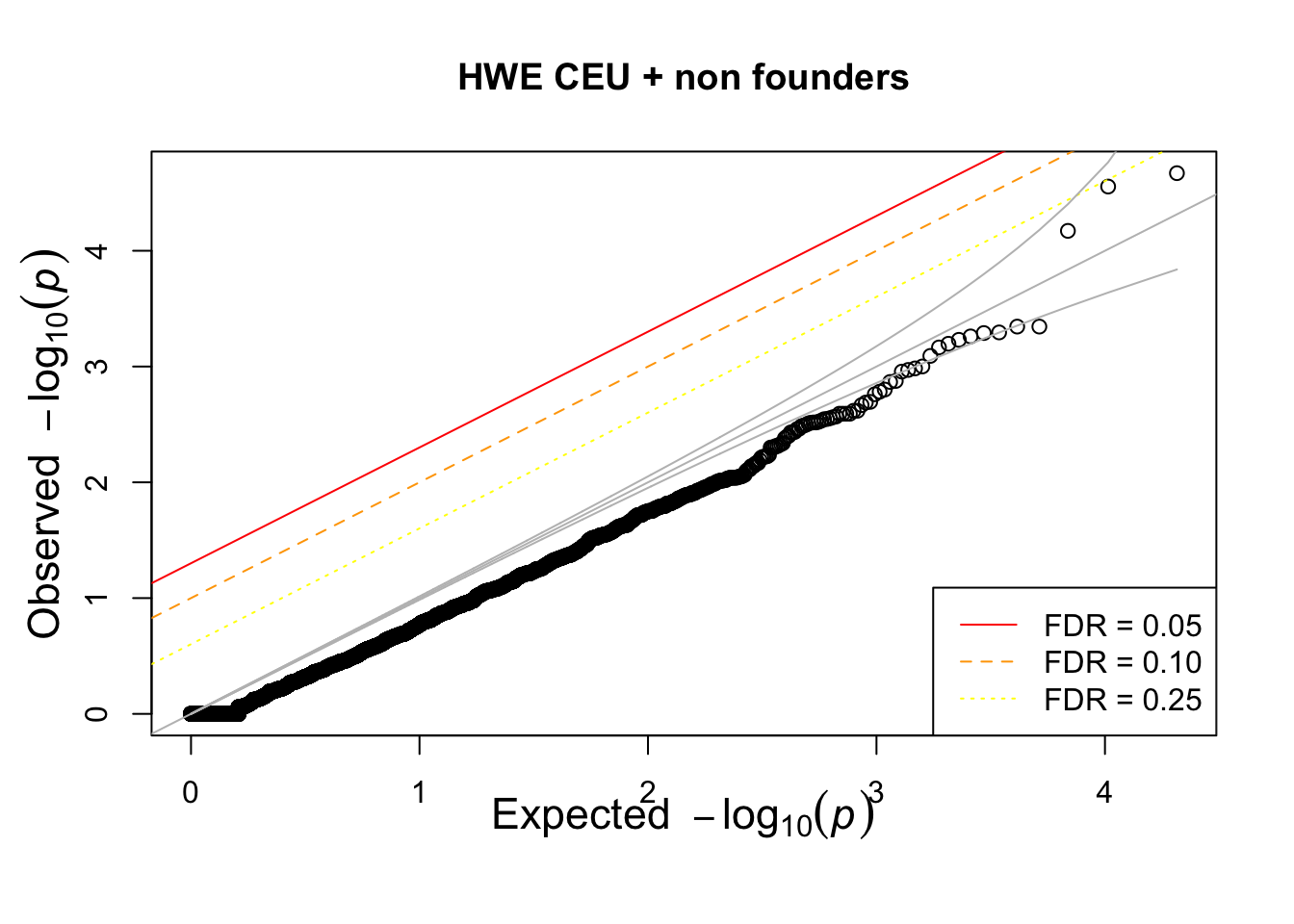
qqplot(-log10(CEUhwe_nf$P),-log10(CEUhwe_nf$P),main="all vs founders only" );abline(0,1) 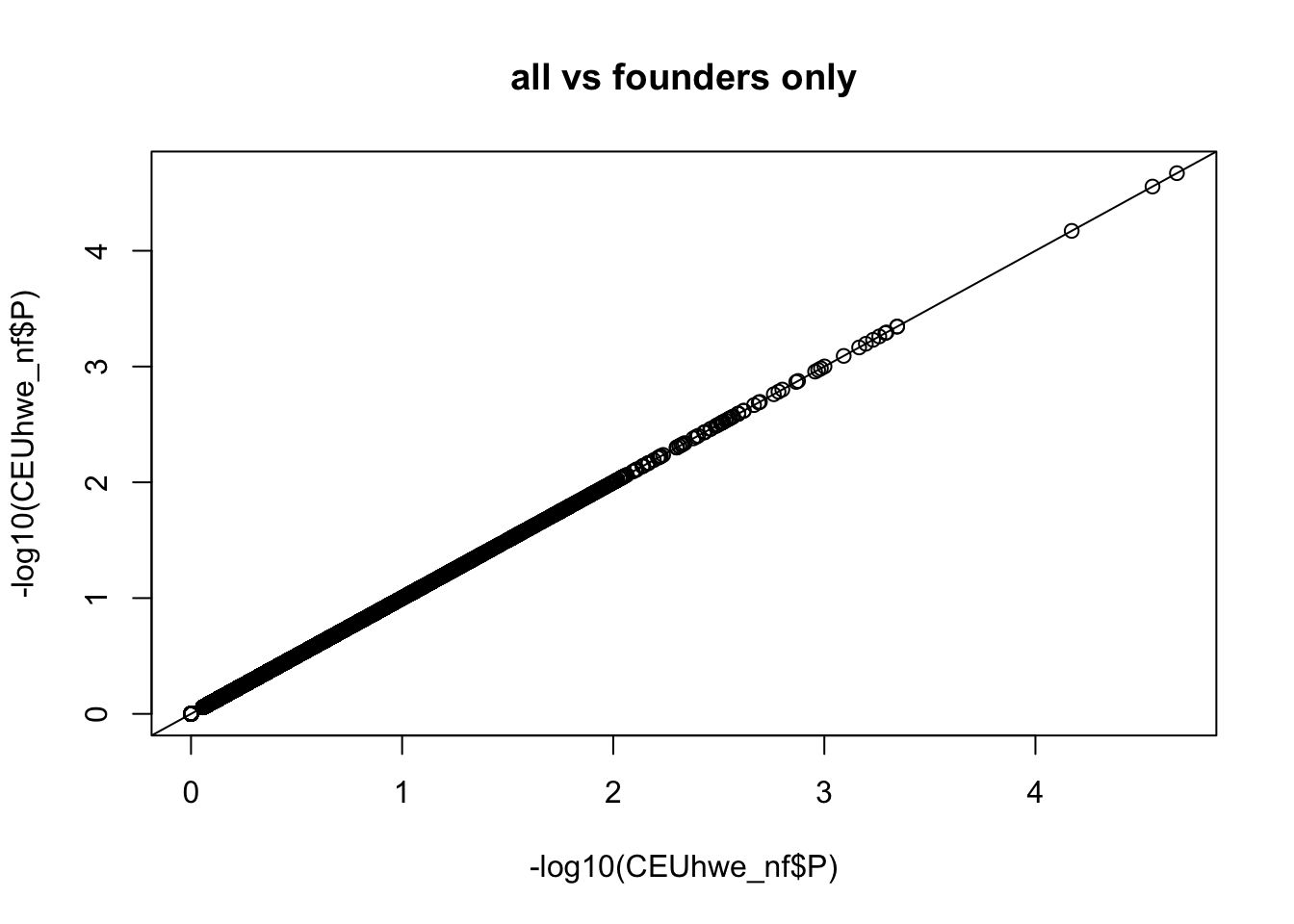
GWAS on a growth phenotype in HapMap samples
## read igrowth
igrowth = read_tsv("https://raw.githubusercontent.com/hakyimlab/igrowth/master/rawgrowth.txt")Parsed with column specification:
cols(
IID = col_character(),
sex = col_double(),
pop = col_character(),
experim = col_double(),
meas.by = col_double(),
serum = col_character(),
growth = col_double()
)## fix FID from igrowth file
igrowth = popinfo %>% select(-pheno) %>% inner_join(igrowth %>% select(IID,growth), by=c("IID"="IID"))
write_tsv(igrowth,path=glue::glue("{work.dir}igrowth.pheno"))
igrowth %>% ggplot(aes(population,growth)) + geom_violin(aes(fill=population)) + geom_boxplot(width=0.2,col='black',fill='gray',alpha=.8) + theme_bw(base_size = 15)Warning: Removed 130 rows containing non-finite values (stat_ydensity).Warning: Removed 130 rows containing non-finite values (stat_boxplot).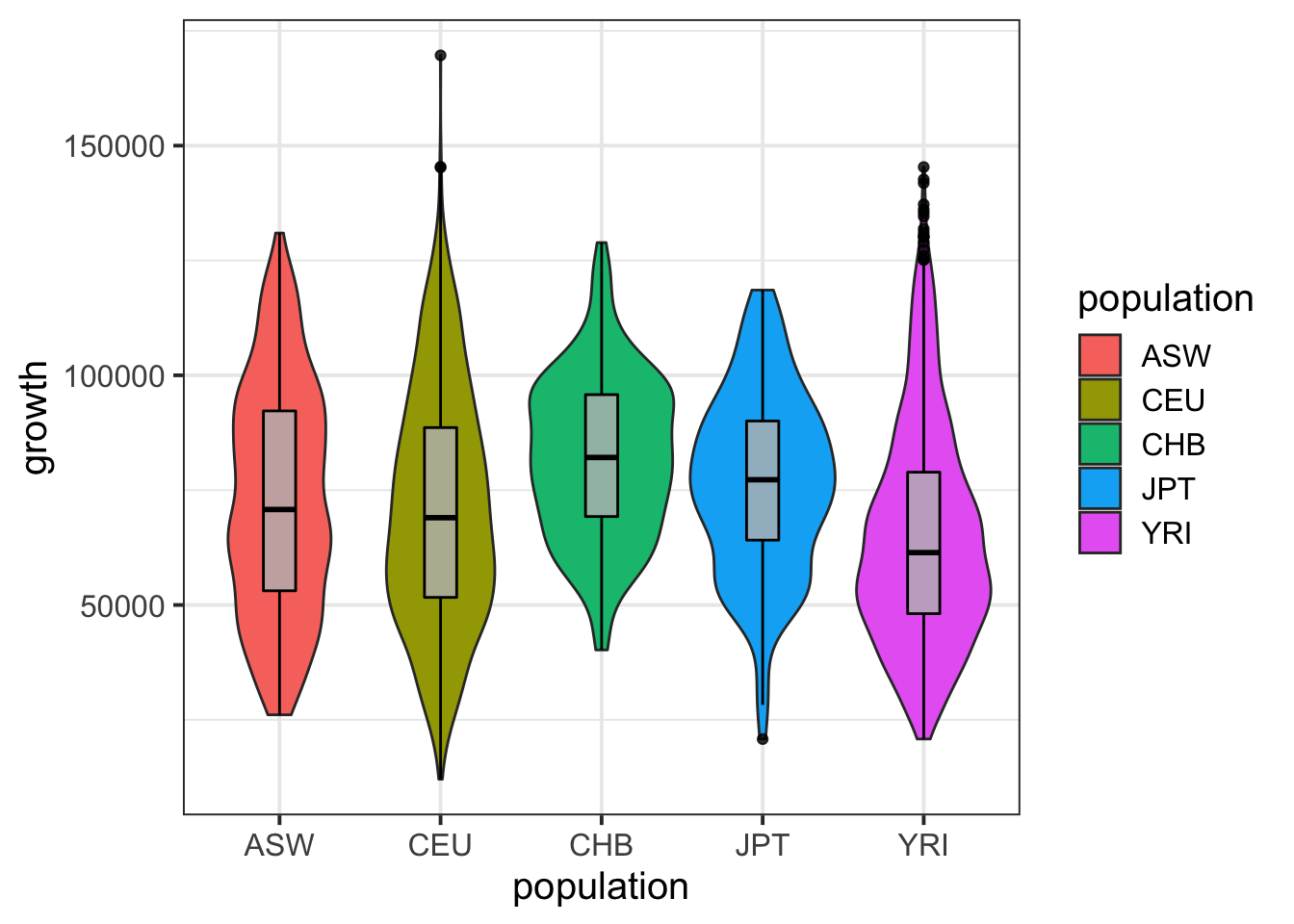
summary( lm(growth~population,data=igrowth) )
Call:
lm(formula = growth ~ population, data = igrowth)
Residuals:
Min 1Q Median 3Q Max
-58821 -18093 -2242 15896 98760
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 73080.8 938.2 77.894 < 2e-16 ***
populationCEU -2190.1 1175.4 -1.863 0.0625 .
populationCHB 9053.1 2043.9 4.429 9.73e-06 ***
populationJPT 3476.8 2034.8 1.709 0.0876 .
populationYRI -7985.2 1137.2 -7.022 2.61e-12 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 24160 on 3591 degrees of freedom
(130 observations deleted due to missingness)
Multiple R-squared: 0.0345, Adjusted R-squared: 0.03342
F-statistic: 32.08 on 4 and 3591 DF, p-value: < 2.2e-16system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --linear --pheno {work.dir}igrowth.pheno --pheno-name growth --maf 0.05 --out {work.dir}output/igrowth"))
igrowth.assoc = read.table(glue::glue("{work.dir}output/igrowth.assoc.linear"),header=T,as.is=T)
hist(igrowth.assoc$P)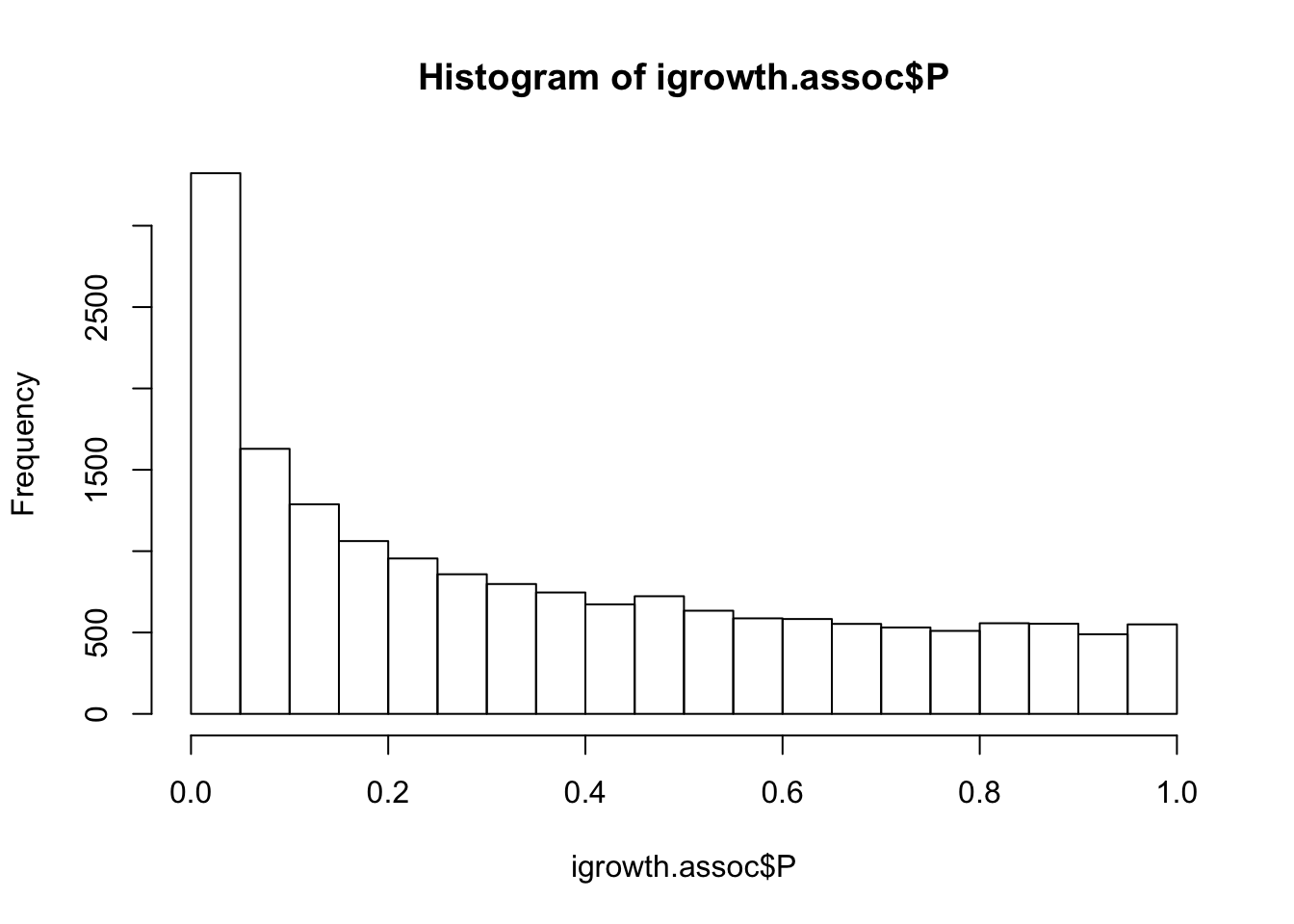
qqunif(igrowth.assoc$P)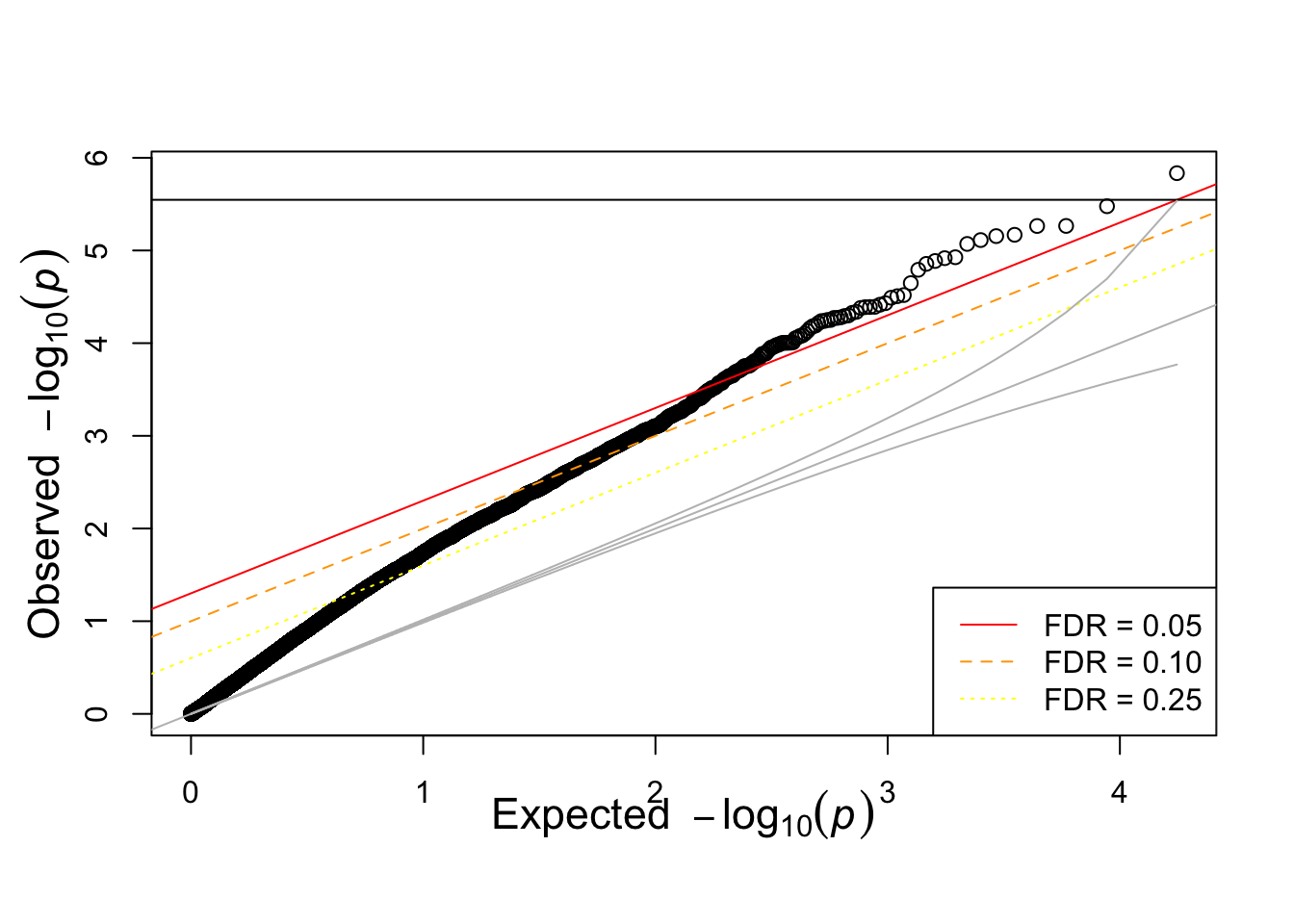
library(qqman)For example usage please run: vignette('qqman')Citation appreciated but not required:Turner, S.D. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. biorXiv DOI: 10.1101/005165 (2014).manhattan(igrowth.assoc, chr="CHR", bp="BP", snp="SNP", p="P" )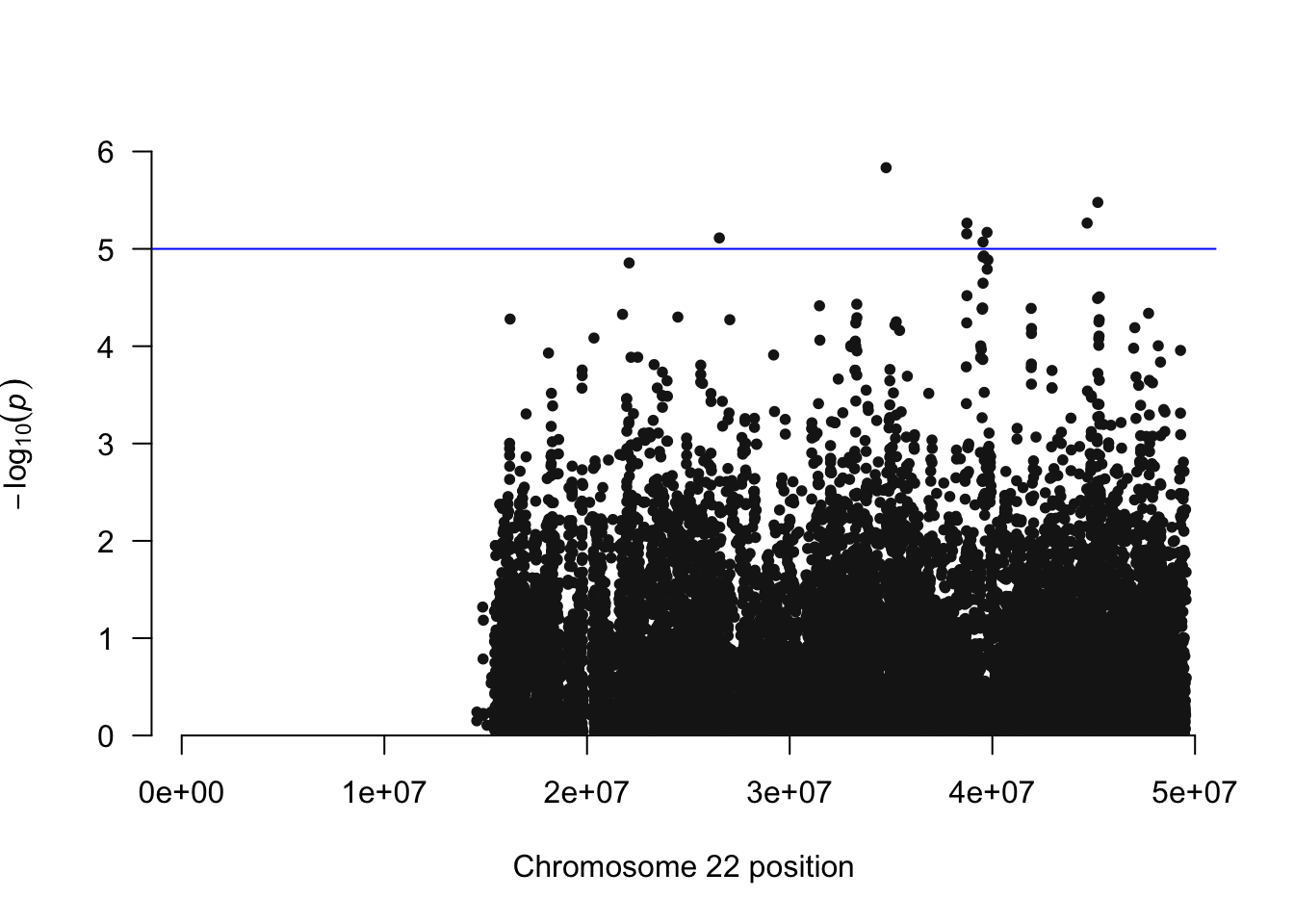
Simulate phenotype
set.seed(10) ## to get the same simulated values each time
simpheno = popinfo %>% mutate(pheno=rnorm(nrow(popinfo)))
write_tsv(simpheno, path=glue::glue("{work.dir}sim.pheno"))
## run association with plink
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --linear --pheno {work.dir}sim.pheno --pheno-name pheno --maf 0.05 --out {work.dir}output/simpheno") )
simpheno.assoc = read.table(glue::glue("{work.dir}output/simpheno.assoc.linear"),header=T,as.is=T)
hist(simpheno.assoc$P)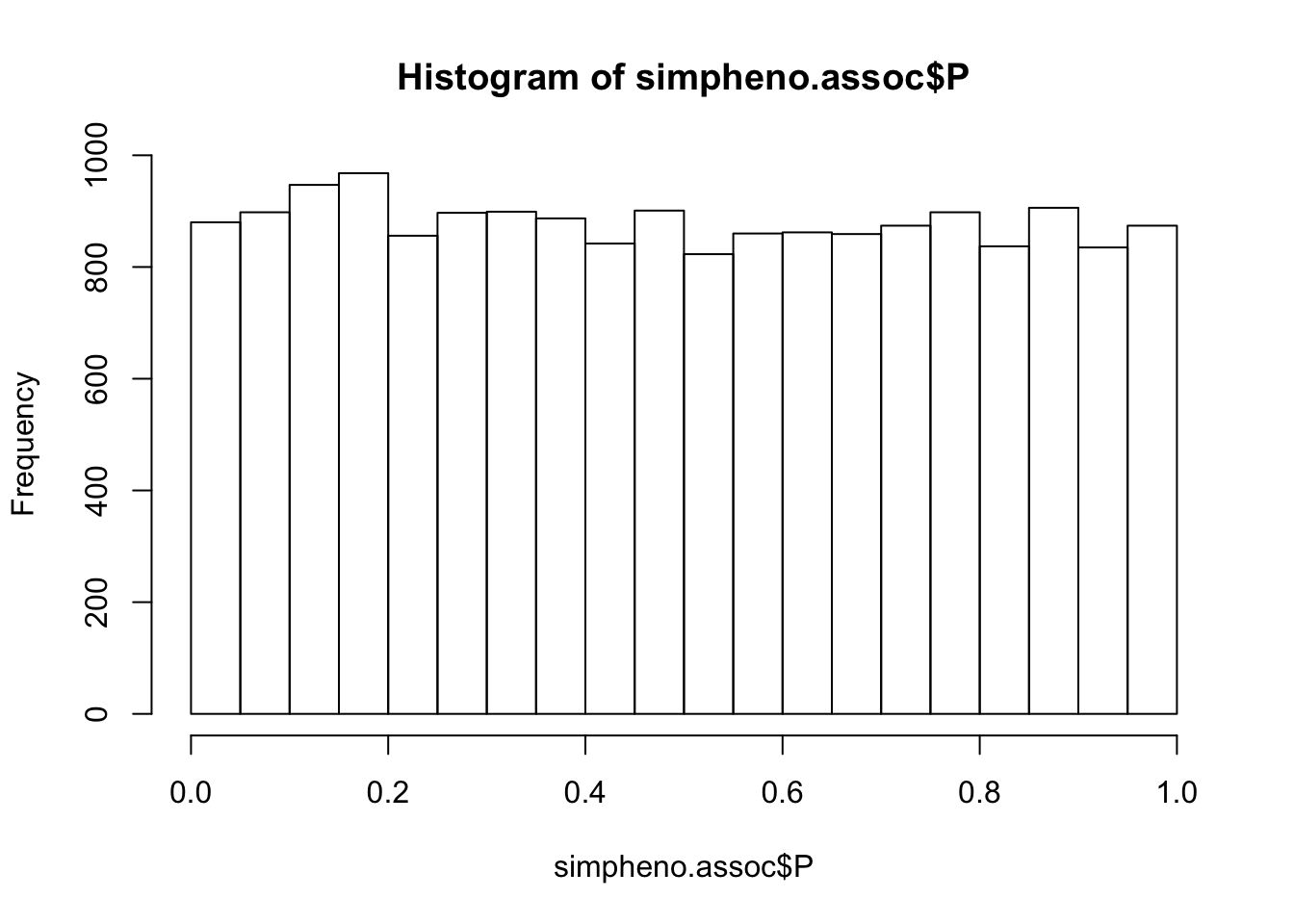
qqunif(simpheno.assoc$P)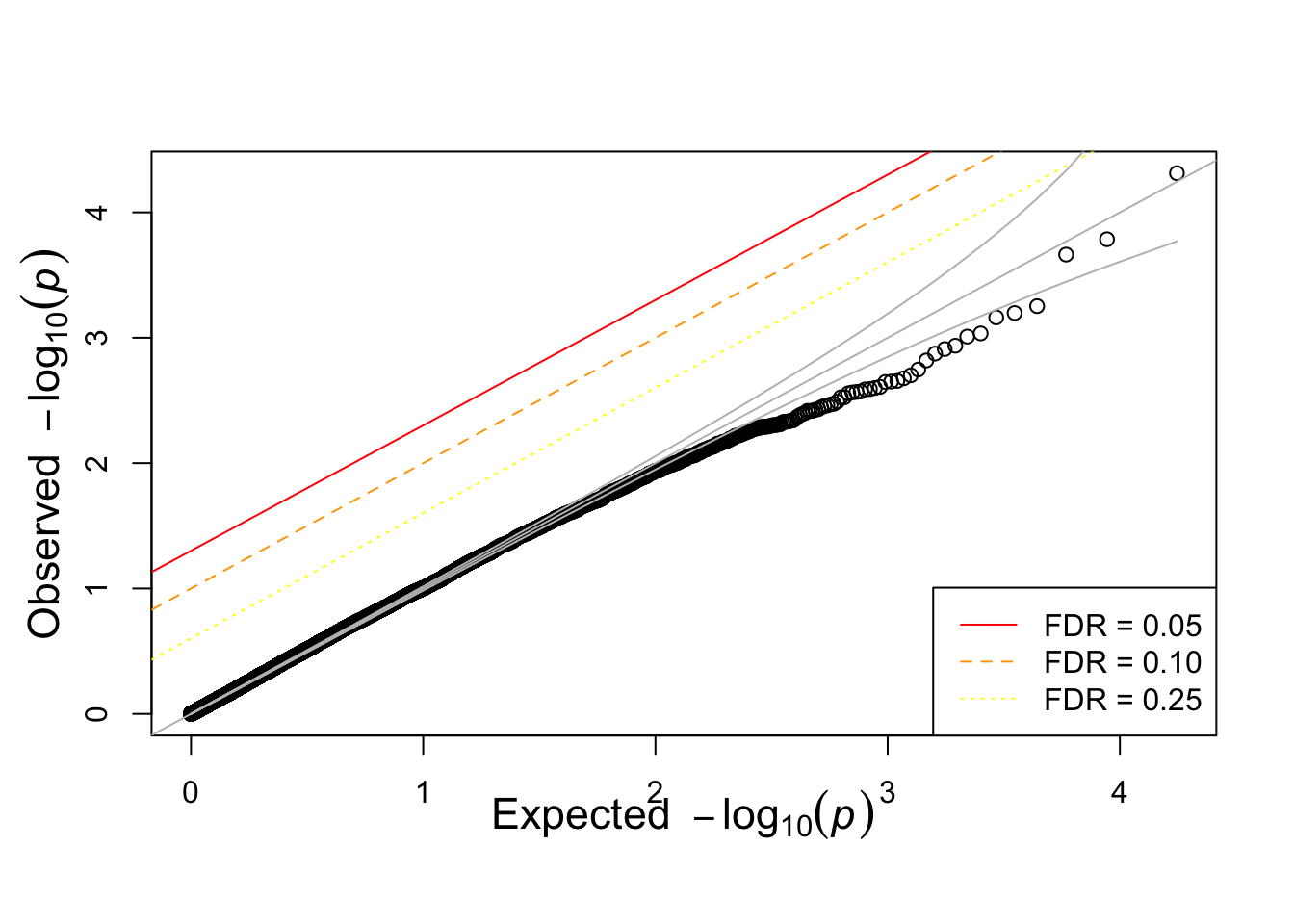
manhattan(simpheno.assoc, chr="CHR", bp="BP", snp="SNP", p="P" )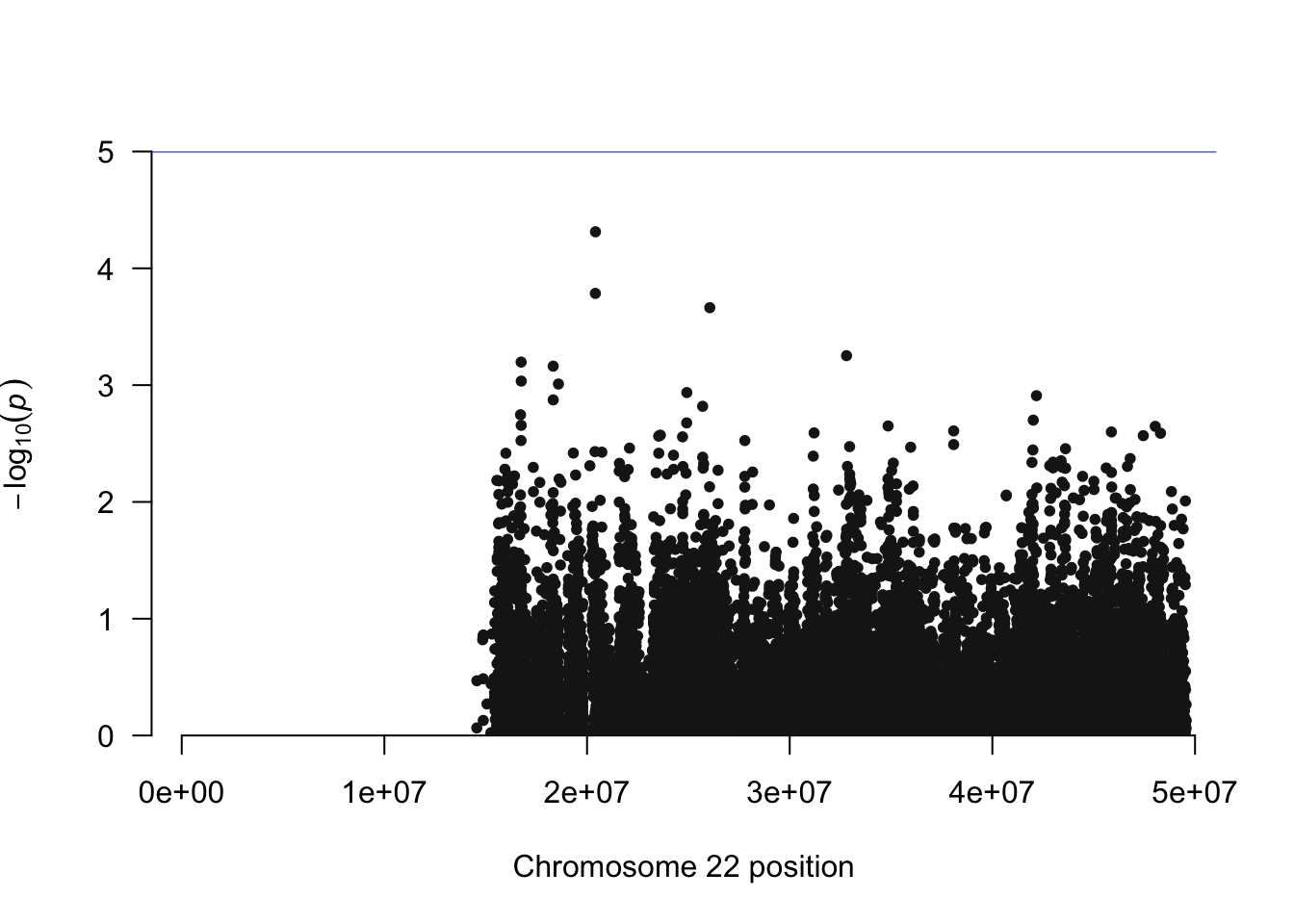
PCA calculation using plink
## generate PCs using plink
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --pca --out {work.dir}output/pca"))
## read plink calculated PCs
pcplink = read.table(glue::glue("{work.dir}output/pca.eigenvec"),header=F, as.is=T)
names(pcplink) = c("FID","IID",paste0("PC", c(1:(ncol(pcplink)-2))) )
pcplink = popinfo %>% left_join(superpop,by=c("population"="Population")) %>% inner_join(pcplink, by=c("FID"="FID", "IID"="IID"))
## plot PC1 vs PC2
pcplink %>% ggplot(aes(PC1,PC2,col=population,shape=SuperPop)) + geom_point(size=3,alpha=.7) + theme_bw(base_size = 15)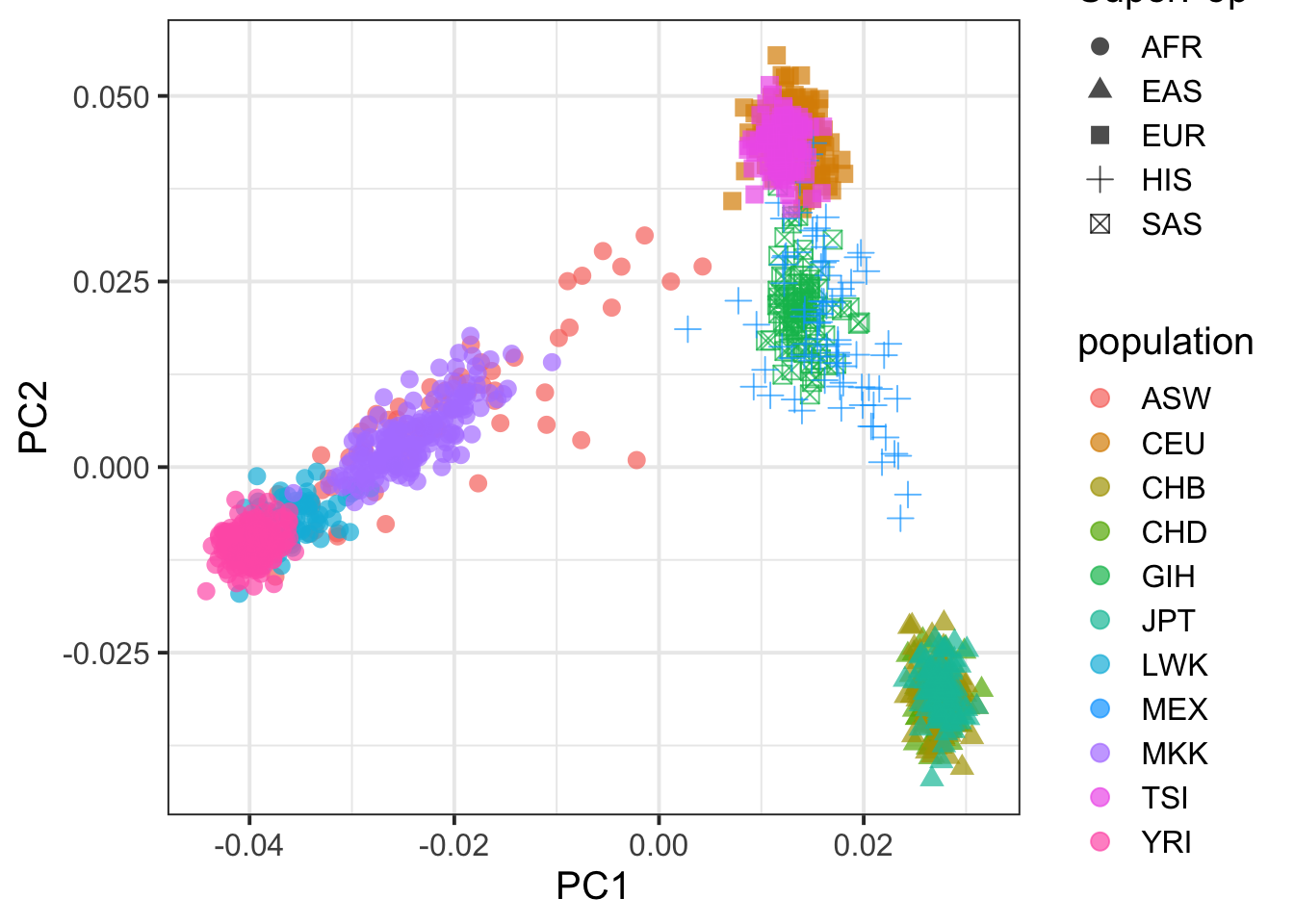
manhattan(simpheno.assoc, chr="CHR", bp="BP", snp="SNP", p="P" ,main="Simulated Phenotype")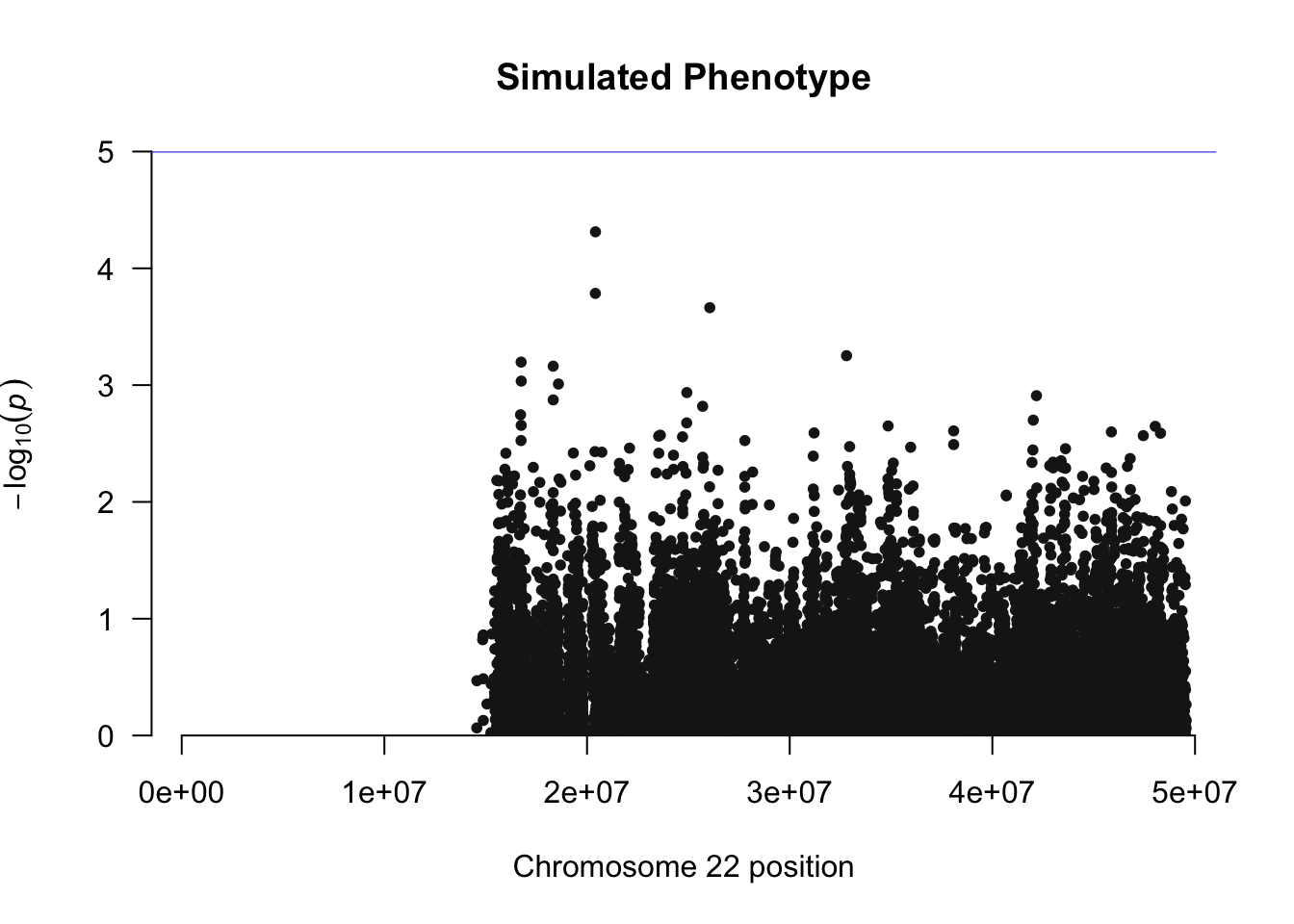
runnig igrowth GWAS using PCs
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --linear --pheno {work.dir}igrowth.pheno --pheno-name growth --covar {work.dir}output/pca.eigenvec --covar-number 1-4 --maf 0.05 --out {work.dir}output/igrowth-adjPC"))
igrowth.assoc = read.table(glue::glue("{work.dir}output/igrowth-adjPC.assoc.linear"),header=T,as.is=T)
indadd = igrowth.assoc$TEST=="ADD"
titulo = "igrowh association adjusted for PCs"
hist(igrowth.assoc$P[indadd],main=titulo)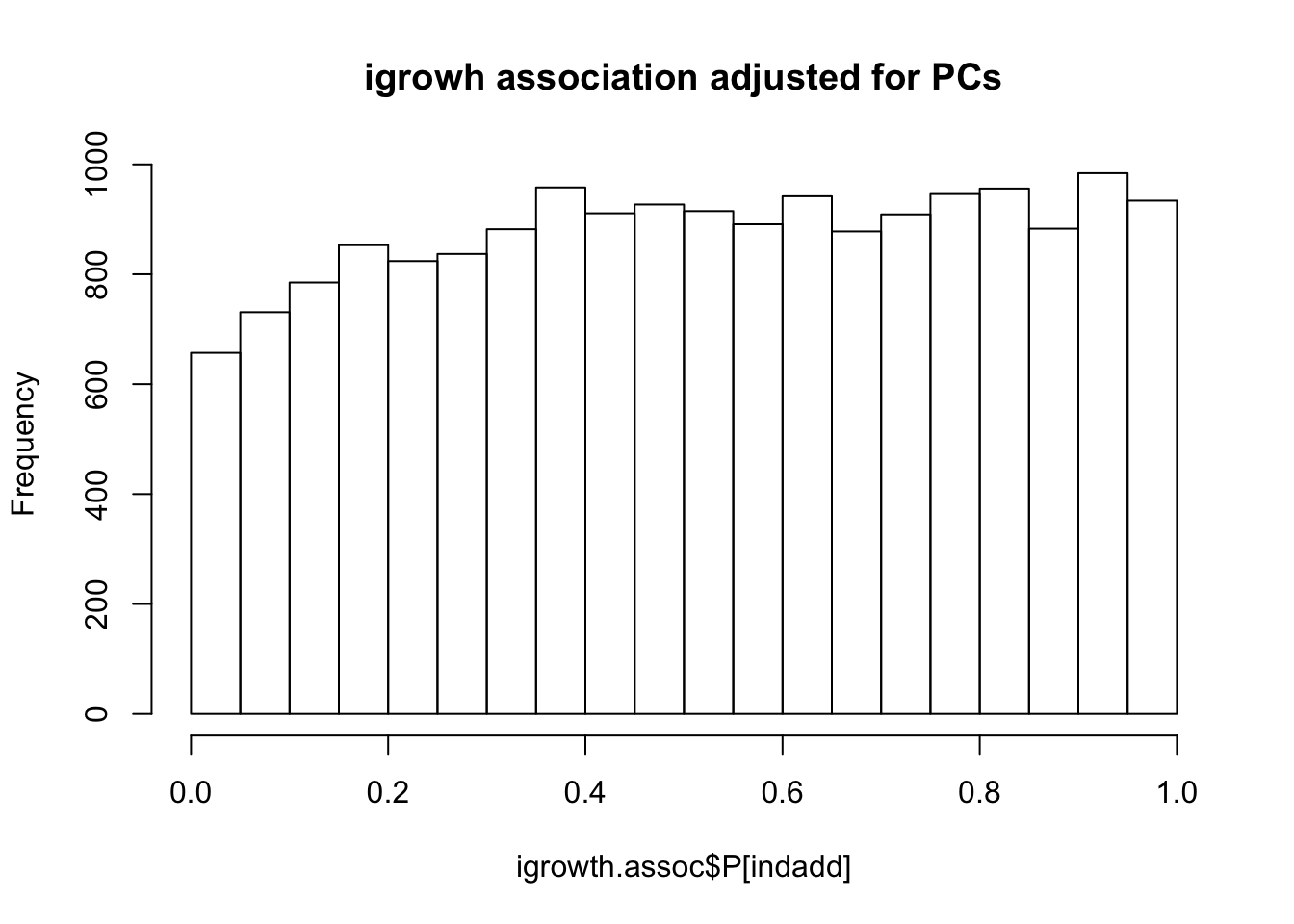
qqunif(igrowth.assoc$P[indadd],main=titulo)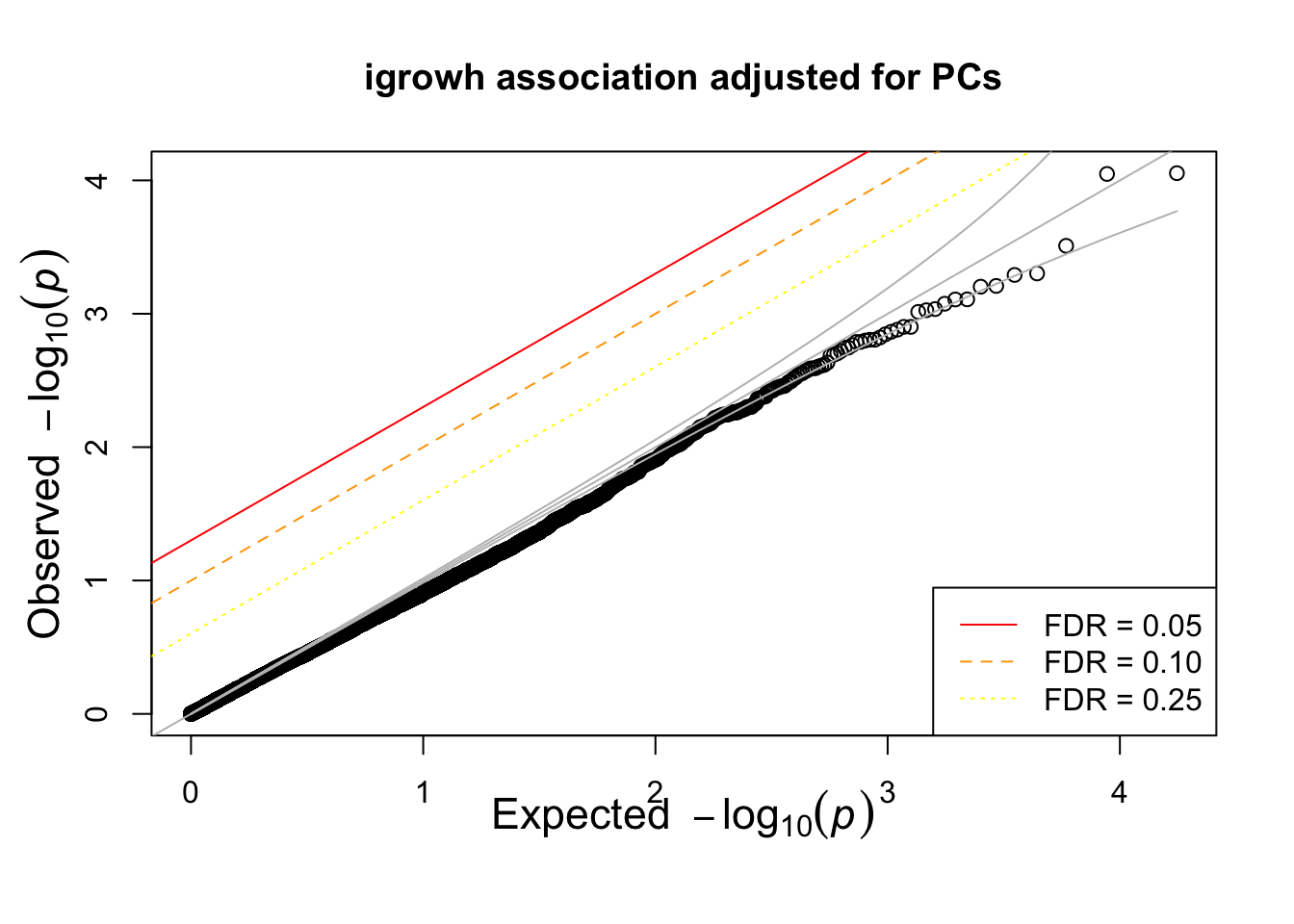
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] qqman_0.1.4 forcats_0.4.0 stringr_1.4.0 dplyr_0.8.1
[5] purrr_0.3.2 readr_1.3.1 tidyr_0.8.3 tibble_2.1.2
[9] ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 xfun_0.7 haven_2.1.0 lattice_0.20-38
[5] colorspace_1.4-1 generics_0.0.2 vctrs_0.1.0 htmltools_0.4.0
[9] yaml_2.2.0 utf8_1.1.4 rlang_0.4.1 pillar_1.4.1
[13] glue_1.3.1 withr_2.1.2 calibrate_1.7.5 modelr_0.1.4
[17] readxl_1.3.1 plyr_1.8.4 munsell_0.5.0 gtable_0.3.0
[21] workflowr_1.3.0 cellranger_1.1.0 rvest_0.3.4 evaluate_0.14
[25] labeling_0.3 knitr_1.23 curl_3.3 fansi_0.4.0
[29] broom_0.5.2 Rcpp_1.0.2 scales_1.0.0 backports_1.1.4
[33] jsonlite_1.6 fs_1.3.1 hms_0.4.2 digest_0.6.19
[37] stringi_1.4.3 grid_3.6.0 rprojroot_1.3-2 cli_1.1.0
[41] tools_3.6.0 magrittr_1.5 lazyeval_0.2.2 crayon_1.3.4
[45] pkgconfig_2.0.2 zeallot_0.1.0 MASS_7.3-51.4 xml2_1.2.0
[49] lubridate_1.7.4 assertthat_0.2.1 rmarkdown_1.13 httr_1.4.0
[53] rstudioapi_0.10 R6_2.4.0 nlme_3.1-139 git2r_0.25.2
[57] compiler_3.6.0